Open Journal of Pathology
Vol.07 No.03(2017), Article ID:77746,13 pages
10.4236/ojpathology.2017.73005
Exploration of Mutation and DNA Methylation of Polo-Like Kinase 1 (PLK1) in Colorectal Cancer
Wayne Ng1,2*, Joo-Shik Shin1,2, Bin Wang2,3, Cheok Soon Lee1,2
1Discipline of Pathology, Western Sydney University, Sydney, Australia
2Centre for Oncology Education and Research Translation, Ingham Institute for Applied Medical Research, Sydney, Australia
3South Western Sydney Clinical School, University of New South Wales (UNSW), Sydney, Australia

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 17, 2017; Accepted: July 16, 2017; Published: July 19, 2017
ABSTRACT
Polo-like kinase 1 (PLK1) is a serine/threonine kinase that plays critical roles in cell cycling and DNA damage response. Overexpression of PLK1 is associated with the poorer prognosis of cancers, including colorectal cancer (CRC). Although the downstream pathways of the overexpression that lead to oncogenesis have been extensively studied, little is known about the factors that cause the overexpression of PLK1 in CRC. DNA methylation was reported to be affecting the expression of PLK1 in some cancers. The study aims to investigate the contribution of genetic mutation and DNA methylation of the PLK1 gene to the overexpression of PLK1 in CRC. The study involves data mining from Catalogue of Somatic Mutations in Cancer (COSMIC) and UniProtKB, Sanger sequencing on DNA from the cell lines HCT116, SW48, Colo320DM and T84 to analyse the possible mutation of PLK1. Other than that methylation status of the PLK1 promoter in CRC are also analysed by using mass spectrometry (MS) and pyrosequencing. Data from the COSMIC show the low incidences of PLK1 mutation for CRC (3.02%) with 46 mutations identified. One of the mutation p.R337Q (c.1010G > A) is located in the D-box which is an important motif for protein ubiquitination and eventually proteasomal degradation. Hence this mutation may potentially result in stabilisation of the PLK1 protein. Mutations are detected at the upstream silencer region, the promoter region and Exon1 in HCT116 but are not located at the protein binding or functioning site. Similarly, the same mutation at promoter region is detected in SW48. Differential trends of changes in methylation status of PLK1 in the IR treated CRC cell lines detected by MS reveal the possible association between the methylation and the radiosensitivity. Furthermore, pyrosequencing shows that PLK1 methylation status in tumour tissues with high expression of PLK1 is not significantly different from those with no PLK1 expression. In conclusion, mutation of PLK1 gene is infrequent in CRC and the expression of PLK1 is unlikely to be dependent on DNA methylation in the promoter region of PLK1 in the CRC.
Keywords:
PLK1, CRC, Mutation, DNA Methylation, Sequencing

1. Introduction
Polo-like kinase is a serine/threonine kinase that promotes mitotic entry during cell division [1] . The overexpression of PLK1 is associated with poorer prognosis in many cancers [2] , including colorectal cancer (CRC) [3] . The expression of PLK1 is variable in both normal and tumour tissues. This had therefore led to the massive development of PLK1 inhibitors, trying to target the PLK1 to improve the treatment outcomes [4] [5] [6] [7] . The development of the drugs was however often halted at Phase I trials, due to inconsistent outcomes and the massive side effects. In addition, separate studies have also reported differential associations between PLK1 expression and radiosensitivity of the tumour [8] [9] . Since PLK1 is interacting within the complicated cell cycle network and DNA damage response that the activity or expression can be regulated by various factors, the upstream protein or factors may have contributed to the complexity of the development of PLK1 as a target for antitumour. Understanding the mechanisms that result in overexpression PLK1 is therefore critical.
Several studies [10] [11] [12] [13] have mapped out some proteins that are interacting with the promoter region of PLK1 to regulate its transcription. Mutation of PLK1 gene that had been reported [14] is mainly linked to the lower stability of PLK1 in the cells and the majority of the mutation is not systematically reviewed for the contribution on PLK1 overexpression. DNA methylation of the CpGs in PLK1 promoter regions has also been reported to be associated with the expression of PLK1 in hepatocellular carcinoma [15] and haematological malignancies [16] . Furthermore, the reported link between DNA methylation and irradiation (IR) exposure also makes it an interesting aspect to study [17] .
The study aims to investigate the contribution of mutation to overexpression of PLK1 by analysing the detected mutations of PLK1 from the Catalogue of Somatic Mutations in Cancer (COSMIC) database [18] and Sanger sequencing. Secondly, the study explores the DNA methylation of PLK1 in relation to PLK1 expression.
2. Materials and Methods
2.1. Cell Culture and Irradiation
All the CRC cell lines were obtained from Sigma-Aldrich, Castle Hill, NSW, and Australia. HCT116 and SW48 that possess high microsatellite instability (MSI), as well as Colo320DM and T84 that are microsatellite stable (MSS), were selected for the in vitro studies [19] [20] . Among the cell lines, SW48 is known to be radiosensitive T84 is radioresistant while radiosensitivity of HCT116 and Colo- 320DM remain intermediate [21] [22] [23] [24] . These allow the exploration of mutation and methylation status of PLK1 gene with the linkage of MSI status and radiosensitivity in CRC. HCT116 and SW48 were cultured in DMEM (Thermo Fisher Scientific, Melbourne, VIC, Australia) supplemented with 10% fetal calf serum (FBS, Sigma-Aldrich, Castle Hill, NSW, Australia) and 1% penicillin and streptomycin (Thermo Fisher Scientific, Melbourne, VIC, Australia). Colo320DM was cultured in RPMI1640 in 10% FBS and 1% penicillin and streptomycin. T84 was grown in DMEM/F12 (Sigma-Aldrich, Castle Hill, NSW, Australia) with the same supplements. All cell lines were cultured under the conditions of 37˚C and 5% CO2. The cells were subjected to IR of 0.5Gy, 2Gy and 5Gy using Elekta Synergy (Elekta, Stockholm, Sweden).
2.2. Patient Tissue Selection
This study received ethics approval from the Sydney South West Area Health Service Human Research Ethics Committee (HREC/12/LPOOL/102). The patients were chosen from rectal cancer patient cohort based on the PLK1 expression of the patients as determined by immunohistochemical (IHC) staining on tissue microarray (TMA) which had been reported as a separate study [3] . Tumour tissues with high PLK1 expression (IHC score of 12, n = 10) were compared with tumour tissues with no PLK1 expression (IHC score of 0, n = 10).
2.3. DNA Extraction and Bisulphite Conversion
To generate cDNA, total RNA was extracted by RNEasy kit (Qiagen, Melbourne, VIC, Australia) and converted to cDNA by reverse transcription by QuantiTect® Reverse Transcription kit (Qiagen, Melbourne, VIC, Australia). DNA was extracted from the CRC cell lines using QIAmp DNA Mini Kit (Qiagen, Melbourne, VIC, Australia) while DNA and RNA of FFPE tissues sections were extracted using AllPrep DNA/RNA FFPE Kit (Qiagen, Melbourne, VIC, Australia). DNA was bisulphite converted to differentiate methylated and non-methylated C in the DNA using EpiTect Bisulfite kit (Qiagen, Melbourne, VIC, Australia).
2.4. Sanger Sequencing
cDNA and DNA were subjected to amplification for different regions with the relevant primer sets by GoTaq® G2 Hot Start Polymerase (Promega, Sydney, NSW, Australia) kit and purified by Qiaquick PCR purification (Qiagen, Melbourne, VIC, Australia) kit before they were sent off for sequencing at Australia Genome Research Facility (AGRF), Melbourne, VIC, Australia. The sequencing data of cDNA were compared with NCBI transcript reference sequence of NM_ 005030.5 whereas DNA sequence data were compared with NCBI gene reference sequence of NC_000016.10 using CLC Main Workbench (Qiagen, Melbourne, VIC, Australia).
The primers for cDNA sequencing are GAGCGGTGCGGAGGCTCT, TGCA TAAAGCCAAGGAAAGG, ATCGAGGTCTGCAGCGCAGCT, CACACTGCA GACATGGCAC, CACCAAGGTTTTCGATTGCT and CTGCAGCATGTCAC TGAGGT. The primers for DNA sequencing are TGGTGGTGTGCACCTGT AGT, AACCAGGTGTAAGCCTCCCA, CTTGTATACAACATTGCACATGG, TCCTTCACCTGCCTTGCAGC, GCACTGCTCTGGGAGCTTG, TGATGCA ACGAAGCTGTCTGG, TCTTCCAACCTTCCCTCCCT, GTCACTGCAGCAC TCATGCTC, GTGCCATGTCTGCAGTGTG and TGCATAAAGCCAAGGA AAGG. The annealing temperature of the primers is 52˚C.
2.5. Mass Spectrometry Methylation Detection
Extracted DNA was sent to Australia Genome Research Facility (AGRF) for analysis by using Sequenom MassArray (Agena Bioscience Inc., San Diego, CA, USA). The primers for PLK1_15 amplicon are AGGAAGAGAGTGTTTGTAATT TTAGTTGTTAGGGAGG and CAGTAATACGACTCACTATAGGGAGAAGG CTTTAAAAAAACACAATACCAAAACCC. The primers for PLK1_16 amplicon are AGGAAGAGAGTTTTTTTAATTTTAGGATGTGTAGGAA and CAG TAATACGACTCACTATAGGGAGAAGGCTTAAAATAACCTAAAAAACCAAACCC.
2.6. Pyro Sequencing
The primer sets are Hs_PLK1_01_PM Pyro Mark CpG assay (PM00063350) and Hs_PLK1_02_PM Pyro Mark CpG assay (PM00063350) from Qiagen (Melbourne, VIC, Australia). Bisulfite converted DNA was amplified with PyroMark PCR Kit (Qiagen, Melbourne, VIC, Australia) and purified by Qiaquick PCR Purification Kit (Qiagen, Melbourne, VIC, Australia) which was then analysed by PyroMark Q24 (Qiagen, Melbourne, VIC, Australia).
2.7 Statistical Analysis
In the pyrosequencing study, the percentages of methylation in each CpG site were determined and compared between high PLK1 and no PLK1 tissues by two-way ANOVA with a p < 0.05 to be considered as significant.
3. Results
3.1. COSMIC Data Search and DNA Sequence Analysis of PLK1 in CRC
The detected mutations and reported polymorphism [25] from COSMIC (top) and Sanger sequencing (bottom) are noted in Figure 1.
Data deposited to COSMIC shows 0.39% mutation rate in PLK1 in a sample size of 38,036 specimens. The mutations detected in CRC specimens (1523 samples) were 8 times more frequent (3.02%) than the overall rate. 46 CRC samples contained a mutation in PLK1 with the mutation occurred more than once in 5 positions and 2 within intronic regions. The mutation of PLK1 are detected only in CRC samples with adenocarcinoma, in which 3.31% (n = 724) in the colon, 1.36% (n = 221) in the rectum, 0.8% (n = 125) at caecum and 2.03% (n = 344) without site mentioned.
The mutations of PLK1 in CRC extracted from COSMIC and the potential functional influences of the mutations are determined by comparing with the gene ontology from UniProtKB (P53350) [26] . Twelve of the mutations are located in the region where the protein kinase domain is coded in which the kinase activity can be affected by amino acid alteration. Three of these mutations (c.596C > T, c.660C > T and c.661G > A) appear at the activation loop of the gene. Three mutations occur in POLO box 1 (pp. 417-480) and eight mutations are located in POLO box 2 (pp. 515-584). One mutation (c.1010G > A/p.R337Q) is identified at D-box motif (pp. 337-340) that targets the protein for proteasomal degradation in anaphase. This is the only site (p.337) that is listed as a site of mutagenesis on UniProtKB. Alteration on the site potentially interferes the ubiquitination and degradation of the protein. The correlation between the mutation and expression could not be compared due to the insufficient matching sample with the expression data.
The DNA sequences of the different amplified regions that cover the cDNA sequence of PLK1 gene were determined in each cell line; the sequences are then combined and matched to NCBI reference sequence NM_005030.5. The forward and reverse strands were sequenced and compared to enable validation. In HCT116 a heterozygous mutation is observed at position c. 47 (A > G, Figure 1). The cDNA sequence of PLK1 gene in SW48, Colo320DM and T84 did not show any variant.
The data generated from Sanger sequencing for the regulatory region is compared to the reference sequence from NCBI (RefSeq: 5347, NC_000016.10). The enhancer, silencer, promoter, core promoter regions and the polymorphism rs27770 were defined according to Akdeli et al. [25] . In order to ensure the

Figure 1. Mutations in PLK1 gene. Boxes on the line represent the exons with Ex1 being exon1 till exon 10 as Ex10, in white being untranslated region and in black being coding region. Mutations extracted from COSMIC data (Top) and mutations detected in Sanger sequencing (bottom) are shown with the CDS (coding DNA sequence) positions indicated. Also noted is the reported polymorphism rs27770 that altered mRNA activity of PLK1 is indicated at 3’ UTR of exon 10.
consistency of the regions with the previous publications, the transcription starting sites are defined according to the previous publication (c. -78C) even though the latest update from NCBI showed that mRNA starts at A (c. -161) prior to the aforementioned C.
In HCT116, heterozygous deleterious mutations of C and T are detected at position c.-1650 (silencer region) and c.-1399 (promoter region) respectively (Figure 1). Similarly, SW48 possesses the same T deletion at position c.-1399. No variant is detected in Colo320DM and T84 cell lines.
The polymorphism rs27770 at the 3’untranslated region of exon 10 does not show any variant in the cell lines. All the cell lines possess a base A on the position.
The pilot study is designed for exploratory purpose in these four cell lines hence statistical analysis is not required.
3.2. In Vitro PLK1 Promoter Region DNA Methylation Exploration in CRC
DNA methylation of CpG islands at the promoter and exon 1 regions of PLK1 (Figure 2) were analysed. The two long stretches of sequences were analysed by bisulphite mass spectrometry (bright green and yellow). In general, Mass spectrometry of the fragmented bisulphite converted DNA of PLK1 gene from CRC lines shows high methylation of CpGs in MSPLK1_15 region (Table 1, bright green region in Figure 2) and low methylation of CpGs in MSPLK1_16 region (Table 1, yellow region in Figure 2). Silent signal is detected in CpG1, CpG3,

Figure 2. Reference sequence of the PLK1 regulatory region for DNA methylation study. Bright green and yellow highlighted regions were investigated in MS DNA methylation study. While the bright green region is MSPLK1_15 amplicon region, yellow region is MSPLK1_16 amplicon. The shorter strands (light blue and pink) are analysed by pyrosequencing with the light blue region being pyroPLK1_2 and dark green region being pyroPLK1_1. The numbers represent the reference position of C in CpG island of the particular analysed region. C in red is the transcription-starting site and ATG in red represents the start codon.

Table 1. DNA methylation statuses (fraction of methylated CpG) of promoter region of PLK1 in CRC cell lines.
a. The methylation status (fraction of methylated CpG) of the CpG islands was detected by mass spectrometry. Samples are named according to the sequence of the cell line (C, H, S and T refer to Colo320DM, HCT116, SW48 and T84 respectively), IR dose (0, 0.5, 2 and 5 Gy) and time point of harvest after IR treatment, (4 h). C_CON represents the conversion control, CON_1 is negative control and CON_2 is the chemical bisulphite converted control. Silent Signal was detected on CpG1, CpG3, CpG4, CpG5 and CpG8 in amplicon MSPLK1_15 and CpG8 in amplicon MSPLK1_16. CpG5 and CpG7 in amplicon MSPLK1_16 are having the same mass therefore the status becomes duplicated. Furthermore mass for CpG9 in amplicon MSPLK1_15 was too low for analysis whereas vice versa for CpG13/14 in amplicon MSPLK1_16. Data with suggested trends are in bold.
CpG4, CpG5, CpG8 and analysis could not be performed for CpG9 due to fragments with low mass, hence is not presented. The methylation in MSPLK1_15 region of Colo320DM tends to reduce when the cells were exposed to IR treatments. The reduction of methylation is dose dependent and occurs across the analysed CpGs. In contrast, SW48 is seen to increase the methylation at CpG10 upon receiving IR exposure. Other than that no obvious trend is observed in methylation of the other CpG.
In MSPLK1_16 region of SW48, CpG1 and CpG5/CpG7 tend to increase in methylation when exposed to IR treatment (Table 1). Methylation of the other sites and cell lines are not affected by IR. Fragments containing CpG5 and CpG7 are sharing the same mass, thus the methylation status cannot be differentiated. CpG8 and CpG13/14 status are not available due to silent signal and high mass respectively. The exploratory study shows differential trends between samples hence statistical analysis is not needed.
3.3. Correlation between PLK1 Expression and Methylation Status in Promoter Region of PLK1
Pyrosequencing was performed to analyse the regions near the start codon (light blue and pink in Figure 2).
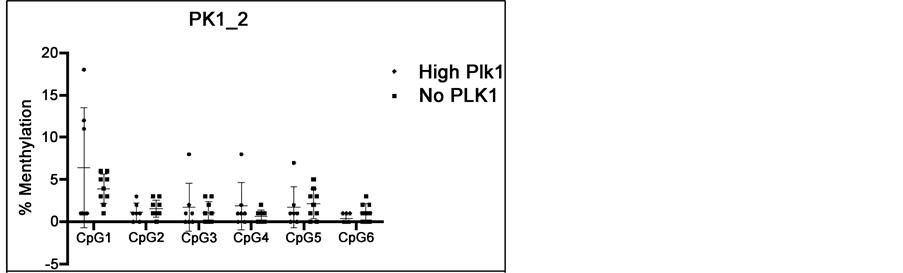
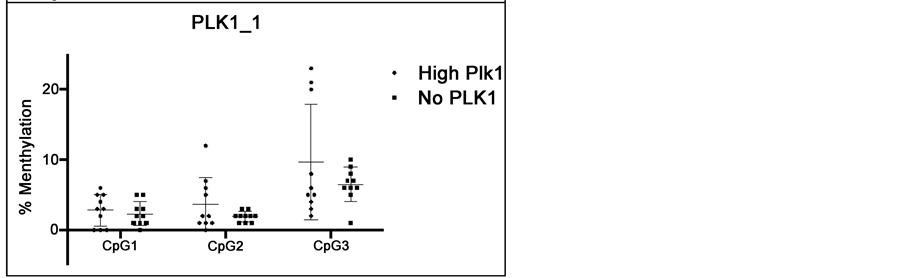
The percentages of methylation in each analysed CpG from pyrosequencing are plotted in Figure 3 to allow comparison between tissues with high PLK1 and no PLK1 expression. The methylation of CpGs in both the regions is low in general with the maximum methylation being 23% and less than 10% in the majority of the samples. The methylation statuses vary in samples with high PLK1 with outliers of higher methylation consistently detected in each CpG. In contrast, methylation statuses in no PLK1 specimens are more consistent and the average values are generally lower than in PLK1 high samples. However, the differences are not significant between PLK1 high and no PLK1 specimens in any of the CpG site.
4. Discussions
According to the data from COSMIC [18] , the mutation of PLK1 gene is not common in CRC even though the occurrence is slightly higher than the other cancers. The occurrence of somatic mutation in CRC is common for the oncogenesis. In addition, the MSI status in the 15% of CRC [27] is causing the tumour to be prone to mutation. It is noted that the mutation events of PLK1 are
 (a)
(a)
 (b)
(b)
Figure 3. Comparison of methylation in CpG island between CRC tissues with high and no expression of PLK1. (a) CpG sites at pyroPLK1_2 region (Figure 2) that are prior to start codon; (b) CpG sites at pyroPLK1_1 region (Figure 2) that are down stream of start codon in exon 1. Tissues with IHC score of 12 and 0 in PLK1 expression were chosen as high and no PLK respectively for this analysis. The differences are not significant (p > 0.05).
only found in the colorectum with adenocarcinoma. Although adenocarcinoma is the main histological type of CRC, the lack of mutation in adenoma also suggests that the mutation PLK1 is related to late stage oncogenesis.
While referring to UniProtKB database [26] for the updated functional do main or motif of the PLK1 protein in relation to mutagenic influences, the majority of the mutations of PLK1 is detected in kinase and POLO boxes domain. Mutations in these regions are generally related to malfunction of the protein which means the disruption of the PLK1 functions, leading to improper cell division.
The missense mutation that was identified on D-box motif according to UniProtKB is potentially contributing to changes of PLK1 expression. The mutation that changes the arginine to alanine at the same position has been reported to have an impact on degradation of PLK1 protein during anaphase of mitosis [28] . Degradation of PLK1 protein is dependent on ubiquitylation and proteasomal activity via the interactions with AP complex [28] . Although the missense mutation replaces the amino acid to glutamine instead of alanine, the degradation mechanism can be disrupted. Failure to degrade the protein can cause accumulation of PLK1 and hence the overexpression. However, it is insufficient to draw a solid conclusion with the limited findings and more functional studies are needed to validate the contribution of the mutation at this site towards the expression of PLK1.
Although the aforementioned mutations may have an impact on the overexpression of PLK1, the overall frequency of PLK1 mutation in CRC is very low. Therefore, mutation is less likely to correlate with the overexpression of PLK1 which appears to be at a much higher rate. Furthermore, the innumerable random mutations that span across the gene also indicate the lack of mutation hotspot.
Both the data from COSMIC and Sanger sequencing are showing the heterozygous mutation at c.47A > G (p.D16 > G) of the gene in HCT116 but the site of mutation is not related to the functionality of PLK1. The sequencing data from cDNA and DNA upstream to the coding region further demonstrates the unlikely mutational event in PLK1 gene. Although variants are detected in HCT116 and SW48 around the border between promoter and silencer region, the deletions of the nucleotides are located at the repetitive sequence with no evidence of it being a protein binding site. Hence, in line with the COSMIC findings, the mutation is rare in PLK1 gene. Therefore this pilot study further suggests the association between mutation and overexpression of PLK1 in CRC is unlikely.
The responses of DNA methylation at PLK1 promoter to IR treatments in the CRC cell lines possess differential trends. The radiosensitive SW48 shows the trend of increase in DNA methylation in a few of the tested CpG sites upon exposure to IR. Although the levels of methylation are dose dependent, all the other assessed CpG sites do not show similar trend. Furthermore, DNA methylation status of the PLK1 promoter in HCT116 and T84 are not affected by the IR treatment. In contrast, CpG sites at the promoter region in PLK1 of Colo320DM are demethylated in response to the IR treatment in a dose-dependent manner. These differential trends are possibly linked with the radiosensitivity of the cell lines. In highly radiosensitive cells, the induction of methylation may be due to the lower survival of the cells after receiving IR. On the other hand, the unaffected methylation or even demethylation in the more radioresistant cell lines in response to IR exposure may be linked to the better survival of the cells. Furthermore, differential changes of DNA methylation status after IR exposure have been reported at genome wide level [29] [30] . Pogribny et al. suggested that the DNA hypomethylation upon IR exposure are specific to sex, tissue and doses. In addition, gene ontology analysis of the genome DNA methylation data of MDA- MB-231 breast cancer cells [17] shows hypomethylation of cell cycle genes when subjected to low dose (2Gy) but hypermethylation of those genes occur at high dose (5Gy). The study also suggests that the changes in DNA methylation due to IR exposure are transient and time point dependent.
Comparison of DNA methylation status in PLK1 between tissues with high PLK1 expression and those with no PLK1 expression confirms that expression of the PLK1 is generally independent of DNA methylation of PLK1 gene since the DNA methylation is low in both high and no PLK1 CRC tissues. However, the outliers are consistently noted on methylation status of the CpG sites in the high PLK1 expressing tissue. The outliers are higher in DNA methylation, leading to more widespread of the level of DNA methylation in high PLK1 samples than those without PLK1 expression. This may implicate that the relationship between DNA methylation and expression of the PLK1 is more complicated than being independent of each other.
The aforementioned DNA methylation analysis of PLK1 gene in human hepatocellular carcinoma [15] also revealed the lack of methylation in tumour tissues and normal tissues even though the PLK1 expression was higher in tumour tissues. The analysis in mouse model however showed differential findings in which the hypomethylation was detected in tumour tissues with high PLK1 expression. In addition, the same group also demonstrated the correlation between DNA methylation of the PLK1 promoter region and the PLK1 expression in haematological malignancies [16] . The contradictory results suggest the role of the DNA methylation may be more complicated in determining PLK1 expression.
5. Conclusions
The genetic mutation of PLK1 gene is rare in CRC although the occurrence is slightly higher than other cancers. Furthermore, no hotspot is observed in the detected mutations. Mutation c.1010A > G (p.R337Q) is potentially interfering the degradation of PLK1, leading to the increase of stability and expression of PLK1. Mutations c.-1650C > del and c.-1399T > del at the border of silencer/promoter regions and c.47A > G at exon 1 are detected in HCT116. Moreover, c.-1650C > del is also detected in SW48. Nonetheless, they are unlikely to impose any effect on the expression of PLK1.
Mass spectrometry shows differential trends of DNA methylation of the PLK1 promoter region in IR-treated CRC cell lines which may be linked to the radiosensitivity of the cell lines. The CpGs of PLK1 gene across the samples are overall low in methylation and are not significantly different between high and undetected PLK1 expressing CRC tissues. Expression of PLK1 is likely to be independent of the DNA methylation at the promoter of the gene.
Acknowledgements
WN thanks the Centre for Oncology Education and Research Translation (CONCERT) which is a translational research centre wholly funded by Cancer Institute NSW for funding the PhD scholarship, Mr. Roy Medich on his kind donation to fund the project, mr. Askar Abubakar for providing technical support..
Cite this paper
Ng, W., Shin, J.-S., Wang, B. and Lee, C.S. (2017) Exploration of Mutation and DNA Methylation of Polo-Like Kinase 1 (PLK1) in Colorectal Cancer. Open Journal of Pathology, 7, 45-57. https://doi.org/10.4236/ojpathology.2017.73005
References
- 1. Archambault, V. and Glover, D.M. (2009) Polo-Like Kinases: Conservation and Divergence in Their Functions and Regulation. Nature Reviews Molecular Cell Biology, 10, 265-275.
https://doi.org/10.1038/nrm2653 - 2. Takai, N., Hamanaka, R., Yoshimatsu, J. and Miyakawa, I. (2005) Polo-Like Kinases (Plks) and Cancer. Oncogene, 24, 287-291.
https://doi.org/10.1038/sj.onc.1208272 - 3. Tut, T.G., Lim, S.H., Dissanayake, I.U., Descallar, J., Chua, W., Ng, W., de Souza, P., Shin, J.S. and Lee, C.S. (2015) Upregulated Polo-Like Kinase 1 Expression Correlates with Inferior Survival Outcomes in Rectal Cancer. PLoS ONE, 10, 1-11.
https://doi.org/10.1371/journal.pone.0129313 - 4. Palmisiano, N.D. and Kasner, M.T. (2015) Polo-Like Kinase and Its Inhibitors: Ready for the Match to Start? American Journal of Hematology, 90, 1071-1076.
https://doi.org/10.1002/ajh.24177 - 5. Chopra, P., Sethi, G., Dastidar, S.G. and Ray, A. (2010) Polo-Like Kinase Inhibitors: An Emerging Opportunity for Cancer Therapeutics. Expert Opinion on Investigational Drugs, 19, 27-43.
https://doi.org/10.1517/13543780903483191 - 6. Yim, H. (2013) Current Clinical Trials with Polo-Like Kinase 1 Inhibitors in Solid Tumors. Anticancer Drugs, 24, 999-1006.
https://doi.org/10.1097/CAD.0000000000000007 - 7. Strebhardt, K. (2010) Multifaceted Polo-Like Kinases: Drug Targets and Antitargets for Cancer Therapy. Nature Reviews Drug Discovery, 9, 643-660.
https://doi.org/10.1038/nrd3184 - 8. Rodel, F., Keppner, S., Capalbo, G., Bashary, R., Kaufmann, M., Rodel, C., Strebhardt, K. and Spankuch, B. (2010) Polo-Like Kinase 1 as Predictive Marker and Therapeutic Target for Radiotherapy in Rectal Cancer. American Journal of Pathology, 177, 918-929.
https://doi.org/10.2353/ajpath.2010.100040 - 9. Cebrian, A., Gomez Del Pulgar, T., Fernandez-Acenero, M.J., Borrero-Palacios, A., Del Puerto-Nevado, L., Martinez-Useros, J., Marin-Arango, J.P., Carames, C., Vega-Bravo, R., Rodriguez-Remirez, M., Manzarbeitia, F. and Garcia-Foncillas, J. (2016) Decreased PLK1 Expression Denotes Therapy Resistance and Unfavourable Disease-Free Survival in Rectal Cancer Patients Receiving Neoadjuvant Chemoradiotherapy. Pathology—Research and Practice, 212, 1133-1137.
https://doi.org/10.1016/j.prp.2016.09.012 - 10. Martin, B.T. and Strebhardt, K. (2006) Polo-Like Kinase 1: Target and Regulator of Transcriptional Control. Cell Cycle, 5, 2881-2885.
https://doi.org/10.4161/cc.5.24.3538 - 11. Alvarez, B., Martinez, A.C., Burgering, B.M. and Carrera, A.C. (2001) Forkhead Transcription Factors Contribute to Execution of the Mitotic Programme in Mammals. Nature, 413, 744-747.
https://doi.org/10.1038/35099574 - 12. Uchiumi, T., Longo, D.L. and Ferris, D.K. (1997) Cell Cycle Regulation of the Human Polo-Like Kinase (PLK) Promoter. Journal of Biological Chemistry, 272, 9166-9174.
https://doi.org/10.1074/jbc.272.14.9166 - 13. Tategu, M., Nakagawa, H., Sasaki, K., Yamauchi, R., Sekimachi, S., Suita, Y., Watanabe, N. and Yoshid, K. (2008) Transcriptional Regulation of Human Polo-Like Kinases and Early Mitotic Inhibitor. Journal of Genetics and Genomics, 35, 215-224.
https://doi.org/10.1016/S1673-8527(08)60030-2 - 14. Simizu, S. and Osada, H. (2000) Mutations in the Plk Gene Lead to Instability of Plk Protein in Human Tumour Cell Lines. Nature Cell Biology, 2, 852-854.
https://doi.org/10.1038/35041102 - 15. Ward, A., Morettin, A., Shum, D. and Hudson, J.W. (2011) Aberrant Methylation of Polo-Like Kinase CpG Islands in Plk4 Heterozygous Mice. BMC Cancer, 11, 71.
https://doi.org/10.1186/1471-2407-11-71 - 16. Ward, A., Sivakumar, G., Kanjeekal, S., Hamm, C., Labute, B.C., Shum, D. and Hudson, J.W. (2015) The Deregulated Promoter Methylation of the Polo-Like Kinases as a Potential Biomarker in Hematological Malignancies. Leukemia & lymphoma, 56, 2123-2133.
https://doi.org/10.3109/10428194.2014.971407 - 17. Antwih, D.A., Gabbara, K.M., Lancaster, W.D., Ruden, D.M. and Zielske, S.P. (2013) Radiation-Induced Epigenetic DNA Methylation Modification of Radiation-Response Pathways. Epigenetics, 8, 839-848.
https://doi.org/10.4161/epi.25498 - 18. Forbes, S.A., Beare, D., Boutselakis, H., Bamford, S., Bindal, N., Tate, J., Cole, C.G., Ward, S., Dawson, E., Ponting, L., Stefancsik, R., Harsha, B., Kok, C.Y., Jia, M., Jubb, H., Sondka, Z., Thompson, S., De, T. and Campbell, P.J. (2017) COSMIC: Somatic Cancer Genetics at High-Resolution. Nucleic Acids Research, 45, 777-783.
https://doi.org/10.1093/nar/gkw1121 - 19. Oliveira, C., Pinto, M., Duval, A., Brennetot, C., Domingo, E., Espin, E., Armengol, M., Yamamoto, H., Hamelin, R., Seruca, R. and Schwartz Jr, S. (2003) BRAF Mutations Characterize Colon but Not Gastric Cancer with Mismatch Repair Deficiency. Oncogene, 22, 9192-9196.
https://doi.org/10.1038/sj.onc.1207061 - 20. Ahmed, D., Eide, P.W., Eilertsen, I.A., Danielsen, S.A., Eknaes, M., Hektoen, M., Lind, G.E. and Lothe, R.A. (2013) Epigenetic and Genetic Features of 24 Colon Cancer Cell Lines. Oncogenesis, 2, e71.
https://doi.org/10.1038/oncsis.2013.35 - 21. Dunne, A.L., Price, M.E., Mothersill, C., McKeown, S.R., Robson, T. and Hirst, D.G. (2003) Relationship between Clonogenic Radiosensitivity, Radiation-Induced Apoptosis and DNA Damage/Repair in Human Colon Cancer Cells. British Journal of Cancer, 89, 2277-2283.
https://doi.org/10.1038/sj.bjc.6601427 - 22. Amundson, S.A., Do, K.T., Vinikoor, L.C., Lee, R.A., Koch-Paiz, C.A., Ahn, J., Reimers, M., Chen, Y., Scudiero, D.A., Weinstein, J.N., Trent, J.M., Bittner, M.L., Meltzer, P.S. and Fornace Jr, A.J. (2008) Integrating Global Gene Expression and Radiation Survival Parameters across the 60 Cell Lines of the National Cancer Institute Anticancer Drug Screen. Cancer Research, 68, 415-424.
https://doi.org/10.1158/0008-5472.CAN-07-2120 - 23. Frykholm, G., Glimelius, B., Richter, S. and Carlsson, J. (1991) Heterogeneity in Antigenic Expression and Radiosensitivity in Human Colon Carcinoma Cell Lines. In Vitro Cellular & Developmental Biology, 27, 900-906.
https://doi.org/10.1007/BF02631115 - 24. Wang, Q., Wang, Y., Du, L., Xu, C., Sun, Y., Yang, B., Sun, Z., Fu, Y., Cai, L., Fan, S., Fan, F. and Liu, Q. (2014) shRNA-Mediated XRCC2 Gene Knockdown Efficiently Sensitizes Colon Tumor Cells to X-Ray Irradiation in Vitro and in Vivo. International Journal of Molecular Sciences, 15, 2157-2171.
https://doi.org/10.3390/ijms15022157 - 25. Akdeli, N., Riemann, K., Westphal, J., Hess, J., Siffert, W. and Bachmann, H.S. (2014) A 3'UTR Polymorphism Modulates mRNA Stability of the Oncogene and Drug Target Polo-Like Kinase 1. Molecular Cancer, 13, 87.
https://doi.org/10.1186/1476-4598-13-87 - 26. The UniProt Consortium (2017) UniProt: The Universal Protein Knowledgebase. Nucleic Acids Research, 45, D158-D169.
https://doi.org/10.1093/nar/gkw1099 - 27. Shin, J.S., Tut, T.G., Yang, T. and Lee, C.S. (2013) Radiotherapy Response in Microsatellite Instability Related Rectal Cancer. The Korean Journal of Pathology, 47, 1-8.
https://doi.org/10.4132/KoreanJPathol.2013.47.1.1 - 28. Lindon, C. and Pines, J. (2004) Ordered Proteolysis in Anaphase Inactivates Plk1 to Contribute to Proper Mitotic Exit in Human Cells. Journal of Cell Biology, 164, 233-241.
https://doi.org/10.1083/jcb.200309035 - 29. Pogribny, I., Koturbash, I., Tryndyak, V., Hudson, D., Stevenson, S.M., Sedelnikova, O., Bonner, W. and Kovalchuk, O. (2005) Fractionated Low-Dose Radiation Exposure Leads to Accumulation of DNA Damage and Profound Alterations in DNA and Histone Methylation in the Murine Thymus. Molecular Cancer Research, 3, 553-561.
https://doi.org/10.1158/1541-7786.MCR-05-0074 - 30. Zielske, S.P. (2015) Epigenetic DNA Methylation in Radiation Biology: On the Field or on the Sidelines? Journal of Cellular Biochemistry, 116, 212-217.
https://doi.org/10.1002/jcb.24959