Open Journal of Pathology
Vol.4 No.1(2014), Article ID:41694,4 pages DOI:10.4236/ojpathology.2014.41001
Clinical Pathological Correlation Quiz: A 7-Month-Old Girl with Congenital Neutropenia and Increased Bone Morrow “Blasts”
![]()
1Division of Hematology/Oncology, Nationwide Children’s Hospital, Columbus, USA; 2Department of Pathology and Laboratory Medicine, Wexner Medical Center at the Ohio State University, Columbus, USA; 3Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, Columbus, USA.
Email: *Samir.Kahwash@nationwidechildrens.org
Received September 24th, 2013; revised October 24th, 2013; accepted October 31st, 2013
ABSTRACT
Persistent Neutropenia in a child presents a wide differential diagnosis, spanning the spectrum of benign disorders (such as congenital neutropenia, cyclic neutropenia, etc.) to malignant processes (such as myelodysplasia or acute leukemia). The morphologic findings in the bone marrow of a patient with neutropenia may show considerable overlap with those seen in hematopoietic malignancies. We present herein a case that demonstrates this overlap and discuss features that can help in avoiding diagnostic pitfalls.
Keywords:Neutropenia; Hematogones; ELANE Mutation; Acute Leukemia
1. Clinical History
The patient is a 7-month-old female who was presented with cheek cellulitis and a fever. She had a history of labial abscess at 2 months of age. The initial complete blood count (CBC) upon admission revealed a normal white blood cell count with an absolute neutrophil count (ANC) of 207/µL, no anemia, and a mild thrombocytosis (platelet count 509,000/µL). Additional testing included a reticulocyte count of 4.2%, unremarkable Band Tlymphocytes by flow cytometry, normal immunoglobulin levels by serum protein electrophoresis, normal lactate dehydrogenase (LDH) and uric acid levels, normal pancreatic function tests, and anti-neutrophil antibody (ANA) negativity. Subsequent CBCs over a 5-day period, listed in Table 1, are notable for worsening absolute neutropenia.
Despite appropriate antibiotic therapy, the cheek cellulitis did not resolve. The lack of clinical improvement, as well as persistent neutropenia, prompted a bone marrow evaluation (Figure 1).
Cell immunophenotyping by flow cytometry showed increased cells in the lymphoblast/myeloblast region (Figure 2).
2. Questions
1) What is the most likely diagnosis?
2) What tests would you recommend to further establish the diagnosis?
3. Answers
1) Congenital neutropenia characterized by myeloid hypoplasia with maturation arrest at the promyelocyte stage and an increase in hematogones.
2) Testing for HAX1 (HCLS1-associated protein X-1) and ELANE (Elastase, Neutrophil Expressed or ELA2) gene mutations was recommended. The patient was found to be positive for ELANE mutation. Serial CBCs demonstrated persistent neutropenia without a cyclic pattern, confirming the diagnosis of severe congenital neutropenia (SCN) with ELANE mutation.
4. Discussion
4.1. Bone Marrow Findings (Figures 1 and 2)
Bone marrow evaluation showed 60% cellularity and myeloid hypoplasia with maturation arrest at the promyelocyte stage. Early myeloid precursors were relatively increased; however, the M:E ratio was still decreased (0.7:1). Megakaryocytes were mildly increased in number with normal morphology. An iron stained aspirate smear showed abundant iron deposits with no ring sideroblasts. There was no evidence of granulomatous inflammation or metastatic tumor. Immunophenotypic analysis by flow cytometry detected increased cells in the blast region, which were most consistent with hematogones.
Immunohistochemical staining for TdT, CD10, CD19, and CD34 showed scattered positive cells. In situ hybridization staining for Epstein-Barr virus-encoded small RNA (EBER) was negative. Immunoperoxidase staining was negative for cytomegalovirus (CMV) and parvovirus. Cytogenetic analysis revealed a normal karyotype, and FISH was negative for monosomy 5, 5q deletion, monosomy 7, 7q deletion, trisomy 8, and MLL gene rearrangement.
4.2. Severe Congenital Neutropenia (SCN)
Severe congenital neutropenia (SCN) was initially described by Kostmann as an autosomal recessive disorder in an isolated population in Sweden [1]. SCN is clinically characterized by severe neutropenia (ANC < 200/µL) and resultant bacterial and fungal infections from early infancy. Untreated patients may suffer from chronic gingivitis, oral ulcers, skin abscesses, recurrent pneumonia, or septicemia [1]. Two important entities in the differential diagnosis include (1) autoimmune neutropenia of infancy, which is associated with a positive ANA and lacks granulocytic maturation arrest on the bone marrow specimen, and (2) cyclic neutropenia, which is diagnosed by serial ANC measurements approximately 2 - 3 days/ week for 6 weeks evaluating for a cyclic pattern of neutropenia.
SCN can be sporadic or inherited, and many underlying genetic aberrations have been implicated in SCN [2,3]. The most common genetic abnormality detected in both sporadic and autosomal dominant cases is a mutation in the neutrophil elastase gene (ELANE), which is detected in ~60%, of SCN patients [4]. ELANE mutations lead to severe neutropenia via a stress response in the endoplasmic reticulum, which provokes activation of the unfolded protein response (UPR), ultimately leading to apoptosis of myeloid progenitor cells [1]. Mutations in the HAX1 gene are frequently implicated in autosomal recessive SCN with variable degrees of neurologic

Table 1 . Patient’s CBC data over the initial hospitalization period.
ANC = Absolute Neutrophil Count.

Figure 1. Bone marrow biopsy and aspirate smear. A: Cellular bone marrow biopsy with mildly increased megakaryocytes, H & E (200×); B: Bone marrow biopsy with myeloid hypoplasia and relatively increased erythroid precursors, H & E (500×); C: Aspirate smear showing myeloid maturation arrest at promyelocyte stage, Wright-Giemsa (500×); D: Aspirate smear with relatively increased promyelocytes, some of which show cytoplasmic vacuolization, Wright-Giemsa (1000×); E: TdT immunohistochemistry shows scattered lymphoblasts in the biopsy (200×).
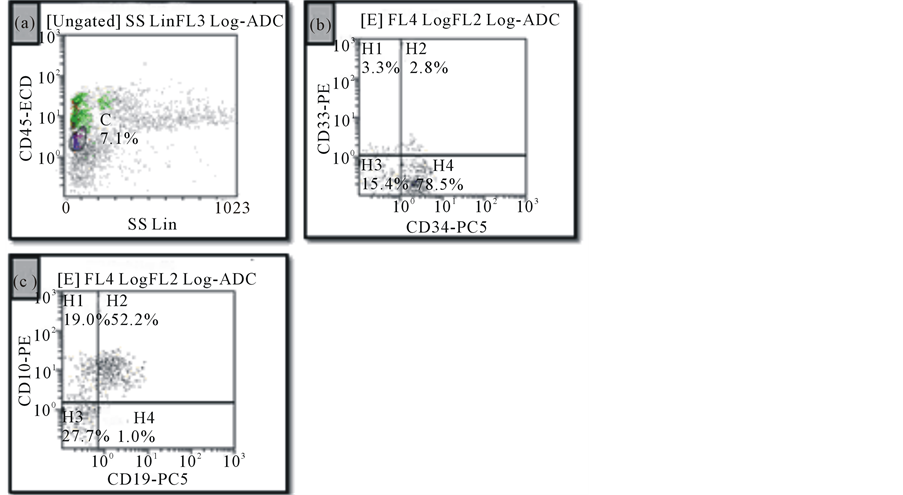
Figure 2. Flow cytometric analysis of bone marrow specimen. (a): Increased events in the lymphoblast region showing dim CD45 staining and low side scatter; (b) and (c): The majority of these events show heterogeneous expression of CD34, CD10, and CD19 and lack CD33, consistent with hematogones.
impairment. Lastly, SCN can be seen in association with additional congenital abnormalities due to underlying disorders of ribosomal dysfunction (Shwachman-Diamond syndrome and dyskeratosis congenita); of metabolism (reticular dysgenesis, Barth syndrome, glycogen storage disease type 1b, glucose-6-phosphatase catalytic subunit 3 syndrome, and Pearson syndrome); of vesicular transport (Chediak-Higashi syndrome, Cohen syndrome, Griscelli syndrome type II, Hermansky-Pudlak syndrome type II, and p14 deficiency); and disorders of immune function (cartilage-hair hypoplasia, hyper-IgM syndrome, Schimke immuno-osseous dysplasia, WHIM syndromeWHIM is an acronym for Warts, Hypogammaglobulinemia, Infections and Myelokathexis; and Wiskott-Aldrich syndrome) [1].
4.3. Clinical Management and Disease Progression
Data collected by the Severe Chronic Neutropenia International Registry (SCNIR) demonstrate that more than 95% of patients with SCN respond to recombinant human (rHu) G-CSF [1,2]. The goal is an ANC of ≥1000/µL, at which point the occurrence of bacterial infections is markedly reduced. Additionally, prophylactic antibiotics are useful for decreasing the risks of bacterial infections [3]. SCN patients have a significant risk of developing a myelodysplastic syndrome (MDS) and/or acute myeloid leukemia (AML) [1,2]. The cumulative incidence of leukemia among patients with SCN has ranged from 10% - 20% [1]. It is unclear if this neoplastic progression reflects the natural disease course of SCN following sequential gain of mutations or if it is secondary to chronic G-CSF therapy [1]. Development of MDS/ AML is frequently associated with additional genetic abnormalities such as monosomy 7 and a G-CSF receptor mutation [5,6]. At least quarterly CBCs and annual BM aspirations with cytogenetics is recommended in SCN patients to evaluate for progression to MDS/AML [1].
Hematopoietic stem cell transplantation (HSCT) remains the only curative therapy for SCN [7]. The absolute indications for HSCT in SCN are a failure to respond to G-CSF or the development of MDS/leukemia. Patients who require higher doses of G-CSF with poor neutrophil response and those who harbor the Gly185Arg mutation in the ELANE gene should also be considered for HSCT with the best available donor [7].
4.4. Comments on Common Bone Marrow Findings in SCN and Potential Pitfalls
The bone marrow in SCN demonstrates normal or slightly decreased cellularity and normal megakaryocytes [1,2]. There is usually granulocytic maturation arrest at the promyelocyte-myelocyte stage. The absolute number of promyelocytes is increased, and promyelocytes can have morphologically atypical nuclei as well as cytoplasmic vacuoles. An eosinophilia and monocytosis are common bone marrow and peripheral blood findings [1]. Additional peripheral blood findings include mild anemia, thrombocytosis, and elevated IgG levels. The case presented here had an increased number of bone marrow precursor B cells, or hematogones. This finding has previously been described in SCN, specifically in Shwachman-Diamond syndrome and ELANE-mutated SCN [8,9]. Although the exact clinical significance is unknown, an increase in hematogones may represent a regenerative or compensatory mechanism in the setting of severe neutropenia [9]. Furthermore, an increased hematogone population in the setting of SCN is important to recognize as a potential diagnostic pitfall since hematogones can simulate blasts. Immunophenotyping and cytogenetic/ FISH analyses are useful in differentiating SCN with increased hematogones from a hematopoietic malignancy such as MDS, AML, and ALL.
5. Conclusion
There is a significant clinical and pathologic overlap between congenital neutropenia and hematopoietic malignancies. Awareness of this fact, along with performing a complete workup that includes immunophenotyping and cytogenetic studies, will help avoid potential diagnostic pitfalls.
REFERENCES
- L. A. Boxer, “How to Approach Neutropenia,” Hematology: The Education Program of the American Society of Hematology, Vol. 2012, No. 1, 2012, pp. 174-182.
- K. Welte C. Zeidler and D. C. Dale, “Severe Congenital Neutropenia,” Seminars in Hematology, Vol. 43, No. 3, 2006, pp. 189-195. http://dx.doi.org/10.1053/j.seminhematol.2006.04.004
- K. Boztug and C. Klein “Novel Genetic Etiologies of Severe Congenital Neutropenia,” Current Opinion in Immunology, Vol. 21, No. 5, 2009, pp. 472-480. http://dx.doi.org/10.1016/j.coi.2009.09.003
- M. S. Horwitz, S. J. Corey, H. L. Grimes and T. Tidwell, “ELANE Mutations in Cyclic and Severe Congenital Neutropenia: Genetics and Pathophysiology,” Hematology/Oncology Clinics of North America, Vol. 27, No. 1, 2013, pp. 19-41. http://dx.doi.org/10.1016/j.hoc.2012.10.004
- M. Shiohara T. Shigemura S. Saito M. Tanaka R. Yanagisawa K. Sakashita H. Asada E. Ishii K. Koike M. Chin M. Kobayashi and K. Koike “Ela2 Mutations and Clinical Manifestations in Familial Congenital Neutropenia,” Journal of Pediatric Hematology/Oncology, Vol. 31, No. 5, 2009, pp. 319-324. http://dx.doi.org/10.1097/MPH.0b013e3181984dbe
- C. A. Tschan, C. Pilz, C. Zeidler, K. Welte and M. Germeshausen, “Time Course of Increasing Numbers of Mutations in the Granulocyte Colony-Stimulating Factor Receptor Gene in a Patient with Congenital Neutropenia Who Developed Leukemia,” Blood, Vol. 97, No. 6, 2001, pp. 1882-1884. http://dx.doi.org/10.1182/blood.V97.6.1882
- J. A. Connelly, S. W. Choi and J. E. Levine, “Hematopoietic Stem Cell Transplantation for Severe Congenital Neutropenia,” Current Opinion in Hematology, Vol. 19, No. 1, 2012, pp. 44-51. http://dx.doi.org/10.1097/MOH.0b013e32834da96e
- K. Welte and L. Boxer, “Severe Chronic Neutropenia: Pathophysiology and Therapy,” Seminars in Hematology, Vol. 34, No. 4, 1997, pp. 267-278.
- N. Klupp I. Simonitsch C. Mannhalter and G. Amann “Emergence of an Unusual Bone Marrow Precursor BCell Population in Fatal Shwachman-Diamond Syndrome,” Archives of Pathology & Laboratory Medicine, Vol. 124, No. 9, 2000, pp. 1379-1381.
NOTES
*Corresponding author.