Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.2A(2013), Article ID:31347,9 pages DOI:10.4236/ojemd.2013.32A001
Mitochondrial Translation in Health and Disease
1Instituto do Cerebro—Hospital Israelita Albert Einstein, São Paulo, Brazil
2Departamento de Microbiologia—Instituto de Ciências Biomedicas—Universidade de São Paulo, São Paulo, Brazil
Email: mariohb@usp.br
Copyright © 2013 Claudia C. Ferreiro-Barros, Mario H. Barros. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 1, 2013; revised April 8, 2013; accepted May 10, 2013
Keywords: Mitochondrial Diseases; Respiratory Chain
ABSTRACT
Mitochondrial disorders have become the most common cause of inborn errors of metabolism. Impairments in mitochondrial protein synthesis are one of the causes of these diseases, which are clinically and genetically heterogeneous. The mitochondrial translation machinery decodes 13 polypeptides essential for the oxidative phosphorylation process. Mitochondria protein synthesis depends on the integrity of mitochondrial rRNAs and tRNAs genes, and at least one hundred of nuclear encoded products. Diseases caused by mutations in mitochondrial genes as well as in ribosomal proteins, translational factors, RNA modifying enzymes, and all other constituents of the translational machinery have been described in patients with combine respiratory chain deficiency, and are the object of this review.
1. Introduction
Most of the energy needed by human cells is provided by the mitochondrial oxidative phosphorylation (OXPHOS) process. OXPHOS couples energy of the electrochemical proton gradient generated by the respiratory complexes (complex I, III and IV) across the mitochondrial inner membrane to produce ATP through ATP synthase (complex V). Impairments in OXPHOS are generically recognized as mitochondrial disorders and have become the most common cause of inborn errors of metabolism [1,2] with a prevalence estimated at 1/5000 birth [3]. Mitochondrial disorders are clinically and genetically heterogeneous. The high incidence and broad heterogeneity stems from the dual genetic origin of the respiratory complexes. Indeed, respiratory complexes (I, III, IV and V) and mitochondrial ribosomes are the best examples of cellular components whose assembly depends on two genomes; mitochondrial DNA (mtDNA) contributes with 13 polypeptides in the assembly of respiratory complexes whereas 74 proteins constituents of these complexes are nuclear encoded. During the evolutionary process mitochondria lost most of mtDNA to the nucleus, however a small molecule persist to exit in nearly all eukaryotes. It is assumed that changes in the mitochondrial genetic code have prevented further loss of mtDNA to the nucleus. In the mitochondrial translation system AUA codes for methionine, UGA codes for tryptophan, AGA and AGG are used as stop codons. Mammalian mtDNA encoded proteins comprehend: ND1-ND6 subunits of complex I, cytochrome b in complex III, COX1, COX2 and COX3, subunits of complex IV and ATP6 and ATP8 in complex V. Moreover, mtDNA encodes 12S rRNA, 16S rRNA and 22 tRNAs [4].
Therefore, according to their inheritance, mitochondrial disorders have been divided in two groups [5]: 1) Mutations in the mtDNA. 2) Mutations in the nuclear DNA.
In contrast to nuclear genes, mtDNA molecules are present in hundreds or thousands of copies in each cell and are maternally inherited. A deleterious mutation in mtDNA usually affects some but not all genomes, such that cells and tissues will have two populations of mtDNA, a situation known as heteroplasmy. If the number of mutant mtDNA molecules reaches a certain threshold, tissue dysfunction and clinical signs will be manifest. This threshold between health and disease will be lower in tissues with high energetic demand such as brain, muscle and heart [6]. Moreover, mitochondria are not equally distributed at cell division and so the proportion of mutant mtDNA may shift in the daughter cells.
mtDNA mutations have been described since 1988 when the first pathogenic mutations were discovered [7]. Abnormalities in mtDNA inheritance and maintenance are also transmitted as mendelian traits because mtDNA depends on nuclear gene products for its expression and stability, and deficiencies in these products lead to depletion and deletions of mtDNA. These disorders have been classified as defects of intergenomic communication because mutations of the nuclear DNA disrupt the integrity of mtDNA [8].
Nevertheless, mitochondrial disorders can arise from more than 1200 different nuclear genes mutations that directly or indirectly affect OXPHOS [9]. For instance, more than three hundred proteins are directly required as auxiliary and chaperones that help the assembly of mature complexes [10]. Mitochondria are also important for the proper exchange of metabolites across the organelle membranes, in the biosynthesis of aminoacids, vitamins, lipids, prostetic groups (metals, heme, iron-sulfur clusters), ion homeostasis, and many other intermediates which are also required for OXPHOS and cell viability in general, deficiencies in any part of this intricate metabolism may cause disease.
2. Mitochondrial Gene Expression
The regulation of mtDNA expression is completely dependent on nuclear genes but the mechanisms are not fully understood [11,12]. Nevertheless, it can be regulated at different levels: mtDNA copy number; transcripttion initiation; mRNA stability; translation initiation; and coordination of respiratory complexes assemblies and translation of newly synthesized polypeptides [13].
2.1. Mitochondrial Transcription
Human mtDNA genes are arranged an end to end disposition with little intergenic regions, their transcription occurs in both mtDNA strands generating polycistronic RNAs, which are processed into rRNAs, tRNAs and mRNAs [4]. Mitochondrial gene expression relies on three promoters to generate the respective polycistronic RNAs. Mitochondrial RNA polymerase is responsible for transcribing the mitochondrially encoded genes, it requires transcription factors such as TFAM, TFB1M and TFB2M [14,15]. TFAM is essential both for transcription initiation [15-18] and it is present in mitochondrial nucleoid controlling mtDNA copy number control [19,20]. TFAM packages mtDNA into a compact protein-DNA structure termed the nucleoid [20]. Mature mRNAs are oligo or polyadenylated at the 3’ end. Terminal 3’ oligoadenylation of mitochondrial mRNAs is essential, because processing of several mitochondrial open reading frames from the primary polycistronic transcript leaves a truncated terminal codon that requires adenylation for completion [21]. Polyadenylation is necessary to mRNAs stability and to direct them to mitochondrial ribosomes for translation [22,23]. In contrast to bacterial mRNAs, mitochondrial transcripts lack Shine-Dalgarno sequences.
2.2. Mitochondrial Translation
The mitochondrial translation machinery involves mtDNA encoded RNAs and a large number of nuclear products that can be categorized into proteins necessary for ribosomes assembly; aminoacyl tRNA synthetases; tRNA modifying enzymes; initiation factors (IF2-mt and IF- 3-mt); elongation factors (EFG1, EFG2, EFT, and EFTu); termination and release factors, (mtRF1a, mtRF1, C12orf65 and ICT1); and ribosomal recycling factors (mtRRF1, mtRRF2) [24].
Mitochondrial mRNAs translation can be divided into four major steps: initiation, elongation, termination and ribosome recycling. The initial translation process starts at 5’ end with the initiation codon for N-formylmethionine [25]. Two initiation factors have been identified in mammalian mitochondria: IF-2 (IF-2mt) stimulates formylmethionine-tRNA binding to the small ribosomal subunit (28S) in the presence of mRNA [26,27], whereas IF-3 (IF-3mt) promotes the dissociation of mitochondrial 55S ribosomes and stimulates initiation complex formation [28].
2.2.1. tRNA Metabolism
Although mitochondria have a complete set of tRNAs, they recognize a broad set of codons [29]. Nevertheless, mitochondrial tRNAs need to be processed and edited [30] before their recognition by specifics aminoacyl-tRNA synthetases present in the organelle [31]. Aminoacyl tRNA synthetases catalyze acylation of tRNA with their cognates amino acids, but in some cases non-discriminating attachment can occur [32]. For instance, the synthesis of glutaminyl-tRNA involves a non discriminating glutamyl tRNA synthetase that charge tRNAGln with glutamic acid, generating glutamyl-tRNAGln. The transamidation of glutamyl-tRNAGln into glutaminyl-tRNAGln is present in prokaryotes and eukaryotes organelles [33-37]. Mitochondrial tRNAs are also processed and matured by RnaseP and RNaseZ, respectively involved in the processing of 5’ and 3’ ends [30], as well as editing enzymes, such as PUS1, MTO1, TRMU and MTFMT. PUS1 is a pseudouridine synthase; pseudouridylation facilitates base-pairing within the t-RNA secondary structure [38], MTO1 catalyzes the 5-carboxymethylaminomethylation of the wobble uridine base on tRNALys, tRNAGlu, tRNAGln, which improves the fidelity in the protein synthesis process [39]. Likewise, TRMU is responsible for the 2-thiolation of the wobble U in tRNALys, tRNAGlu, tRNAGln [40]. MTFMT is responsible for the formylation of methionine-tRNAMet and therefore requested for translation initiation [41].
2.2.2. Ribosomes Biogenesis
The processed mRNAs are translated in mitochondrial ribosomes, interacting with the mitochondrial inner membrane to ensure the insertion of the newly synthesize polypeptides [42]. In yeast several proteins have already been shown to facilitate this interaction [24]. Nevertheless, mitochondrial ribosomes have preserved some prokaryotic characteristics such as sensitivity to amynoglycosides chloroamphenicol and erythromycin [43]. Moreover, about half of mitochondrial ribosome proteins have bacterial homologues [44]. However, in human, they present lower rRNA and higher protein content; because of that, even with a 55S sedimentation coefficient they are bigger than 70S bacterial ribosomes. The small and large subribosomal particles have sedimentation coefficients of 28S and 39S, respectively [45]. The small 28S subunit is constituted of 12S rRNA and 30 proteins while the large 39S subunit constitutes of 16S rRNA and 48 proteins [46,47]. Not unlike the respiratory complexes, assembly of mitochondrial ribosomes depends on the coordination of nuclear gene products and mtDNA products a process that is also assisted by other chaperones and regulatory proteins, however little is known about them, in yeast the mitochondrial GTPases 1, 2 and 3 (Mtg1, Mtg2, and Mtg3) assist the assembly of large and small subunit [48-51]. Human Mtg1, ObhH1, C7orf30 are involved in the assembly of the large subunit [51,52], whereas Era-like1 and C4orf14 in the assembly of the small subunit [53,54].
2.2.3. Translational Process
Mitochondrial translation process also requires specific proteins that recognize their respective mRNAs to activate translation [55]. In yeast translational activators recognize 5’-UTRs of mitochondrial mRNA in a substrate specific manner, promoting a new level for mitochondrial protein synthesis regulation. Curiously, these activators can be part of feedback control loops, allowing translation only when the assembly of the respective respiratory complex is being correctly performed [13].
The elongation factors: EFTu, EFTs, EFG are necessary for the sequential addition of amino acids to the growing polypeptide chain. EFTu associate with all aminoacyl-tRNA substrates, discriminating those mischarged [56]. The aminoacyl-tRNA is delivered to the ribosome “A” site complexed with EFTu and GTP; after codon-anticodon interaction and consequent GTP hydrolysis, EFTu-GDP is recycled by EFTs. EFGs catalyzes the translocation step in which the deacylated tRNA in the “P” site is moved to the exit site (“E”) [24]. Only two stop codons terminate translation in human mitochondria, either UAG and UAA are recognized by the release factors RF1a/RF1L, which promote hydrolysis of the ester bond between the tRNA the newly synthesized polypeptide [24]. Three putative ribosomes recycling factors RRF1, RRF2 and C12orf65 have been identified in mitochondria being responsible for the release of ribosomes from mRNA [57].
3. Mitochondrial Protein Synthesis Deficiencies
Mitochondrial protein synthesis deficiencies stems from mutations in any part of the mitochondrial translational machinery discussed above, including tRNAs, rRNAs (mtDNA encoded), as well as from mutations in nuclear genes involved in the process.
3.1. MtDNA Mutations: tRNA and rRNA Genes
Complete loss of any of the mitochondrial rRNAs or tRNAs should be lethal. Milder mutations causing decreased stability of tRNAs or rRNAs, should decrease mitochondrial translation and cause disease [58].
Mutations in all mitochondrial tRNAs have been associated with disease, and represent a large proportion of mitochondrial disorders. More than 150 mutations in tRNA gene have already been described [59,60]. For instance, tRNA mutations account for most of mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS) and for syndrome myloclonus epilepsy with ragged red fibers known as MERRF cases [58].
MELAS is a progressive neurodegenerative disorder with typical onset between the ages of 2 and 15, it can occur in infancy, or as late as adulthood. Stroke-like episodes, often accompanied by seizures, are the hallmark symptom of MELAS and cause partial paralysis, loss of vision, and focal neurological defects. The disease is maternally inherited and there are at least 17 different mutations that can cause MELAS, by far the most prevalent is the A3243G mutation in the tRNALeu(UUR) gene, which is responsible for about 80% of the cases and it is also present in patients with progressive external ophtalmoplegia (PEO) [58]. The A3243 mutation is located within a tridecamer sequence, which binds the transcription terminator factor mTERF. The binding of mTERF at this site increases the transcription of 12S and 16S rRNA genes [61].
Mutations in tRNALEU(UUR) reduce both the aminoacylation of tRNALEU(UUR) as well as affect posttranslational modifications in the wobble position of tRNALEU(UUR) reducing the transcription levels of mitochondria-encoded proteins. The reduced aminoacylation affects the decoding of UUG and UUA while the wobble defect specifically diminishes UUG deconding. Curiously, the wobble defect specifically alters the expression of ND6 mitochondria-encoded protein [62].
MERRF syndrome is characterized by progressive myoclonus, cerebellar ataxia, and muscle weakness. Although a few cases of MERRF are sporadic, most cases are maternally inherited due to A8344G missense mutation in tRNALys, [58]. Absence of ophtalmoplegia, pigmentary retinopathy and heart block and lack of myloclonus distinguish MELAS from MERRF.
MELAS, MERRF and other systemic neuromuscular syndromes such as Kearns-Sayre, frequently have hearing loss as one of their clinical signs. Nevertheless, the typical mutation found in MELAS cases can lead to hearing loss without any other neurological symptom present [63]. Curiously, while for most mtDNA mutations the homoplasmic state would be lethal as discussed above, homoplasmy of mutant mtDNA is observed in maternally inherited hearing loss. The homoplasmic A1555G mutation in the mitochondrial 12S rRNA gene has been found to be associated with non-syndromic deafness and aminoglycoside-induced deafness [64]. In the absence of aminoglycosides, the A1555G mutation produces a clinical phenotype that ranges from severe congenital deafness through moderate progressive hearing loss of later onset to completely normal hearing [64]. Therefore the nuclear background should be determinant in the expression of the different phenotypes; accordingly, the activity of mitochondrial RNA polymerase transcription factor TFBM1 has been suggested to alter the penetrance of the deafness phenotype [65].
3.2. Nuclear Mutations: Proteins Involved in the Translation Machinery
Nuclear gene products account for all proteins involved in the translation machinery. However, the number of mitochondrial translation impairments that lead to disease caused by genetically defined defects of nuclear genes is still small (Table 1).
3.2.1. Ribosome Biogenesis and Proteins
Altogether the mitochondrial ribosome subunits are constituted with 80 proteins [46,47], only 3 have been firmly linked to mitochondrial disease, MRPS16, MRPS22 and MRPL3 [60]. 12S rRNA surrounds MRPS16 protein in the ribosome small subunit. Consequently truncated versions of MRPS16 diminish the mitochondrial content of 12S rRNA. Patients with mutated MRPS16 was described with agenesis of corpous callosum , dysmorphism and fatal neonatal lactic acidosis with decreased complex I and IV activity in muscle [66]. MRPS22 has no bacterial homolog, but likewise MRPS16 it is located in the ribosome small subunit and affects 12S rRNA stability. One patient with MRPS22 homozygous mutation presented antenatal skin oedema, hypotonia, cardiomyopathy and tubulopathy [67].
Recently it was shown that patients with mutations in the RMND1 gene present impaired mitochondrial translation [68-70]. Defective RMND1 lead to lower steady state level of ribosome subunits MRPL13, MRPL32, MRPS2, as well as lower content of 16S rRNA indicating a role for RMND1 in the assembly or maintenance of mitochondrial ribosome [68,70].
3.2.2. Aminoacyl-tRNA Synthetases
Aminoacyl tRNA synthetases catalyze acylation of tRNA with their cognates amino acids in both cytoplasm and
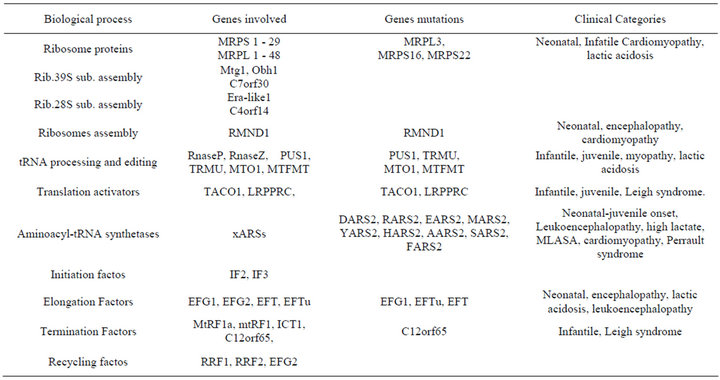
Table 1. Nuclear genes associated with the mitochondrial translational machinery.
mitochondria compartments. Mitochondria use 19 different Aminoacyl t-RNA synthetases, two of them are also present in the cytosol (glycyl-tRNA synthetase and lysil-tRNA synthetase), and the one missing is the glutaminyl-tRNA synthetase. As explained before, the synthesis of glutaminyl-tRNA involves a non discriminating glutamyl tRNA synthetase that charge tRNAGln with glutamic acid, generating glutamyl-tRNAGln which is modified to glutaminyl-tRNA after a transamidation reaction [35]. Patients with deleterious mutation in mitochondrial arginyl-tRNA synthetase (RARS2), aspartyl-tRNA synthetase (DARS2), seryl-tRNA synthetase (SARS2), tyrosyl-tRNA synthetase (YARS2) and histidyl-tRNA synthetase (HARS2) have already been described [71-75]. Their clinical conditions are very heterogeneous, with symptoms ranging from pontocerebellar hypoplasia, leukoencephalopathy, hyperuricemia, pulmonary hypertension, renal failure, alkalosis, ovarian dysgenesis, sensorineural hearing loss, HUPRA and MLASA syndromes [76].
3.2.3. Translational Activators
TACO1 and LRPPRC are the only mitochondrial translational activators already described in human. Both are COX1 activators and have been identified in human patients with Leigh syndrome [77,78]. Considering the number of translational activators for mitochondrial encoded polypeptides already described in other organisms such as Saccharomyces cerevisiae as well as their importance in coordinate translation and the modular assembly of the respiratory complexes (Costanzo and Fox, 1988; Herrmann et al., 2011) it is expected that our knowledge of human translational activators will increase in the next years with the massive parallel sequencing of patients.
3.2.4. Translational Factors
Patients with mutations in the elongation factors EFG1, EFTs, EFTu, [79-82] and in the termination factor C12orf65 have already been described [57]. Mutations of these genes are relatively rare and result in globally decreased mitochondrial translation in cultured skin fibroblasts of patients [59]. Curiously, an EFG1 mutation was associated with combined decrease of activity in the respiratory complexes in fibroblasts but not in muscle [83]. C12orf65 mutations were detected in patients with encephalomyopathy and the gene product seem to play a role in recycling abortive peptidyl-tRNA species release from the ribosome during the elongation phase of translation [57].
3.2.5. tRNA Modifying Enzymes
Nuclear products that affect mitochondrial tRNAs processing can also cause disease, patients with mutations in PUS1, TRMU, MTO1 and MTFMT have been recently described [38,41,84-86]. PUS1 mutations were identified in patients with mitochondrial myopathy and sideroblastic anemia (MLASA) [84]. TRMU and MTO1 mutations affect the penetrance of A1555G mutation in the mitochondrial 12S rRNA gene [65]. However, mutations in these two genes can also cause more severe clinical conditions. TRMU patients with fatal infantile liver failure [41] and MTO1 patients with cardiomyopathy and lactic acidosis have been described recently [85]. Finally MTFMT mutations were identified in children with Leigh syndrome and impaired mitochondrial translation initiation [86].
4. Conclusion and Prospects
Impairment of mitochondrial protein synthesis diseases is an important medical area to be studied. Defects in mitochondrial translation produce a diverse clinical phenoltype, with the most severe presenting in infancy as metabolic disorders. In the last years, the proteomic studies and high throughput gene sequencing analyses have succeeded in identifying new factors involved in mitochondrial translation.
5. Acknowledgements
This work was supported by grants and fellowships from the Fundação de Amparo a Pesquisa de São Paulo (FAPESP) (CF-grant 2010/51924-0; MB-grant 2011/07 366-5), Conselho Nacional de Desenvolvimento Cient- ífico e Tecnológico (CNPq).
REFERENCES
- E. J. Tucker, A. G. Compton, S. E. Calvo and D. R. Thorburn, “The Molecular Basis of Human Complex I Deficiency,” IUBMB Life, Vol. 63, No. 9, 2011, pp. 669- 677.
- L. M. Cree, D. C. Samuels and P. F. Chinnery, “The Inheritance of Pathogenic Mitochondrial DNA Mutations,” Biochimica et Biophysica Acta, Vol. 1792, No. 12, 2009, pp. 1097-1102. doi:10.1016/j.bbadis.2009.03.002
- D. R. Thorburn, “Mitochondrial Disorders: Prevalence, Myths and Advances,” Journal of Inherited Metabolic Disease, Vol. 27, No. 3, 2004, pp. 349-362. doi:10.1023/B:BOLI.0000031098.41409.55
- S. Anderson , A. T. Bankier, B. G. Barrell, M. H. de Bruijn, A. R. Coulson, J. Drouin, I. C. Eperon, D. P. Nierlich, B. A. Roe, F. Sanger, P. H. Schreier, A. J. Smith, R. Staden and I. G. Young, “Sequence and Organization of the Human Mitochondrial Genome,” Nature, Vol. 290, No. 5806, 1981, pp. 457-465. doi:10.1038/290457a0
- S. DiMauro, E. Bonilla, M. Davidson, M. Hirano and E. A. Schon, “Mitochondria in Neuromuscular Disorders,” Biochimica et Biophysica Acta, Vol. 1366, No. 1-2, 1998, pp. 199-210. doi:10.1016/S0005-2728(98)00113-3
- S. DiMauro, E. Bonilla and D. C. De Vivo, “Does the Patient Have a Mitochondrial Encephalomyopathy?” Journal of Child Neurology, Vol. 14, No. 1, 1999, pp. S23- S35.
- I. J. Holt, A. E. Harding and J. A. Morgan-Hughes, “Deletions of Muscle Mitochondrial DNA in Patients with Mitochondrial Myopathies,” Nature, Vol. 331, No. 6158, 1988, pp. 717-719. doi:10.1038/331717a0
- M. Hirano and T. H. Vu, “Defects of Intergenomic Communication: Where Do We Stand?” Brain Pathology, 2000 Vol. 10, No. 3, 2000, pp. 451-461. doi:10.1111/j.1750-3639.2000.tb00277.x
- C. Scharfe, H. H. Lu, J. K. Neuenburg, E. A. Allen, G. C. Li, T. Klopstock, T. M. Cowan, G. M. Enns and R. W. Davis, “Mapping Gene Associations in Human Mitochondria Using Clinical Disease Phenotypes,” PLoS Computational Biology, Vol. 5, 2009, Article ID: e1000374. doi:10.1371/journal.pcbi.1000374
- A. Tzagoloff and C. L. Dieckmann, “PET Genes of Saccharomyces Cerevisiae,” Microbiology Review, Vol. 54, No. 3, 1990, pp. 211-225.
- M. Falkenberg, N. G. Larsson and C. M. Gustafsson, “DNA Replication and Transcription in Mammalian Mitochondria,” Annual Review of Biochemistry, Vol. 76, 2007, pp. 679-699. doi:10.1146/annurev.biochem.76.060305.152028
- D. A. Clayton, “Transcription and Replication of Animal Mitochondrial DNAs,” International Review of Cytology, Vol. 141, 1992, pp. 217-232. doi:10.1016/S0074-7696(08)62067-7
- J. M. Herrmann, M. W. Woellhaf and N. Bonnefoy, “Control of Protein Synthesis in Yeast Mitochondria: The Concept of Translational Activators,” Biochimica et Biophysica Acta, Vol. 1833, No. 2, 2013, pp. 286-294. doi:10.1016/j.bbamcr.2012.03.007
- R. P. Fisher and D. A. Clayton, “A Transcription Factor Required for Promoter Recognition by Human Mitochondrial RNA Polymerase. Accurate Initiation at the Heavyand Light-Strand Promoters Dissected and Reconstituted in Vitro,” Journal of Biological Chemistry, Vol. 260, No. 20, 1985, pp. 11330-11338.
- M. Falkenberg, M. Gaspari, A. Rantanen, A. Trifunovic, N. G. Larsson and C. M. Gustafsson, “Mitochondrial Transcription Factors B1 and B2 Activate Transcription of Human mtDNA,” Nature Genetics, Vol. 31, No. 3, 2002, pp. 289-294. doi:10.1038/ng909
- H. B. Ngo, J. T. Kaiser and D. C. Chan, “The Mitochondrial Transcription and Packaging Factor Tfam Imposes a U-Turn on Mitochondrial DNA,” Nature Structural and Molecular Biology, Vol. 18, No. 11, 2011, pp. 1290-1296. doi:10.1038/nsmb.2159
- A. Rubio-Cosials, J. F. Sidow, N. Jiménez-Menéndez, P. Fernández-Millán, J. Montoya, H. T. Jacobs, M. Coll, P. Bernadó and M. Solà, “Human Mitochondrial Transcription Factor A Induces a U-Turn Structure in the Light Strand Promoter,” Nature Structural and Molecular Biology, Vol. 18, No. 11, 2011, pp. 1281-1289. doi:10.1038/nsmb.2160
- Y. Shi, A. Dierckx, P. H. Wanrooij, S. Wanrooij, N. G. Larsson, L. M. Wilhelmsson, M. Falkenberg and C. M. Gustafsson, “Mammalian Transcription Factor A is a Core Component of the Mitochondrial Transcription Machinery,” Proceedings of the National Academy of Science of USA, Vol. 109, No. 41, 2012, pp. 16510-16515. doi:10.1073/pnas.1119738109
- M. I. Ekstrand, M. Falkenberg, A. Rantanen, C. B. Park, M. Gaspari, K. Hultenby, P. Rustin, C. M. Gustafsson and N. G. Larsson, “Mitochondrial Transcription Factor A Regulates mtDNA Copy Number in Mammals,” Human Molecular Genetics, Vol. 13, No. 9, 2004, pp. 935-944.
- C. Kukat, C. A. Wurm, H. Spåhr, M. Falkenberg, N. G. Larsson and S. Jakobs, “Super-Resolution Microscopy Reveals that Mammalian Mitochondrial Nucleoids Have a Uniform Size and Frequently Contain a Single Copy of mtDNA,” Proceedings of the National Academy of Science of USA, Vol. 108, No. 33, 2011, pp. 13534-13539. doi:10.1073/pnas.1109263108
- D. Ojala, S. Crews, J. Montoya, R. Gelfand and G. Attardi, “A Small Polyadenylated RNA (7 S RNA), Containing a Putative Ribosome Attachment Site, Maps near the Origin of Human Mitochondrial DNA Replication,” Journal of Molecular Biology, Vol. 150, No. 2, 1981, pp. 303-314. doi:10.1016/0022-2836(81)90454-X
- T. Nagaike, T. Suzuki, T. Katoh and T. Ueda, “Human Mitochondrial mRNAs Are Stabilized with Polyadenylation Regulated by Mitochondria-Specific Poly(A) Polymerase and Polynucleotide Phosphorylase,” Journal of Biological Chemistry, Vol. 280, No. 20, 2005, pp. 19721- 19727. doi:10.1074/jbc.M500804200
- M. Wydro, A. Bobrowicz, R. J. Temperley and R. N. Lightowlers, “Chrzanowska-Lightowlers ZM. Targeting of the Cytosolic Poly(A) Binding Protein PABPC1 to Mitochondria Causes Mitochondrial Translation Inhibition,” Nucleic Acids Research, Vol. 38, No. 11, 2010, pp. 3732-3742. doi:10.1093/nar/gkq068
- B. E. Christian and L. L. Spremulli, “Mechanism of Protein Biosynthesis in Mammalian Mitochondria,” Biochimica et Biophysica Acta, Vol. 1819, No. 9-10, 2012, pp. 1035-1054. doi:10.1016/j.bbagrm.2011.11.009
- J. Montoya, D. Ojala and G. Attardi, “Distinctive Features of the 5’-Terminal Sequences of the Human Mitochondrial mRNAs,” Nature, Vol. 290, No. 5806, 1981, pp. 465-470. doi:10.1038/290465a0
- H. X. Liao and L. L. Spremulli, “Identification and Initial Characterization of Translational Initiation Factor 2 from Bovine Mitochondria,” Journal of Biological Chemistry, Vol. 265, No. 23, 1990, pp.13618-13622.
- L. Ma and L. L. Spremulli, “Cloning and Sequence Analysis of the Human Mitochondrial Translational Initiation Factor 2 cDNA,” Journal of Biological Chemistry, Vol. 270, No. 4, 1995, pp. 1859-1865. doi:10.1074/jbc.270.4.1859
- E. C. Koc and L. L. Spremulli, “RNA-Binding Proteins of Mammalian Mitochondria,” Mitochondrion, Vol. 2, No. 4, 2003, pp. 277-291. doi:10.1016/S1567-7249(03)00005-9
- S. G. Bonitz, R. Berlani, G. Coruzzi, M. Li, G. Macino, F. G. Nobrega, M. P. Nobrega, B. E. Thalenfeld and Tzagoloff, “Codon Recognition Rules in Yeast Mitochondria,” Proceedings of the National Academy of Science of USA, Vol. 77, No. 6, 1980, pp. 3167-3170. doi:10.1073/pnas.77.6.3167
- E. Vilardo, C. Nachbagauer, A. Buzet, A. Taschner, J. Holzmann and W. A. Rossmanith, “A Subcomplex of Human Mitochondrial RNase P Is a Bifunctional Methyltransferase—Extensive Moonlighting in Mitochondrial tRNA Biogenesis,” Nucleic Acids Research, Vol. 40, No. 22, 2012, pp. 11583-11593. doi:10.1093/nar/gks910
- A. Tzagoloff, D. Gatti and A. Gampel, “Mitochondrial Aminoacyl-tRNA Synthetases,” Progress in Nucleic Acid Research and Molecular Biology, Vol. 39, 1990, pp. 129- 158. doi:10.1016/S0079-6603(08)60625-X
- M. Wilcox and M. Nirenberg, “Transfer RNA as a Cofactor Coupling Amino Acid Synthesis with That of Protein,” Proceedings of the National Academy of Science of USA, Vol. 61, 1968, pp. 229-236. doi:10.1073/pnas.61.1.229
- J. Lapointe, L. Duplain and M. Proulx, “A Single Glutamyl-tRNA Synthetase Aminoacylates tRNAGlu and tRNAGln in Bacillus Subtilis and Efficientlymisacylates Escherichia Coli tRNAGln1 in Vitro,” Journal of Bacteriology, Vol. 165, 1986, pp. 88-93.
- C. Pujol, M. Bailly, D. Kern, L. Maréchal-Drouard, H. Becker and A. M. Duchêne, “Dual-Targeted tRNA-Dependent Amidotransferase Ensures Both Mitochondrial Andchloroplastic Gln-tRNAGln Synthesis in Plants,” Proceedings of the National Academy of Science of USA, Vol. 105, No. 17, 2008, pp. 6481-6485. doi:10.1073/pnas.0712299105
- M. Frechin, B. Senger, M. Brayé, D. Kern, R. P. Martin and H. D. Becker, “Yeast Mitochondrial Gln-tRNA(Gln) is Generated by a GatFAB-Mediated Transamidation Pathway Involving Arc1p-controlled Subcellular Sorting of Cytosolic GluRS,” Genes and Development, Vol. 23, 2009, pp.1119-1130. doi:10.1101/gad.518109
- A. Nagao, T. Suzuki, T. Katoh, Y. Sakaguchi and T. Suzuki, “Biogenesis of Glutaminyl-mt tRNAGln in Human Mitochondria,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 106, No. 38, 2009, pp. 16209-16214. doi:10.1073/pnas.0907602106
- M. H. Barros, M. Rak, J. A. Paulela and A. Tzagoloff. “Characterization of Gtf1p, the Connector Subunit of Yeast Mitochondrial tRNA-Dependent Amidotransferase,” The Journal of Biological Chemistry, Vol. 286, No. 38, 2011, pp. 32937-32947. doi:10.1074/jbc.M111.265371
- J. R. Patton, Y. Bykhovskaya, E. Mengesha, C. Bertolotto, N. Fischel-Ghodsian, “Mitochondrial Myopathy and Sideroblastic Anemia (MLASA): Missense Mutation in the Pseudouridine Synthase 1 (PUS1) Gene Is Associated with the Loss of tRNA Pseudouridylation,” The Journal of Biological Chemistry, Vol. 280, No. 20, 2005, pp. 19823-19828. doi:10.1074/jbc.M500216200
- X. Wang, Q. Yan and M. X. Guan, “Combination of the Loss of cmnm5U34 with the Lack of s2U34 Modifications of tRNALys, tRNAGlu, and tRNAGln Altered Mitochondrial Biogenesis and Respiration,” The Journal of Biological Chemistry, Vol. 395, No. 5, 2010, pp. 1038-1048. doi:10.1016/j.jmb.2009.12.002
- Q. Yan, Y. Bykhovskaya, R. Li, E. Mengesha, M. Shohat, X. Estivill, N. Fischel-Ghodsian and M. X. Guan, “Human TRMU Encoding the Mitochondrial 5-Methylaminomethyl-2-thiouridylate-methyltransferase Is a Putative Nuclear Modifier Gene for the Phenotypic Expression of the Deafness-Associated 12S rRNA Mutations,” Biochemical and Biophysical Research Communications, Vol. 342, No. 4, 2006, pp. 1130-1136. doi:10.1016/j.bbrc.2006.02.078
- A. Zeharia, A. Shaag, O. Pappo, A. M. Mager-Heckel, A. Saada, M. Beinat, O. Karicheva, H. Mandel, N. Ofek, R. Segel, D. Marom, A. Rötig, I. Tarassov and O. Elpeleg, “Acuteinfantile Liver Failure Due to Mutations in the TRMU Gene,” The American Journal of Human Genetics, Vol. 85, No. 3, 2009, pp. 401-417. doi:10.1016/j.ajhg.2009.08.004
- M. Liu and L. Spremulli, “Interaction of Mammalian Mitochondrial Ribosomes with the Inner Membrane,” The Journal of Biological Chemistry, Vol. 275, No. 38, 2000, pp. 29400-29406. doi:10.1074/jbc.M002173200
- A. Tzagoloff, “Genetic and Translational Capabilities of the Mitochondrion,” BioScience, Vol. 27, No. 1, 1977, pp. 18-23. doi:10.2307/1297789
- P. Smits, J. A. Smeitink, L. P. van den Heuvel, M. A. Huynen and T. J. Ettema, “Reconstructing the Evolution of the Mitochondrial Ribosomal Proteome,” Nucleic Acids Research, Vol. 35, No. 14, 2007, pp. 4686-4703. doi:10.1093/nar/gkm441
- T. W. O’Brien, “Properties of Human Mitochondrial Ribosomes,” IUBMB Life, Vol. 55, No. 9, 2003, pp. 505- 513. doi:10.1080/15216540310001626610
- E. C. Koc, W. Burkhart, K. Blackburn, A. Moseley, H. Koc and L. L. Spremulli, “A Proteomics Approach to the Identification of Mammalian Mitochondrial Small Subunit Ribosomal Proteins,” The Journal of Biological Chemistry, Vol. 275, No. 42, 2000, pp. 32585-32591. doi:10.1074/jbc.M003596200
- E. C. Koc, W. Burkhart, K. Blackburn, M. B. Moyer, D. M. Schlatzer, A. Moseley and L. L. Spremulli, “The Large Subunit of the Mammalian Mitochondrial Ribosome. Analysis of the Complement of Ribosomal Proteins Present,” The Journal of Biological Chemistry, Vol. 276, No. 47, 2001, pp. 43958-43969. doi:10.1074/jbc.M106510200
- A. Barrientos, D. Korr, K. J. Barwell, C. Sjulsen, C. D. Gajewski, G. Manfredi, S. Ackerman and A. Tzagoloff, “MTG1 Codes for a Conserved Protein Required for Mitochondrialtranslation,” Molecular Biology of the Cell, Vol. 14, No. 6, 2003, pp. 2292-2302. doi:10.1091/mbc.E02-10-0636
- K. Datta, J. L. Fuentes and J. R. Maddock, “The Yeast GTPase Mtg2p Is Required for Mitochondrial Translation and Partially Suppresses an rRNA Methyltransferase Mutant, mrm2,” Molecular Biology of the Cell, Vol. 16, No. 2, 2005, pp. 954-963. doi:10.1091/mbc.E04-07-0622
- M. F. Paul, G. M. Alushin, M. H. Barros, M. Rak and A. Tzagoloff, “The Putative GTPase Encoded by MTG3 Functions in a Novel Pathway for Regulating Assembly of the Small Subunit of Yeast Mitochondrial Ribosomes,” The Journal of Biological Chemistry, Vol. 287, No. 29, 2012, pp. 24346-24355. doi:10.1074/jbc.M112.363309
- T. Kotani, S. Akabane, K. Takeyasu, T. Ueda and N. Takeuchi, “Human G-Proteins, ObgH1 and Mtg1, Associate with the large Mitochondrial Ribosome Subunit and Are Involved in Translation and Assembly of Respiratory Complexes,” Nucleic Acids Research, Vol. 41, No. 6, 2013, pp. 3713-3722. doi:10.1093/nar/gkt079
- J. Rorbach, P. A. Gammage and M. Minczuk, “C7orf30 Is Necessary for Biogenesis of the Large Subunit of the Mitochondrial Ribosome,” Nucleic Acids Research, Vol. 40, No. 9, 2012, pp. 4097-4109. doi:10.1093/nar/gkr1282
- S. Dennerlein, A. Rozanska, M. Wydr, Z. M. Chrzanowska-Lightowlers and R. N. Lightowlers, “Human ERAL1 Is a Mitochondrial RNA Chaperone Involved in the Assembly of the 28S Small Mitochondrial Ribosomal Subunit,” Biochemical Journal, Vol. 430, No. 3, 2010, pp. 551-558. doi:10.1042/BJ20100757
- J. He , H. M. Cooper, A. Reyes, M. Di Re, L. Kazak, S. R. Wood, C. C. Mao, I. M. Fearnley, J. E. Walker and I. J. Holt, “Human C4orf14 Interacts with the Mitochondrial Nucleoid and Is Involved in the Biogenesis of the Small Mitochondrial Ribosomal Subunit,” Nucleic Acids Research, Vol. 40, No. 13, 2012, pp. 6097-6108. doi:10.1093/nar/gks257
- M. C. Costanzo and T. D. Fox, “Control of Mitochondrial Gene Expression in Saccharomyces cerevisiae,” Annual Review of Genetics, Vol. 24, No. 1, 1990, pp. 91-113. doi:10.1146/annurev.ge.24.120190.000515
- M. Stanzel, A. Schön and M. Sprinzl, “Discrimination against Misacylated tRNA by Chloroplast Elongation Factor Tu,” European Journal of Biochemistry, Vol. 219, No. 1-2, 1994, pp. 435-439. doi:10.1111/j.1432-1033.1994.tb19956.x
- H. Antonicka, E. Ostergaard, F. Sasarman, W. Weraarpachai, F. Wibrand, A. M. Pedersen, R. J. Rodenburg, M. S. van der Knaap, J. A Smeitink, et al., “Mutations in C12orf65 in Patients with Encephalomyopathy and a Mitochondrial Translation Defect,” The American Journal of Human Genetics, Vol. 87, No. 1, 2010, pp. 115-122. doi:10.1016/j.ajhg.2010.06.004
- N. G. Larsson and D. A. Clayton, “Molecular Genetic Aspects of Human Mitochondrial Disorders,” Annual Review of Genetics, Vol. 29, No. 1, 1995, pp. 151-178. doi:10.1146/annurev.ge.29.120195.001055
- A. Rötig, “Human Diseases with Impaired Mitochondrial Protein Synthesis,” Biochimica et Biophysica Acta, Vol. 1807, No. 9, 2011, pp. 1198-1205.
- [61] S. Pearce, C. L. Nezich and A. Spinazzola, “Mitochondrial Diseases: Translation Matters,” Molecular and Cellular Neuroscience, Vol. 55, 2012, pp. 1-12.
- [62] J. A. Morgan-Hughes and M. G. Hanna, “Mitochondrial Encephalomyopathies: The Enigma of Genotype versus Phenotype,” Biochimica et Biophysica Acta, Vol. 1410, No. 2, 1999, pp. 125-145. doi:10.1016/S0005-2728(98)00162-5
- [63] S. W. Schaffer, C. J. Jong, D. Warner, T. Ito and J. Azuma, “Taurine Deficiency and MELAS Are Closely Related Syndromes,” Advances in Experimental Medicine and Biology, Vol. 776, 2013, pp. 153-165. doi:10.1007/978-1-4614-6093-0_16
- [64] W. Reardon, “Genetic Deafness,” Journal of Medical Genetics, Vol. 29, No. 8, 1992, pp. 521-526. doi:10.1136/jmg.29.8.521
- [65] T. R. Prezant, J. V. Agapian, M. C. Bohlman, X. Bu, S. Oztas, W. Q. Qiu, et al., “Mitochondrial Ribosomal RNA Mutationassociated with Both Antibiotic-Induced and NonSyndromic Deafness,” Nature Genetics, Vol. 4, No. 3, 1993, pp. 289-294. doi:10.1038/ng0793-289
- [66] Y. Bykhovskaya, E. Mengesha, D. Wang, H. Yang, X. Estivill, M. Shohat and N. Fischel-Ghodsian, “Phenotype of Non-Syndromic Deafness Associated with the Mitochondrial A1555G Mutation Is Modulated by Mitochondrial RNA Modifying Enzymes MTO1 and GTPBP3,” Molecular Genetics and Metabolism, Vol. 83, No. 3, 2004, pp. 199-206. doi:10.1016/j.ymgme.2004.07.009
- [67] C. Miller, A. Saada, N. Shaul, N. Shabtai, E. Ben-Shalom, A. Shaag, E. Hershkovitz and O. Elpeleg, “Defective Mitochondrial Translation Caused by a Ribosomal Protein (MRPS16) Mutation,” Annals of Neurology, Vol. 56, No. 5, 2004, pp. 734-738. doi:10.1002/ana.20282
- [68] A. Saada, A. Shaag, S. Arnon, T. Dolfin, C. Miller, D. Fuchs-Telem, A. Lombes and O. Elpeleg, “Antenatal Mitochondrial Disease Caused by Mitochondrial Ribosomal Protein (MRPS22) Mutation,” Journal of Medical Genetics, Vol. 44, No. 12, 2007, pp. 784-786. doi:10.1136/jmg.2007.053116
- [69] C. C. Ferreiro-Barros, C. H. Tengan, M. H. Barros, L. Palenzuela, C. Kanki C. Quinzii, J. Lou, N. El Gharaby, A. Shokr, D. C. De Vivo, S. DiMauro and M. Hirano, “Neonatal Mitochondrial Encephaloneuromyopathy Due to a Defect of Mitochondrial Protein Synthesis,” Journal of the Neurological Sciences, Vol. 275, No. 1, 2008, pp. 128-132. doi:10.1016/j.jns.2008.08.028
- [70] B. Garcia-Diaz, M. H. Barros, S. Sanna-Cherchi, V. Emmanuele, H. O. Akman, C. C. Ferreiro-Barros, R. Horvath, S. Tadesse, N. El Gharaby, S. Dimauro, D. C. De Vivo, A. Shokr, M. Hirano and C. M. Quinzii, “Infantile Encephaloneuromyopathy and Defective Mitochondrial Translation Are Due to a Homozygous RMND1 Mutation,” The American Journal of Human Genetics, Vol. 91, No. 4, 2012, pp. 729-736. doi:10.1016/j.ajhg.2012.08.019
- [71] A. Janer, H. Antonicka, E. Lalonde, T. Nishimura, F. Sasarman, G. K. Brown, R. M. Brown, J. Majewski and E. A. Shoubridge, “An RMND1 Mutation Causes Encephalopathy Associated with Multiple Oxidative Phosphorylation Complex Deficiencies and a Mitochondrial Translation Defect,” The American Journal of Human Genetics, Vol. 91, No. 4, 2012, pp. 737-743. doi:10.1016/j.ajhg.2012.08.020
- [72] S. Edvardson, A. Shaag, O. Kolesnikova, J. M. Gomori, I. Tarassov, T. Einbinder, A. Saada and O. Elpeleg, “Deleterious Mutation in the Mitochondrial Arginyl-Transfer RNA Synthetase Gene Is Associated with Pontocerebellar hypoplasia,” The American Journal of Human Genetics, Vol. 81, No. 4, 2007, pp. 857-862. doi:10.1086/521227
- [73] S. Yamashita, N. Miyake, N. Matsumoto, H. Osaka, M. Iai, N. Aida and Y. Tanaka, “Neuropathology of Leukoencephalopathy with Brainstem and Spinal Cord Involvement and High Lactate Caused by a Homozygous Mutation of DARS2,” Brain & Development, Vol. 35, No. 4, 2012, pp. 312-316.
- [74] R. Belostotsky, E. Ben-Shalom, C. Rinat, R. BeckerCohen, S. Feinstein, S. Zeligson, R. Segel, O. Elpeleg, S. Nassar and Y. Frishberg, “Mutations in the Mitochondrial seryl-tRNA Synthetase Cause Hyperuricemia, Pulmonary Hypertension, Renal Failure in Infancy and Alkalosis, HUPRA Syndrome,” The American Journal of Human Genetics, Vol. 88, No. 2, 2011, pp. 193-200. doi:10.1016/j.ajhg.2010.12.010
- [75] L. G. Riley, S. Cooper, P. Hickey, J. Rudinger-Thirion, M. McKenzie, A. Compton, S. C. Lim, D. Thorburn, M. T. Ryan, R. Giegé, M. Bahlo and J. Christodoulou, “Mutation of the Mitochondrial Tyrosyl-tRNA Synthetase Gene, YARS2, Causes Myopathy, Lactic Acidosis, and Sideroblastic Anemia—MLASA syndrome,” The American Journal of Human Genetics, Vol. 87, No. 1, 2010, pp. 52-59. doi:10.1016/j.ajhg.2010.06.001
- [76] S. B. Pierce, K. M. Chisholm, E. D. Lynch, M. K. Lee, T. Walsh, J. M. Opitz, W. Li, R. E Klevit and M.-C. King, “Mutations in Mitochondrial Histidyl tRNA Synthetase HARS2 Cause Ovarian Dysgenesis and Sensorineural Hearing Loss of Perrault Syndrome,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 108, No. 16, 2011, pp. 6543-6548. doi:10.1073/pnas.1103471108
- [77] A, Antonellis and E. D. Green, “The Role of Aminoacyl- tRNA Synthetases in Genetic Diseases,” The American Journal of Human Genetics, Vol. 9, 2008, pp. 87-107. doi:10.1146/annurev.genom.9.081307.164204
- [78] W. Weraarpachai, H. Antonicka, F. Sasarman, J. Seeger, B. Schrank, J. E. Kolesar, H. Lochmuller, M. Chevrette, B. A. Kaufman, et al., “Mutation in TACO1, Encoding a Translational Activator of COX I, Results in Cytochrome c Oxidase Deficiency and Late-Onset Leigh Syndrome,” Nature Genetics, Vol. 41, No. 7, 2009, pp. 833-837. doi:10.1038/ng.390
- [79] F. Xu, C. Morin, G. Mitchell, C. Ackerley and B. H. Robinson, “The Role of the LRPPRC (Leucine-Rich Pentatricopeptide Repeat Cassette) Gene in Cytochrome Oxidase Assembly: Mutation Causes Lowered Levels of COX (Cytochrome c Oxidase) I and COX III mRNA,” Biochemical Journal, Vol. 382, Pt. 1, 2004, pp. 331-336.
- [80] M. J. Coenen, H. Antonicka, C. Ugalde, F. Sasarman, R. Rossi, J. G. Heister, R. F. Newbold, F. J. Trijbels, L. P. van den Heuvel, E. A. Shoubridge and J. A. Smeitink, “Mutant Mitochondrial Elongation Factor G1 and Combined Oxidative Phosphorylation Deficiency,” The New England Journal of Medicine, Vol. 351, No. 20, 2004, pp. 2080-2086. doi:10.1056/NEJMoa041878
- [81] J. A. Smeitink, O. Elpeleg, H. Antonicka, H. Diepstra, A. Saada, P. Smits, et al., “Distinct Clinical Phenotypes Associated with a Mutation in the Mitochondrial Translation Elongation Factor EFTs,” The American Journal of Human Genetics, Vol. 79, No. 5, 2006, pp. 869-877. doi:10.1086/508434
- [82] I. Valente, V. Tiranti, R. M. Marsano, E. Malfatti, E. Fernandez-Vizarra, C. Donnini, et al., “Infantile Encephalopathy and Defective Mitochondrial DNA Translation in Patients with Mutations of Mitochondrial Elongation Factors EFG1 and EFTu,” The American Journal of Human Genetics, Vol. 80, No. 1, 2007, pp. 44-58. doi:10.1086/510559
- [83] H. Antonicka, F. Sasarman, N. G. Kennaway and E. A. Shoubridge, “The Molecular Basis for Tissue Specificity of the Oxidative Phosphorylation Deficiencies in Patients with Mutations in the Mitochondrial Translation Factor EFG1,” Human Molecular Genetics, Vol. 15, No. 11, 2006, pp. 1835-1846. doi:10.1093/hmg/ddl106
- [84] P. Smits, H. Antonicka, P. M. van Hasselt, W. Weraarpachai, W. Haller, M. Schreurs, H. Venselaar, R. J. Rodenburg, J. A. Smeitink and L. P. van den Heuvel, “Mutation in Subdomain G’ of Mitochondrial Elongation Factor G1 Is Associated with Combined OXPHOS Deficiency in Fibroblasts but Not in Muscle,” European Journal of Human Genetics, Vol. 19, No. 3, 2011, pp. 275-279. doi:10.1038/ejhg.2010.208
- [85] Y. Bykhovskaya, K. Casas, E. Mengesha, A. Inbal and N. Fischel-Ghodsian, “Missense Mutation in Pseudouridine Synthase 1 (PUS1) Causes Mitochondrial Myopathy and Sideroblastic Anemia (MLASA),” The American Journal of Human Genetics, Vol. 74, No. 6, 2004, pp. 1303-1308. doi:10.1086/421530
- [86] D. Ghezzi E. Baruffini, T. B. Haack, F Invernizzi, L. Melchionda, C. Dallabona, T. M. Strom, R. Parini, A. B. Burlina, T. Meitinger, H. Prokisch, I. Ferrero and M. Zeviani, “Mutations of the Mitochondrial-tRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis,” The American Journal of Human Genetics, Vol. 90, No. 6, 2012, pp. 1079-1087. doi:10.1016/j.ajhg.2012.04.011
- [87] E. J. Tucker, S. G. Hershman, C. Köhrer, C. A. BelcherTimme, J. Patel, et al., “Mutations in MTFMT Underliea Human Disorder of Formylation causIng Impaired Mitochondrial Translation,” Cell Metabolism, Vol. 14, No. 3, 2011, pp. 428-434.