World Journal of Cardiovascular Diseases
Vol.07 No.01(2017), Article ID:73613,9 pages
10.4236/wjcd.2017.71001
Cardiac Paraganglioma―A Rare Subset of a Rare Tumor
Alecsandra-Anca Tudor1, Joëlle Tschui2, Jürg Schmidli3, Ralph A. Schmid1, Patrick Dorn1*
1Division of General Thoracic Surgery, Inselspital, University Hospital, Bern, Switzerland
2Institute of Pathology, University of Bern, Bern, Switzerland
3Department of Cardiovascular Surgery, Inselspital, University Hospital, Bern, Switzerland

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: November 25, 2016; Accepted: January 16, 2017; Published: January 19, 2017
ABSTRACT
Background: Cardiac paraganglioma is a rare entity of an uncommon neuroendocrine tumor. Clinically, non-secreting tumors are often diagnosed because of their growth effects, secreting tumors present symptoms related to catecholamine. Correct diagnosis of a paraganglioma can be reached by biochemical investigations and imaging. Surgical resection is the treatment of choice and has to be planned carefully and interdisciplinarily. Aim: On the basis of a patient with a vague clinical presentation and an unclear situation after primary investigations, we highlight the diagnostic challenge of this rare subset of paragangliomas. Case presentation: We present the case of a 42- year-old woman whose unspecific symptoms and further investigations revealed a paracardiac mass with unknown local behavior and dignity. Surgical resection and histopathological examination led to the diagnosis of a cardiac paraganglioma. Conclusion: Cardiac paragangliomas are extremely rare, but may be treated curatively by resection after careful preoperative investigation and interdisciplinary planning.
Keywords:
Cardiac Paraganglioma, Paraganglioma

1. Introduction
This report presents the case of a middle-aged female who was referred to our hospital with unspecific clinical symptoms. Primary investigations and the rarity of a cardiac association with the identified tumor, initially led us on the wrong track. However, careful planning of the operation and performing the resection in an interdisciplinary setting was the key to success. Histopathology revealed a benign cardiac paraganglioma.
On the basis of this case, we show the diagnostic challenge of this rare subset of paragangliomas.
2. Case Presentation
A Caucasian, overweight female patient, 42 years old at the time of referral, consulted her family doctor because of intermittent fever, attacks of sweating, dyspnea and dull epigastric pain and also relapse of previously experienced back pain. She was a non-smoker and had no other significant medical history. Clinical check-up and blood test showed no evidence of an infectious disease. As one of the further investigations, a computed tomography (CT) of the chest and abdomen was decided, in order to exclude a pulmonary embolism. Surprisingly, a mediastinal mass of 6.6 × 3.9 cm in size was revealed. It was located in close contact to the tracheal carina and the left atrium. Some enlarged right hilar nodes were seen, but no other suspicious findings were documented. A magnetic resonance imaging (MRI) could not completely exclude an infiltration of the surrounding tissue and organs. Radiologically the mediastinal mass was considered highly suspicious for a lymphoma, other differential diagnosis were rather unlikely, but could not be completely ruled out at the time. For further assessment the patient was referred to our thoracic surgery department. Positron emission tomography/computed tomography (PET/CT) supported the suspicion of a lymphoma limited to that area, no other FDG-avid lesions were seen, neither in hilar or mediastinal lymph node stations, nor in regions expected for any primary tumors (Figure 1). Lymphoma limited to the mediastinum was the first differential diagnosis. Therefore, a diagnostic mediastinal lymphnode biopsy by right-sided thoracoscopy was decided in order to obtain a tissue biopsy and to definitely rule out metastatic disease. Histopathological investigations of the hilar and mediastinal lymph nodes were negative for lymphoma as well as for other malignancies.
Further evaluation with dynamic heart MRI (Figure 2) was performed and did not show involvement of the atrial wall, however, the inhomogeneity and intense vascularization indicated a neurogenic tumor. Surgical excision was planned as the next diagnostic and simultaneously therapeutic step.
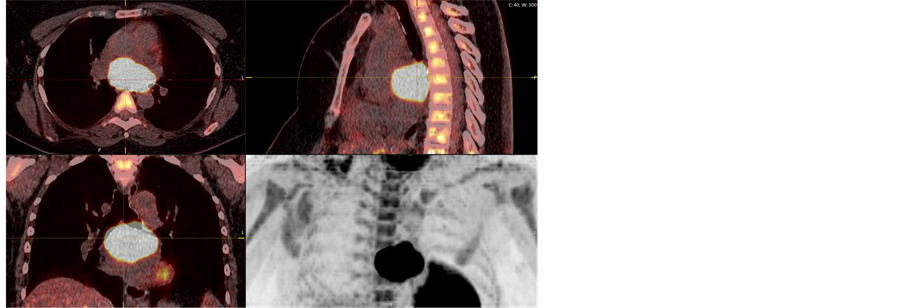
Figure 1. Pre-operative PET/CT.

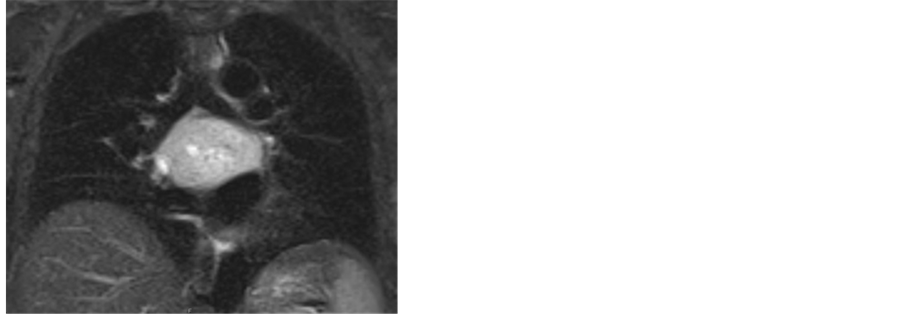
Figure 2. Pre-operative dynamic MRI: T1-weighed coronal section and T2-weighed oblique sagittal section.
We performed an open tumor resection via right lateral thoracotomy. After complete preparation of the situs and localization of the mass, which showed an infiltration of the left atrium of the heart, cardiopulmonary bypass (CPB) was required to perform resection. For better access to the tumor, the superior right pulmonary vein was temporarily disconnected. After removal of the tumor with resection of the adherent left atrial wall, which was infiltrated by the tumor over an area of 3 cm in diameter, the atrial wall was reconstructed with a bovine pericardium patch. The pulmonary vein was re-anastomosed.
The patient was monitored overnight in the intermediate care unit and subsequently transferred to the ward after an uneventful course. An oral anticoagulation was started for prophylaxis of thromboembolic complications. The patient was discharged on the 9th postoperative day. A rehabilitation schedule was organized in an outpatient setting.
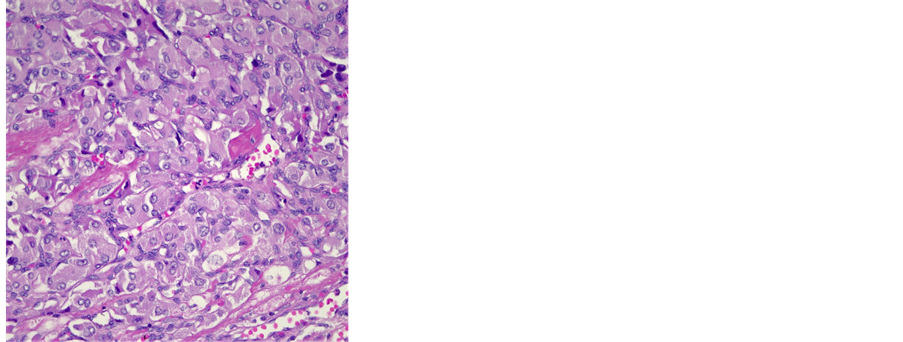
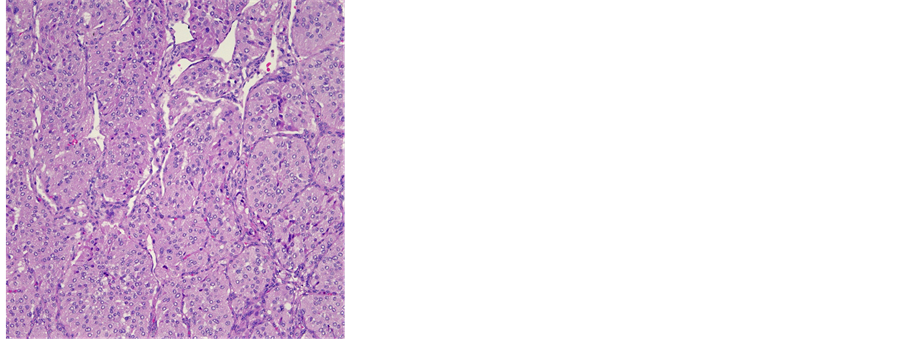
Histopathologically, the gross specimen consisted of a round to ovoid encapsulated mass of 6.4 cm in diameter, with a predominantly homogeneous, focally fibrotic cut surface. Light microscopy showed a solid neoplasm composed of a monomorphous population of round to polygonal cells with amphophilic, finely granular cytoplasm. The lesion’s architecture displayed a conspicuous nested (“Zellballen”) pattern throughout (Figure 3(a), Figure 3(b)). Mitotic activity was virtually absent, and neither extracapsular vascular invasion nor necrosis was noted. Nevertheless, focal encroachment upon the adjacent left atrial musculature was noted at the tumor’s periphery (Figure 3(c)). Immunohistochemistry confirmed expression of the neuroendocrine markers chromogranin A and synaptophysin, while the Zellballen architecture was highlighted by staining for S100 protein in sustentacular cells. Of note, tumor cells exhibited intense staining for SDHB (Figure 3(c)).
Based on these findings, the pathological diagnosis of paraganglioma with focal infiltration of the left atrium was issued.
At our interdisciplinary tumor board, regular follow-up with PET/CT was decided without any further treatments being considered necessary. There was no suspicion for recurrent disease or metastases in the PET/CT at 3 and 9 months
 (a) (b)
(a) (b)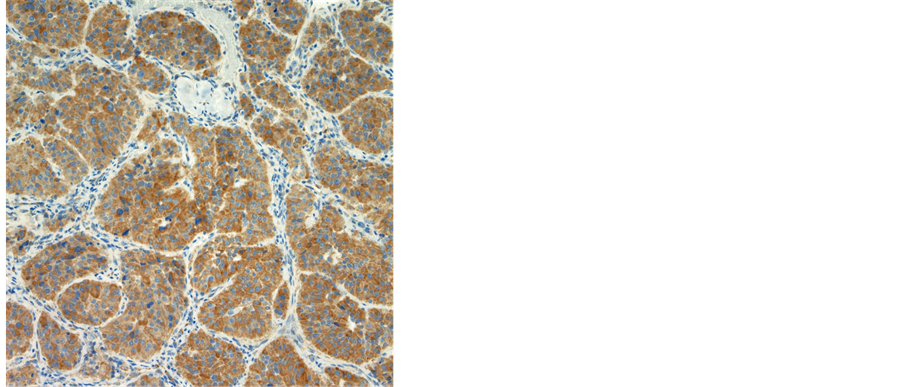 (c)
(c)
Figure 3. (a) Histological features of a paraganglioma. Note the organoid architecture (“zellballen”) arranged around ectatic, sinusoidal blood vessels. The cubic to polygonal tumor cells presents a partially amphophile, partially eosinophilic cytoplasm, moderate anisokaryosis and no mitotic activity (HE × 10); (b) Focal invasion of the left atrium. Several areas were noted, where single cardiac myocytes were surrounded by tumor cells, hence a focal invasion of the heart was postulated (HE × 40); (c) Immunohistochemical reaction with SDHB: 100% of the tumor cells presented a strong cytoplasmatic reaction (×10).
after operation. The patient was in a good general condition during the entire follow-up period. The investigations of cardiac function revealed no abnormalities. We plan to follow the patient in our thoracic surgery clinic every year radiologically and clinically.
3. Discussion
Paragangliomas are rare neuroendocrine tumors, which originate from extra-adrenal, sympathetic and parasympathetic paraganglia [1] . Embryonic origin is the neural crest, as part of the sympathetic nervous system [2] . Their neuroendocrine cells can secrete catecholamines (sympathetic) or have no secretory function (mostly parasympathetic). Very often, paragangliomas are described alongside pheochromocytomas, which are also called intra-adrenal paragangliomas. While functioning equally, the latter arise from the adrenal glands. Both are quite indistinguishable histologically and clinically, and their nomenclature varied and intertwined over the years. The 2004 World Health Organization (WHO) classification defines pheochromocytomas as neuroendocrine tumors of the adrenal gland, and paragangliomas as extra-adrenal sympathetic or parasympathetic tumors originating from the paraganglia. In the literature, the incidence combined for both tumors is estimated and calculated at 0.8 per 100,000 person years, with a markedly higher incidence after autopsy, as a not insignificant number remains asymptomatic [3] [4] . The mean age at diagnosis of benign paragangliomas is around 47 years [5] , varying depending on tumor localization [6] and genetic background (sporadic vs. hereditary) [7] . The latter is also influencing the gender-dependent incidence. While sporadic tumors are more fre- quent in female patients, the hereditary ones are equally distributed among both sexes [8] .
The parasympathetic paragangliomas are predominantly localized at cervical level and at the skull base [9] . These types of paragangliomas are often associated with genetic syndromes, and are generally benign tumors [10] . Consistent genetic associations of paraganglioma have been known to involve in particular multiple endocrine neoplasia syndrome type I (MEN I), and mutation of the su- ccinate dehydrogenase gene (SDHx). In the present case, positive immunoreaction for SDHB argued against an underlying sporadic or germline mutation of succinate dehydrogenase (SDH).
Sympathetic paragangliomas derive from paravertebral sympathetic nerve str- ands. Hence, they can be found anywhere along their path or along the aorta, where additional paraganglia are located [11] . Up to 86% of these tumors secrete catecholamines (almost always norepinephrine), provoking the same clinical symptoms as pheochromocytomas. A correlation with genetic syndromes is seen in around 25% of patients and approximately 20% of these tumors are malignant.
Clinically, non-secreting paragangliomas are often diagnosed because of their growth effect and compression of adjacent structures. In contrast, the secreting tumors present symptoms related to catecholamine hypersecretion. Not rarely, patients present with nonspecific symptoms such as dyspnea, gastrointestinal hemorrhage, back or chest pain and exercise associated nausea and emesis [12] .
Suspected diagnosis of a paraganglioma can be reached by biochemical investigations and imagery. The recommended diagnostic pathway for secretory paraganglioma is a 24-hour urine test for fractioned metanephrines and catecholamines and/or plasma fractioned metanephrines [5] [13] . If this test is positive, imaging should be performed for tumor localization. CT, MRI, PET/CT or somatostatine receptor scintigraphy is possible approaches [14] [15] [16] . Paragangliomas are associated with other genetic syndromes in about 30%, therefore genetic analysis should be considered depending on the individual situation and clinical presentation [17] .
Histologically, paragangliomas are typically very well vascularized tumors, characteristically in close contact to blood vessels or neural areas. Malignant paragangliomas are not common and not easy to define. No reliable markers are known for differentiation of malignant or benign disease. Definite confirmation of malignancy can be obtained only by evidence of metastatic disease, mostly in regional lymph nodes. Additionally, malignant behavior can be predicted by combining different histological parameters. A helpful instrument is called PASS system (pheochromocytoma of the adrenal gland scales score system), which includes 12 different histologic criteria and should reflect the aggressiveness of the tumor. A high proliferative index, as evidenced by an elevated fraction of Ki-67 immunoreactive tumor cell nuclei, has been shown to correlate with increased malignant potential [18] [19] . In one representative study, a proliferative index inferior to 2.5% tended to correlate with a benign course [20] . In the present case, the proliferative fraction was very low (<1% of the tumor cells).
There is no widely accepted official staging system for paragangliomas below the neck.
According to current literature, surgery is the preferred treatment option for management of potentially resectable paragangliomas below the neck region [1] [5] . In endocrine active tumors, a careful perioperative management of the catecholamine homeostasis is important. Not only the change in circulating catecholamines before and after tumor removal can provoke an imbalance of the hemodynamic situation, manipulation of the tumor alone can potentially lead to hemodynamic instability during operation and secondarily to organ dysfunction, particularly kidney failure. Alpha-adrenergic blockage is commonly used to control blood pressure, for both pre-and intraoperatively [2] .
Paragangliomas as primary cardiac tumors are a rare entity and constitute less than 1% of all cardiac tumors [21] . They normally arise from cells of visceral paraganglia in the left artrium or the aorta. Most cardiac paragangliomas are benign, but local invasion can occur and metastases are also described in single cases [22] . Clinical symptoms depend on its functional status or its local influence by compression of adjacent structures. Nowadays, cardiac MRI is used for definitive preoperative planning. Despite advancements in the imaging techniques, it seems sometimes difficult to rule out local invasion before surgery.
Because of their highly developed vascularization, some authors recommend an embolization by selective angiography some days preoperatively [23] .
Possible operative approaches are commonly midline sternotomy, and right or left thoracotomy. Most patients have to be operated on cardiopulmonary bypass and reconstruction of the cardiac wall has to be performed frequently, using either autologous or bovine pericardial patch [24] . When pulmonary veins are partially involved, an extended patchy reconstruction has to be performed to cover defect and to maintain vascular continuity. Cardiac autotransplantation or even orthotopic cardiac transplantation has to be included in the preoperative debate in case of extended resection. When complete resection of the tumor is achieved, long-term prognosis is excellent. Survival of up to 14 years has been reported [25] .
4. Conclusion
Paragangliomas are extremely rare chromaffin cell tumors that may be treated curatively by resection. Because of unspecific symptoms and clinical presentation, the diagnosis of this type of tumor is a challenge. Moreover, as seen in our case, advanced imaging techniques do not predict the surgical strategy for the intervention in all instances. Surgical resection, however, is the treatment of choice and operative techniques suggested include simple excision, excision with reconstruction, or even cardiac autotransplantation or orthotopic cardiac transplantation. Performing careful hemostasis during operation, complete resection and reconstruction can commonly be achieved in a safe manner and with excellent local tumor control and outcome for the patient.
Consent for Publication
Informed consent was obtained from the patient for publication of this case report and any accompanying images.
Cite this paper
Tudor, A.-A., Tschui, J., Schmidli, J., Schmid, R.A. and Dorn, P. (2017) Cardiac Paraganglioma―A Rare Subset of a Rare Tumor. World Journal of Cardiovascular Diseases, 7, 1-9. http://dx.doi.org/10.4236/wjcd.2017.71001
References
- 1. Ramlawi, B., David, E.A., Kim, M.P., Garcia-Morales, L.J., Blackmon, S.H., Rice, D.C., Vaporciyan, A.A. and Reardon, M.J. (2012) Contemporary Surgical Management of Cardiac Paragangliomas. The Annals of Thoracic Surgery, 93, 1972-1976.
- 2. Hui, S., Miao, Q., Luo, A., Liu, J., Yu, C. and Huang, Y. (2016) Unexpected Circulatory Collapse after Cardiac Paraganglioma Resection: Rescue with IntraAortic Balloon Pump and Extracorporeal Membrane Oxygenator. Journal of Cardiothoracic and Vascular Anesthesia, 30, 1057-1060.
https://doi.org/10.1053/j.jvca.2015.11.006 - 3. Beard, C.M., et al. (1983) Occurrence of Pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clinic Proceedings, 58, 802-804.
- 4. McNeil, A.R., et al. (2000) Phaeochromocytomas Discovered during Coronial Autopsies in Sydney, Melbourne and Auckland. Australian & New Zealand Journal of Medicine, 30, 648-652.
- 5. Erickson, D., Kudva, Y.C., Ebersold, M.J., Thompson, G.B., Grant, C.S., van Heerden, J.A. and Young, W.F.J. (2001) Benign Paragangliomas: Clinical Presentation and Treatment Outcomes in 236 Patients. The Journal of Clinical Endocrinology and Metabolism, 86, 5210-5216. https://doi.org/10.1210/jcem.86.11.8034
- 6. Al-Harthy, M., et al. (2009) Comparison of Pheochromocytomas and Abdominal and Pelvic Paragangliomas with Head and Neck Paragangliomas. Endocrine Practice, 15, 194-202.
- 7. O’Riordain, D.S., et al. (1996) Clinical Spectrum and Outcome of Functional Extra Adrenal Paraganglioma. World Journal of Surgery, 20, 916-921.
- 8. Boedeker, C.C., et al. (2007) Malignant Head and Neck Paragangliomas in SDHB Mutation Carriers. Otolaryngology-Head and Neck Surgery, 137, 126.
- 9. Barnes, L., Tse, L.L., Hunt, J.L. and Michaels, L. (2005) Tumours of the Paraganglionic System: Introduction. In: Barnes, L., Eveson, J.W., Reichart, P. and Sidransky, D., Eds., World Health Organization Classification of Tumours. Pathology & Genetics Head and Neck Tumours, IARC Press, Lyon, 362.
- 10. Welander, J., SÖderkvist, P. and Gimm, O. (2011) Genetics and Clinical Characteristics of Hereditary Pheochromocytomas and Paragangliomas. Endocrine-Related Cancer, 18, R253-R276.
https://doi.org/10.1530/ERC-11-0170 - 11. Lee, J.A. and Duh, Q.Y. (2008) Sporadic Paraganglioma. World Journal of Surgery, 32, 683-687.
https://doi.org/10.1007/s00268-007-9360-4 - 12. King, K.S., Darmani, N.A., Hughes, M.S., Adams, K.T. and Pacak K. (2010) Exercise-Induced Nausea and Vomiting: Another Sign and symptom of Pheochro-mocytoma and Paraganglioma. Endocrine, 37, 403-407.
https://doi.org/10.1007/s12020-010-9319-3 - 13. Lenders, J.W., Duh, Q.Y., Eisenhofer, G., Gimenez-Roqueplo, A.P., Grebe, S.K., Murad, M.H., Naruse, M., Pacak, K. and Young, W.F.J. (2014) Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. Endocrine Society. The Journal of Clinical Endocrinology and Metabolism, 99, 1915-1942.
- 14. Stein, P.P. and Black, H.R. (1991) A Simplified Diagnostic Approach to Pheochromocytoma. A Review of the Literature and Report of one Institution’s Experience. Medicine (Baltimore), 70, 46-66.
- 15. Intenzo, C.M., Jabbour, S., Lin, H.C., Miller J.L., Kim, S.M., Capuzzi, D.M. and Mitchell, E.P. (2007) Scintigraphic Imaging of Body neuroendocrine Tumors. Radiographics, 27, 1355-1369.
- 16. Timmers, H.J., Chen, C.C., Carrasquillo, J.A., Whatley, M., Ling, A., Eisenhofer, G., King, K.S., Rao, J.U., Wesley, R.A., Adams, K.T. and Pacak K.J. (2012) Staging and Functional Characterization of Pheochromocytoma and Paraganglioma by 18F-Fluorodeoxyglucose (18F-FDG) Positron Emission Tomography. Journal of the National Cancer Institute, 104, 700-708.
https://doi.org/10.1093/jnci/djs188 - 17. Chen, H., Sippel R.S., O’Dorisio, M.S., Vinik A.I., Lloyd, R.V. and Pacak, K. (2010) The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors: Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas, 39, 775-783.
https://doi.org/10.1097/MPA.0b013e3181ebb4f0 - 18. Kovacs, K., Bell, D., Gardiner G.W., Honey, R.J., Goguen, J. and Rotondo, F. (2005) Malignant Paraganglioma of the Urinary Bladder: Immunohistochemical Study of Prognostic Indicators. Endocrine Pathology, 16, 363-369.
- 19. Brown, H.M., Komorowski, R.A., Wilson, S.D., Demeure, M.J. and Zhu, Y.R. (1999) Predicting Metastasis of Pheochromocytomas Using DNA Flow Cytometry and Immunohistochemical Markers of Cell Proliferation: A Positive Correlation between MIB-1 Staining and Malignant Tumor Behavior. Cancer, 86, 1583-1589.
- 20. Van der Harst, E., et al. (2000) Proliferative Index in Phaeochromocytomas: Does It Predict the Occurrence of Metastases? The Journal of Pathology, 191, 175-180.
- 21. Lam, K.Y., Dickens, P. and Chan, A.C. (1993) Tumors of the Heart. A 20-Year Experience with a Review of 12485 Consecutive Autopsies. Archives of Pathology & Laboratory Medicine, 117, 1027-1031.
- 22. Arai, A., et al. (1998) Cardiac Malignant Pheochromocytoma with Bone Metastases. Journal of Internal Medicine, 37, 940-944.
- 23. Rakovich, G., Ferraro, P., Therasse, E. and Duranceau, A. (2001) Preoperative Embolization in the Management of a Mediastinal Paraganglioma. The Annals of Thoracic Surgery, 72, 601-603.
- 24. Khan, M.F., Datta, S., Chisti, M.M. and Movahed, M.R. (2013) Cardiac Paraganglioma: Clinical Presentation, Diagnostic Approach and Factors Affecting Short and Long-Term Outcomes. International Journal of Cardiology, 166, 315-320.
- 25. Pachter, M.R. (1963) Mediastinal Nonchromaffin Paraganglioma. A Clinicopathologic Study Based on Eight Cases. The Journal of Thoracic and Cardiovascular Surgery, 45, 152-160.