World Journal of Cardiovascular Diseases
Vol.3 No.1A(2013), Article ID:29022,9 pages DOI:10.4236/wjcd.2013.31A015
Cardiac remodeling in postischemic end-stage human hearts: Involvement of extracellular matrix and angiogenesis-related molecules*
![]()
1Department of Translational Medical Sciences, “Federico II” University of Naples, Naples, Italy
2Department of Cardiothoracic Sciences, Second University of Naples c/o Monaldi Hospital, Naples, Italy
3Department of Public Health, “Federico II” University of Naples, Naples, Italy
Email: #loredana.postiglione@unina.it
Received 16 January 2013; revised 1 March 2013; accepted 18 March 2013
Keywords: Heart Failure; Cardiac Remodeling; Vascular Endothelial Growth Factor; Tenascin X; α2-Chain Laminin
ABSTRACT
Background: Extracellular matrix (ECM) participates in heart growth and influences cardiac stem-cell differentiation and migration. The modification of ECM associated with cardiomyopathies is a complex process involving a cohort of proteins. ECM proteins are involved in the regulation of neoangiogenesis in physiological and pathological conditions through their interaction with some angiogenic factors. Our aim was to investigate the role of some angiogenesisrelated ECM proteins in the remodeling heart. Methods: We examined cardiac tissue samples from 21 explanted human hearts and 10 non-failing hearts before transplantation. Each specimen was submitted to morphological and biomolecular analysis. Results: We demonstrated a reduced expression of α2-chain laminin mRNA in pathological samples that could play an important role in the progression of cardiac failure by contributing to sarcolemma modifications. Reduced expression of tenascin cytotactin (TN-C) and TN-X in explanted hearts indicated chronic cardiac damage and an impaired capacity to stimulate new vessel development. The observed type IV collagen increase was not related to neoangiogenesis, as reflected by the decreased expression of vascular endothelial growth factor (VEGF)-A and VEGF receptor-2. The inverse correlation between heart dimension and VEGF-A immunopositivity seems particularly interesting. Conclusions: Our findings suggest that ECM reacts strongly to ischemic damage in failing hearts through some important modifications of its protein composition. Nevertheless, this reaction cannot completely restore myocardium structure if it is not supported by adequate neoangiogenesis. The decrease in some ECM proteins related to vessel development has a negative effect on postischemic neoangiogenesis and clinical outcome.
1. INTRODUCTION
The progression of heart failure (HF), the only cardiovascular disease with an increasing incidence and prevalence [1], is characterized by left ventricular remodeling [2]. Modifications of the extracellular matrix (ECM) associated with cardiomyopathies are very complex, implicating a cohort of proteins [3,4]. Damaged cardiac tissue appears to be able to remodel and regenerate, and the ability of myocytes to proliferate has also been demonstrated [5,6]. Interactions between the ECM and proliferating or differentiating cells play an important and complex role in the control of tissue remodeling in HF [7,8]. This study focused on the production of the ECM proteins most involved in this process [9]; for example, type I collagen (CoI) and fibronectin (FN) are the major components of cardiac connective tissue and their increased production during heart fibrosis is a well-known phenomenon. Less is known about other proteins involved in tissue remodeling and angiogenesis-related processes. For example, type IV collagen (CoIV), a component of the basal lamina of muscle and endothelial cells, is involved in cellular adhesion via integrins and its expression increases during pressure overload or HF [10].
Laminins (LMs), heterotrimeric glycoproteins localized in the basal lamina, perform their function by selfassembling and interacting with other ECM components and with cells via specific integrins [11,12] and a specific LM receptor of 67 KD (67LMR). LMs are composed of three subunits (α, β, γ) organized in a three-dimensional intertwined structure. Among the numerous α-chain isoforms, the α2-chain has been identified only in striated muscles and peripheral nerves [13]; a deficiency in its expression results in muscular dystrophy at birth or in early childhood, accompanied by dilated cardiomyopathy [14] and characterized by the loss of cytoskeleton-ECM linkage [9], with consequent heart dysfunction [15,16].
Tenascins (TNs) constitute another family of cardiac ECM proteins. We have investigated the roles of tenascin cytotactin (TN-C) and TN-X in modulating cell migration and adhesion during angiogenesis. TN-C is strongly expressed in early embryonic development and in pathological conditions such as HF [17], where its expression is related to inflammation and cardiac damage [18]. TNX is expressed during embryogenesis; this protein can stimulate the vascular endothelial growth factor (VEGF) family [19-21] and is associated with blood vessels in many tissues. Their induction is an early and transient response associated with the development of fibrosis in the area surrounding an infarct [19]. In such situations, TN inhibits cardiomyocytes from forming costameric focal contacts but increases the number of cells with noncostameric attachments.
The involvement of these ECM proteins in the regulation of neoangiogenesis in physiological and pathological conditions through interaction with some essential angiogenic factors, such as the VEGF family, has been demonstrated. In adults, ample experimental evidence has shown unequivocally that VEGF is required for the pathological growth of vessels in many conditions, including inflammation, retinopathies, and arthritis [22]. Furthermore, the dependency of tumor expansion on neovascularization has stimulated efforts to suppress VEGF signaling by targeting the ligand or its receptors [23].
VEGF acts through two high-affinity tyrosine kinase receptors, VEGF receptor (VEGFR)-1 [fms-related tyrosine kinase 1 (Flt-1)] and VEGFR-2 [kinase insert domain receptor (KDR)/fetal liver kinase-1 (Flk-1)], both expressed on normal endothelial cells [24] and upregulated during angiogenesis [25]. VEGFR-2 mediates most of the angiogenic functions attributed to VEGF, whereas the role of VEGFR-1 signaling is less clear: it is generally considered as a decoy receptor, at least during development [26]. In addition, autocrine VEGF signaling is required for VEGFR-2 phosphorylation and vascular homeostasis [27], and increased VEGFR-2 expression enhances in vitro angiogenesis [28].
On the basis of these observations, we investigated the expression of angiogenesis-related factors and ECM proteins in healthy and end-stage human cardiac tissues to assess their possible role in heart remodeling.
2. MATERIALS AND METHODS
2.1. Patients and Samples
Cardiac tissue samples were obtained from the explanted hearts of 16 men and 5 women with chronic ischemic cardiopathy presenting as end-stage HF. These patients had been selected for transplantation according to published guidelines. Echocardiographic parameters are reported in Table 1. The mean interval between symptom onset and transplant candidacy was 34 ± 17.18 (range 1 - 60) months, and the mean time on the waiting list was 3.65 ± 2.64 (range 0.26 - 7.48) months. The mean indexed pulmonary vascular resistance was 3.53 ± 1.57 (range 1.0 - 5.8). One patient deteriorated to status I and was maintained on high-dose inotropic support; all other patients were in stable hemodynamic compensation on medical therapy (angiotensin-converting-enzyme inhibitors, diuretics, ß-blockers, digoxin, and warfarin).
Tissue fragments obtained from 10 non-failing human hearts before transplantation were utilized as controls. The donors (aged 21 - 68 years) had died due to causes other than cardiovascular diseases, such as trauma or cerebral hemorrhage. In each case, transmural fragments were obtained from the left atrium and left ventricle. Each collected specimen was divided into three parts and
Table 1. Clinical data.
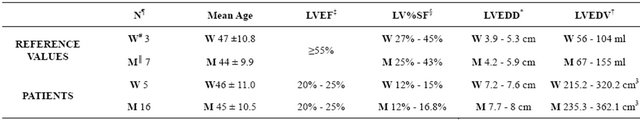
Cardiac tissue samples from explanted hearts of 16 men and 5 women with chronic ischemic cardiopathy; *LVEDD: Left Ventricular End Diastolic Dimension; †LVEDV: Left Ventricular End Diastolic Volume; ‡LVEF: Left Ventricular Ejection Fraction; §LV%SF: Left Ventricular % Shortening Fraction; ║M: Men; ¶N: Controls; #W: Women.
used for paraffin embedding and molecular and cellular biological analyses. Each experiment was performed in triplicate.
All samples from explanted hearts were obtained in accordance with the requirements of the hospital ethics committee. The investigation conformed to the principles outlined in the Declaration of Helsinki [29]. The study was approved by the hospital ethics committee and informed consent was obtained from all transplanted subjects.
2.2. Doppler-Echocardiographic Evaluation
M-mode, two-dimensional, and spectral Doppler-echocardiograph evaluation were performed in all patients before heart transplantation using an ultrasound mechanical system equipped with a 3.5-Hz transducer. System recordings were made with the patients in a left lateral position, according to the standardized protocol of the American Society of Echocardiography. Measurements included inter-ventricular septum end-diastolic dimension, left ventricular posterior end-diastolic wall dimension, left ventricular end-diastolic dimension (LVEDD), and left ventricular end-systolic dimension (LVESD). Left ventricular ejection fraction (LVEF) was calculated as follows: LVEF = (LVEDD3 – LVESD3)/ LVEDD3 [30]. Left ventricular mass (LVM) was calculated according to Devereux et al. [31] and indexed by body surface area (LVMI, g/m2).
2.3. Immunohistochemistry
Fragments obtained from normal and explanted hearts were fixed in 10% buffered formalin, embedded in paraffin, and sectioned. Serial sections (5-µm thickness) of heart specimens were deparaffinized and treated for immunofluorescence and immunoperoxidase staining, as follows.
Sections prepared for immunofluorescence were covered with primary anti-CoIV and anti-TN monoclonal antibodies or with anti-FN and anti-LM polyclonal antibodies (Sigma-Aldrich Corporation, St. Louis, MO, USA) and incubated in a moist chamber at 37˚C for 1 h. After washing in phosphate-buffered saline (PBS), sections were covered with fluoresceinated or rodaminated secondary antibodies (Jackson ImmunoResearch Inc., West Grove, PA, USA) and incubated again under the same conditions. Nuclei were stained with 4’,6-diamidino-2- phenylindole (DAPI) and the sections were mounted in Vecta Shield (Vector Laboratories, Burlingame, CA, USA) and observed under a fluorescence microscope (Leica DM LB Microscope) equipped with an image analyzer.
For immunoperoxidase staining, slides were immersed for 3 min in H2O2 to neutralize endogenous peroxidasethen covered with primary antibodies against VEGF-A (Sigma-Aldrich Corporation) and incubated in a moist chamber at 37˚C for 45 min. After two washes in PBS, sections were covered with peroxidase-conjugated immunoglobulin (AutoProbe III, product no. 08-804x; Biomeda Corporation, Foster City, CA, USA) and incubated in a moist chamber at 37˚C for 45 min, then washed again in PBS and incubated with avidin-biotin peroxidase for 30 min. The dye reaction was developed by incubating for 8 min in a solution of diaminobenzidine tetrahydrochloride containing 0.01% hydrogen peroxide. Slides were finally washed in water, counterstained with hematoxylin, washed in water again, and then mounted in aqueous glycerol solution. Immunopositivity was quantified by three independent investigators blinded to the type of sample, as commonly accepted for semiquantitative morphological evaluations.
2.4. Western Blotting Analysis
Western blotting was used to determine the presence of CoIV, FN, TN, LM, VEGF, and α2-chain LM in heart specimens. Lysates were centrifuged at 14,000 rpm at 4˚C for 5 min. Supernatants were harvested and protein concentrations in cell lysates were determined by a colorimetric assay (Bio-Rad Laboratories Inc., Richmond, CA, USA). For each sample, 100 µg total proteins was incubated at 90˚C for 5 min in Laemmli sample buffer and separated by electrophoresis in 10% sodium dodecyl sulfate-polyacrylamide gels. Gels were electroblotted on polyvinylidene difluouride (PVDF) filters (Millipore, Bedford, MA, USA) and membranes were blocked at room temperature for 30 min. After three passages in washing solution (PBS/0.1% ovalbumin, 0.1% fat-free dry milk, 1% fetal calf serum), membranes were incubated overnight at 4˚C with the specific primary monoclonal antibodies anti-CoIV and anti-FN (Chemicon International Inc., Temencula, CA, USA), anti-TN and anti-LM (Sigma-Aldrich Corporation), and the specific primary polyclonal antibodies anti-VEGF and anti- α2-chain LM (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After four 5-min washes at room temperature with washing solution, membranes were incubated at room temperature for 30 min with a horseradish peroxidase–conjugated secondary antibody (Bio-Rad Laboratories Inc.) diluted to 1:3000 in PBS. After three final washes in Tris-buffered saline Tween-20 (50 mM Tris, pH 7.5; 0.5 M NaCl; 0.2% Tween 20), membranes were stained with an enhanced chemiluminescence detection kit (Amersham, Little Chalfront, UK) and incubated over-night at 4˚C with an anti-α-sarcomeric actin antibody (Sigma-Aldrich Corporation) as an internal standard. Quantitative analysis was performed by scanner densitometry (SnapScan 1212; Agfa-Gevaert N.V.-Mortsel, Belgium).
2.5. Semiquantitative Reverse-Transcriptase Polymerase Chain Reaction Assays
RNA purification. Total RNA was isolated from fragments of cardiac tissue using the phenol-guanidine method. Briefly, samples were homogenized in 4.5 M guanidine thiocyanate, 0.75 M sodium citrate (pH 7.0), and 10% Sarcosyl. After several centrifugations and precipitations in isopropanol, RNA was eluted in sterile, nuclease-free, double-distilled H2O to a total volume of 60 μl. Total RNA concentration was determined in triplicate by absorbance measurements (GeneQuant RNA/ DNA Calculator). The quality of RNA in all specimens was confirmed with a 2% agarose gel, which enabled the visualization of the 28S and 18S bands, markers of RNA integrity.
Reverse-transcriptase polymerase chain reaction. Contaminating DNA was digested with DNase using a DNase Kit (Invitrogen, Grand Island, NY, USA) and 2 μg total RNA was reverse-transcribed with 100 U SuperScript II RNase H reverse transcriptase (RT; Invitrogen) in a volume of 40 μl, using 100 ng random hexamer primers (Roche, Basel, Switzerland) and 10 mM dNTP (Invitrogen) mix, according to the manufacturer’s instructions. The same cDNA product obtained from each sample was used for subsequent polymerase chain reaction (PCR) amplification with the primer sets prepared for the target genes and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) housekeeping gene. The amplification of the GAPDH gene was used as a double internal control. Ratios between the samples and the housekeeping gene were calculated to normalize for initial variations in sample concentration and as a control for reaction efficiency. Primer sequences for RT-PCR were designed using the Primer 3 software (developed by Steve Rozen and Helen J Skaletsky, available at http://www.genome.wi.mit.edu). The reaction mixture contained 3 µl cDNA, 10 - 20 pmol of each primer, 200 mM dNTP, 1.5 mM MgCl2, and 1 unit Taq polymerase with the reaction buffer supplied with the kit.
Amplification was established using a DNA thermal cycler (Perkin-Elmer/Cetus Model 480 DNA Thermal Cycler; Perkin-Elmer Corporation, Norwalk, CT, USA), with 18 - 24 cycles consisting of denaturation at 95˚C for 45 s, annealing at 60˚C for 30 s, and extension at 72˚C for 30 s. At the beginning of the reaction, a 5-min cycle was carried out at 95˚C to activate the Taq polymerase.
The amplified products (12 µl of each sample) were analyzed by electrophoresis in a 2% agarose gel containing ethidium bromide, followed by photography under ultraviolet illumination. Target mRNA levels were estimated by densitometric scanning and normalized against GAPDH loading controls. Densitometric analysis of the PCR products was performed using the ImageJ software (ver. 1.29; developed by Wayne Rasband, available at http://rsb.info.nih.gov/ij/). The densitometric values for the amount of PCR products generated by the increasing volume of the RT reaction were averaged to yield the target genes and GAPDH signals for each sample (densitometric units/μl RT reaction). Data are expressed as the means ± standard deviations (SDs) of the observed values. All PCR products were purified using the QIA quick PCR purification kit (Qiagen Inc., Santa Clarita, CA, USA) and their identities were verified by automated DNA forward and reverse sequencing using dideoxy terminator reaction chemistry for sequence analysis (DNA sequencer Model 373A; Applied Biosystems, Carlsbad, CA, USA).
2.6. Statistical Analysis
Molecular and cellular biological data are presented as means ± SDs. Normally distributed parametric data were analyzed using Student’s t-test, and differences of p < 0.05 or p < 0.01 were considered significant. A regression analysis was also performed to evaluate the relationship between semiquantitative VEGF immunostaining parameters and LVM, as calculated using echocardiogram findings.
3. RESULTS
3.1. Echocardiographic Parameters
As expected, echocardiographic evaluation revealed that the hearts of all patients with end-stage HF were enlarged (Table 1). The LVMI, the major parameter indicating heart enlargement, was 172.7 ± 35.5 g/m2, as compared to a normal value of 135 ± 25.5 g/m2.
3.2. Immunohistochemistry
Immunofluorescence analysis to evaluate immunopositivity in sections of heart specimens due to the expression of ECM proteins demonstrated that LM, FN, and TN immunopositivity was always more evident in pathological specimens, with some difference in the tissue distribution of LM (Table 2). In contrast, immunoperoxidase staining revealed decreased immunopositivity for VEGF-A in specimens from explanted hearts (Figure 1). A significant (p < 0.05) inverse relationship was
Table 2. Semiquantitative immunohistochemical data.
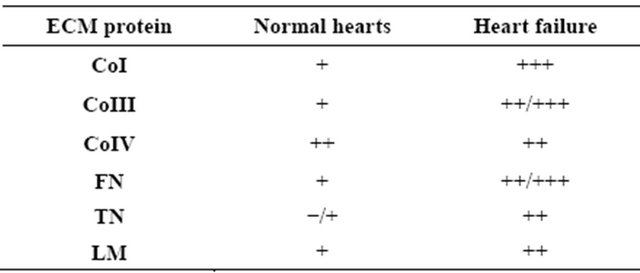
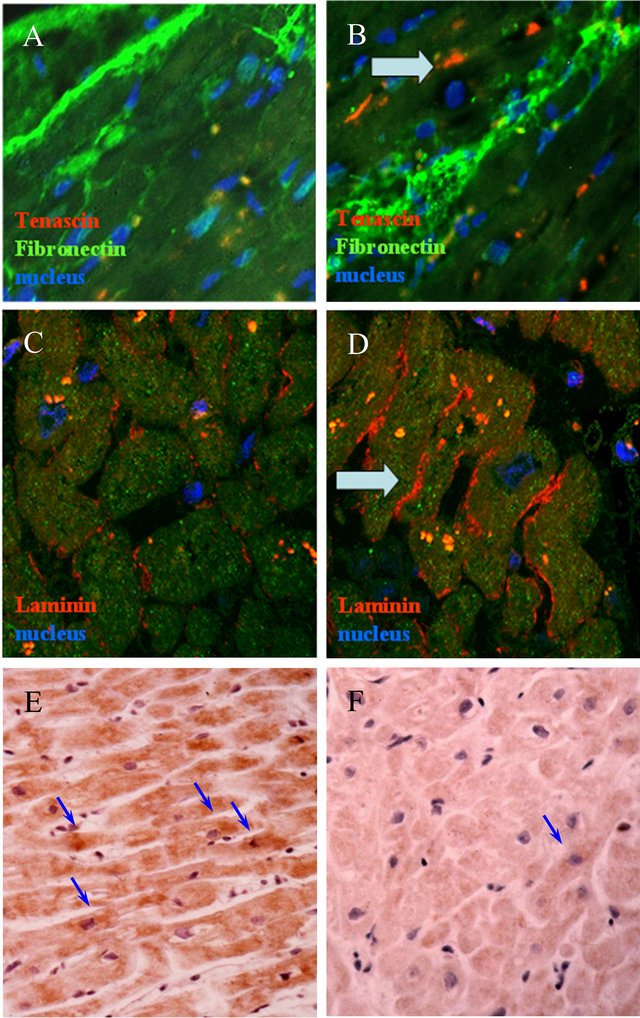
Figure 1. A-Immunofluorescence staining of Tenascin (red) and Fibronectin (yellow): panels A, B; Laminins (red): panels C, D. Sections from non-failing hearts: panels A, C; sections from post-ischemic hearts: panels B, D. Arrows indicate an increased immunopositivity for Tenascin in pathological hearts, as well as an increased and changed distribution of Laminins. Nuclei are stained in blue (magnification: 40×). B-Immunoperoxidase staining of VEGF-A: Panels E: section from nonfailing heart: Panel F: section from post-ischemic heart. Arrows indicate decreased number of VEGF-A+ cells in pathological hearts. Nuclei are stained in blue (magnification: 25×).
found between VEGF-A immunopositivity and the LVMI (r = −0.58; Figure 2).
3.3. Western Blotting Analysis
Western blotting analysis of all cardiac lysates showed bands of variable intensity, with the expected molecular weights of 92 kDa for CoIV, 220 kDa for FN, 200 kDa for TN, 215 kDa for LM, 21 kDa for VEGF, and 400 kDa for α2-chain LM. Quantitative analysis showed that the expression of CoIV and FN was significantly increased in all patients in comparison with control specimens (p < 0.05), whereas no significant difference in TN, LM,
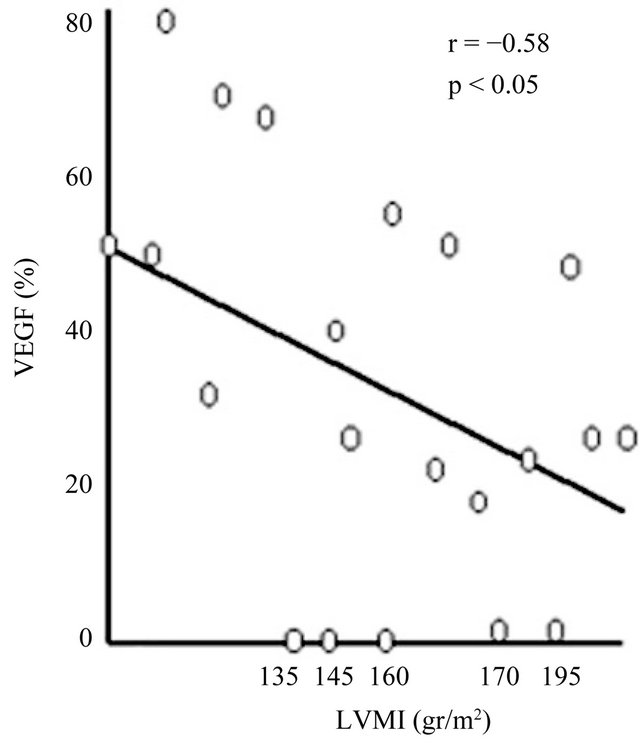
Figure 2. Inverse relationship between VEGF-A expression evaluated as the percentage of stained cardiomyocytes and the Left Ventricular Mass Index (LVMI), calculated as described in the Methods section. The inverse relationship (r = −0.58) was statistically significant (p < 0.05).
VEGF, or α2-chain LM was observed between control and patient samples (Figure 3).
3.4. Semiquantitative Reverse-Transcriptase Polymerase Chain Reaction Assays
RT-PCR analysis of the expression of ECM protein mRNAs (Figure 4) showed increased mRNA expression that confirmed the significant increase in the amounts of FN and CoIV in ischemic hearts observed by western blot analysis. In the same specimens, TN-C, VEGF, and VEGFR-2 mRNA expression was significantly reduced. The level of α2-chain LM mRNA was dramatically reduced in pathological samples when compared with normal specimens. All of these variations were significant (p < 0.05). TN-X mRNA expression was also reduced in pathological samples, but this difference was not significant.
4. DISCUSSION
The ECM participates in the growth and development of the heart, and in its response to pathophysiological stimuli. It is in dynamic equilibrium with the cellular components of the heart and its important role may be mediated by a variety of molecules, such as structural proteins, growth factors [4], metalloproteases, and cytokines. It is well known, for example, that ECM synthesized by fibroblasts can influence cardiac stem-cell differentiation and migration [6] and that environmental factors may
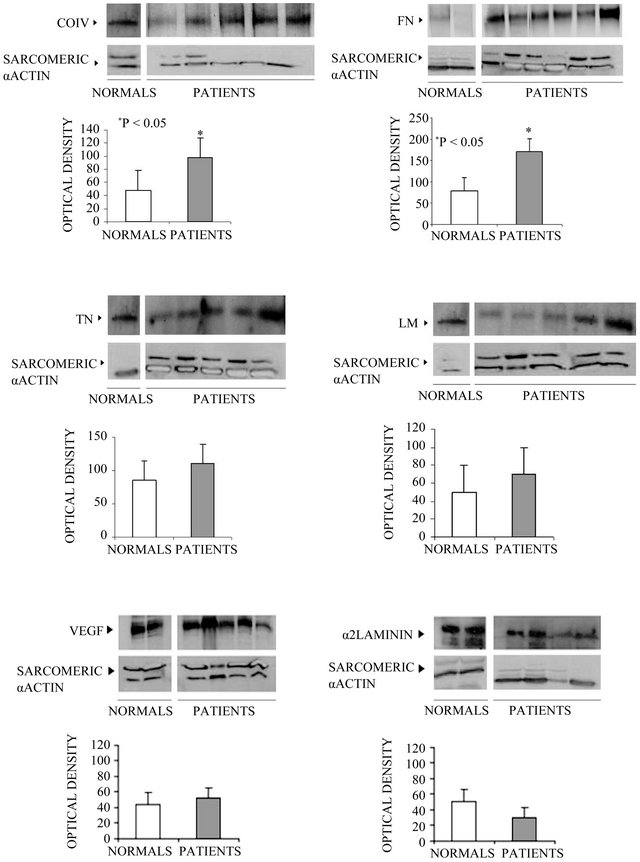
Figure 3. Western blot analysis of type IV Collagen, Fibronectin, Tenascin, Laminin, VEGF and α2 Laminin, in lysates from normal and pathological cardiac tissue fragments. In each panel, the densitometric scanning of the blots is also shown. The difference between normal subjects and patients were considered statistically significant if *p < 0.05. (Each experiment was repeated three times). CoIV = Type IV Collagen; FN = Fibronectin; TN = Tenascin; LM = Laminin; VEGF = Vascular Endothelial Growth Factor.
drive the development of stem cells toward differentiated cell populations, such as cardiomyocytes, smooth muscle cells, and endothelial cells [9].
Less is known about the role of some ECM proteins in inducing and favoring angiogenesis. Our purpose was to study ECM proteins traditionally related to angiogenesis in the remodeling heart, namely CoIV, FN, LM, and TN. The observed increase in some of these proteins confirmed the involvement of ECM in cardiac repair, even though this process was unable to restore physiological
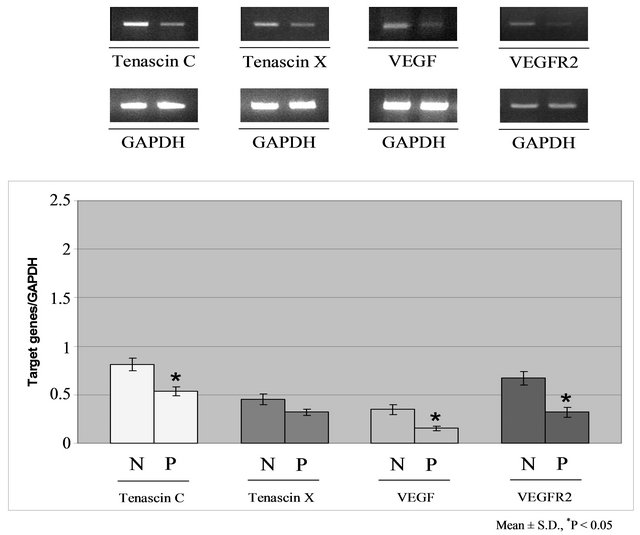
Figure 4. Semiquantitative RT-PCR analysis of mRNA expression for Tenascin-C, Tenascin-X, VEGF and VEGFR2 (upper panel) and for Laminin α2-chain, Fibronectin and type IV Collagen (bottom panel) in cardiac tissues (N = normal subjects; P = patients). The differences were considered statistically significant for *p < 0.05 or **p < 0.01. (Each experiment was repeated three times). VEGF = Vascular Endothelial Growth Factor; VEGFR2 = Vascular Endothelial Growth Factor Receptor 2; GAPDH = Glyceraldehyde-3-phosphate dehydrogenase.
conditions in end-staged hearts.
In particular, we observed by immunofluorescence an increase in TN, FN, and CoIV expression and a more diffuse distribution of LM, which surrounds cardiomyocytes in specimens from pathologic hearts. Western blotting analysis confirmed these data for the whole family of proteins; we observed a strong, significant increase in the expression of CoIV and FN that was further confirmed by semiquantitative RT-PCR. TN and LM also showed a moderate, albeit insignificant, increase.
We then focused our attention on the subtypes of ECM protein that may play a more significant role in human heart remodeling and angiogenesis. We investigated the expression of α2-chain LM, TN-C, and TN-X by RTPCR because these proteins seem to be particularly important in the failing myocardium. TN-C is strongly expressed in the cardiac tissue following inflammatory processes, whereas TN-X is highly expressed in damagedheart and skeletal-muscle ECM and plays a positive role in the regulation of blood vessel development by interacting with VEGF and enhancing its ability to stimulate endothelial cell proliferation.
Our results regarding the expression of LM were particularly interesting. Our immunofluorescence data showed some differences between pathological and control samples in the amount and distribution of the whole family, and western blotting indicated a small increase in protein expression. However, RT-PCR demonstrated a strong, significant reduction in the expression of α2- chain LM mRNA in pathological samples, which may play an important role in the progression of HF by contributing to sarcolemma modification. The α2-chain is present in LM2 and LM4, which are components of the basal lamina of muscle cells such as cardiomyocytes and vascular smooth-muscle cells. Thus, while mRNA expression of the LM family may show a moderate increase as a consequence of the repair process, the suppression of the α2-chain may be one reason for the failure of this process. We also found that mRNA expression of TN-C and TN-X was reduced in explanted hearts, in which chronic damage was paralleled by a reduction of TN-C, which is considered a marker of active inflammatory reactions. The reduction in VEGF and VEGFR-2 expression is probably related to the small decrease in TN-X expression, because this protein can stimulate the VEGF family and is associated with blood vessel neogenesis in many tissues. As the VEGF/VEGFR-2 system is related to enhanced angiogenesis, the small amounts of both proteins reflect the severity of tissue damage in our samples.
The inverse correlation between heart dimension, as evaluated echocardiographically, and VEGF immunopositivity suggests that the reduced neoangiogenic ability of the end-stage heart is related to progressive heart enlargement.
As expected, we also found a significant increase in CoIV and FN levels in end-stage hearts in comparison with control samples. CoIV deposition may be a result of bulk collagen deposition, rather than a contribution to a well-organized basement membrane. The mechanism of quantitative and distributional changes in these non- fibrillar collagens remains unclear, but several lines of evidence have indicated that they play key roles in cardiac cell activation, infarct scar formation, fibrillar collagen organization, and perhaps several other processes during the prolonged phases of myocardial repair. Evidence has also suggested that FN plays an important role in ECM organization and stabilization. Our data confirmed that collagen deposition required FN polymerization into the ECM.
In conclusion, our findings suggest that ECM reacts strongly to ischemic damage in failing hearts through some important modifications of protein amount and distribution. Nevertheless, this reaction cannot completely restore the myocardium structure if it is not supported by an efficacious neoangiogenic process. Our data demonstrate that the reduction in some ECM proteins related to vessel development has a negative effect on postischemic neoangiogenesis, as well as on the clinical outcomes of ischemic patients. In addition, reduced neoangiogenesis seems to be related to the increased dimensions of the heart. Our observations provide a useful foundation for a better understanding of the cardiac microenvironment in healthy and diseased states, particularly regarding the relationship between profibrotic and proangiogenic ECM components.
![]()
![]()
REFERENCES
- Braunwald, E. (2008) Biomarkers in heart failure. New England Journal of Medicine, 358, 2148-2159. doi:10.1056/NEJMra0800239
- Goldsmith, E.C. and Borg, T.K. (2002) The dynamic interaction of the extra cellular matrix in cardiac remodeling. Journal Cardiac Failure, 8, 314-318. doi:10.1054/jcaf.2002.129258
- Cleutjens, P.M.J. and Creemers, E.E.J.M. (2002) Integration of concepts: Cardiac extracellular matrix remodelling after myocardial infarction. Journal Cardiac Failure, 8, 344-348. doi:10.1054/jcaf.2002.129261
- Postiglione, L., Montagnani, S., Rossi, G., et al. (2006) Granulocyte macrophage-colony stimulating factor receptor expression on human cardiomyocytes from endstage heart failure patients. Eurpean Journal of Heart Failure, 8, 564-570. doi:10.1016/j.ejheart.2005.12.007
- Beltrami, A.P., Urbanek, K., Anversa, P., et al. (2001) Evidence that human cardiac myocytes divide after myocardial infarction. New England Journal of Medicine, 344, 1750-1757. doi:10.1056/NEJM200106073442303
- Beltrami, A.P., Barlucchi, L., Anversa, P., et al. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell, 114, 763-776. doi:10.1016/S0092-8674(03)00687-1
- Nadal-Ginard, B., Kajstura, J., Leri, A. and Anversa, P. (2003) Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circulation Research, 92, 139-150. doi:10.1161/01.RES.0000053618.86362.DF
- Urbanek, K., Quaini, F., Anversa, P., et al. (2003) Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proceedings of the National Academy of Sciences USA, 100, 10440-10445. doi:10.1073/pnas.1832855100
- Bendall, J.K., Heymes, T., Ratajczak, P. and Samuel, J.L. (2002) Extracellular matrix and cardiac remodelling. Cardiovascular Research, 95, 1226-1239.
- Shamhart, P.E. and Meszaros, J.G. (2010) Non-fibrillar collagens: Key mediators of post-infarction cardiac remodeling? Journal Molecular Cell Cardiology, 48, 530- 537. doi:10.1016/j.yjmcc.2009.06.017
- Durbeej, M. (2010) Laminins. Cell Tissue Research, 339, 259-268. doi:10.1007/s00441-009-0838-2
- Jones, J.C.R., Dehart, G.W., Gonzales, M. and Goldfinger, L.E. (2000) Laminins: An overview. Microsc Research Technique, 1, 211-213. doi:10.1002/1097-0029(20001101)51:3<211::AID-JEMT1>3.0.CO;2-P
- Ehrig, K., Leivo, I., Argraves, W.S., Ruoslahti, E. and Engvall, E. (1990) Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proceedings of the National Academy of Sciences USA, 87, 3264- 3268. doi:10.1073/pnas.87.9.3264
- Campbell, K.P. (1995) Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell, 80, 675-679. doi:10.1016/0092-8674(95)90344-5
- Di Somma, S., Marotta, M., de Divitiis, O., et al. (2000) Changes in myocardial cytoskeletal intermediate filaments and myocyte contractile dysfunction in dilatative cardiomyopathy: An in vivo study in humans. Heart, 84, 659-667. doi:10.1136/heart.84.6.659
- Oliviero, P., Chassagne, C., Salichon, N., Corbier, A., et al. (2000) Expression of laminin α2-chain during normal and pathological growth of myocardium in rat and human. Cardiovascular Research, 46, 346-355. doi:10.1016/S0008-6363(00)00034-1
- Chiquet-Ehrismann, R. and Chiquet, M. (2003) Tenascins: Regulation and putative functions during pathological stress. Journal of Pathology, 200, 488-499. doi:10.1002/path.1415
- Imanaka-Yoshida, K., Hiroe, M., Yasutomi, Y. and Toyozaki, T., et al. (2002) Tenascin-C is a useful marker for disease activity in myocarditis. Journal of Pathology, 197, 388-394. doi:10.1002/path.1131
- Ikuta, T., Ariga, H. and Matsumoto, K. (2000) Extracellular matrix Tenascin X in combination with vascular endothelial growth factor B enhances endothelial cell proliferation. Genes Cells, 5, 913-927. doi:10.1046/j.1365-2443.2000.00376.x
- Ikuta, T., Ariga, H. and Matsumoto, K. (2001) Effect of Tenascin-X together with vascular endothelial growth factor A on cell proliferation in cultured embrionic hearts. Biological Pharmaceutical Bulletin, 24, 1320-1323. doi:10.1248/bpb.24.1320
- Shalcwijk, J., Zweers, M.C., Steijlen, P.M., Dean, W.B., et al. (2007) A recessive form of the Ehlers-Danlos syndrome caused by Tenascin-X deficiency. New England Journal of Medicine, 345, 1167-1172. doi:10.1056/NEJMoa002939
- Hicklin, D.J. and Ellis L.M. (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. Journal Clinical Oncology, 23, 1011-1027. doi:10.1200/JCO.2005.06.081
- Ferrara, N. and Kerbel, R.S. (2005) Angiogenesis as a therapeutic target. Nature, 438, 967-974. doi:10.1038/nature04483
- Koch, S., Tugues, S., Li, X., Gualandi, L. and ClaessonWelsh, L. (2011) Signal transduction by vascular endothelial growth factor receptors. Biochemical Journal, 437, 169-183. doi:10.1042/BJ20110301
- Dvorak, H.F., Nagy, J.A., Feng, D., Brown, L.F. and Dvorak, A.M. (1999) Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Current Topics in Microbiology and Immunology, 237, 97- 132. doi:10.1007/978-3-642-59953-8_6
- Zeng, H., Dvorak, H.F. and Mukhopadhyay, D. (2001) Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) peceptor-1 down-modulates VPF/ VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinasedependent pathways. Journal of Biological Chemistry, 276, 26969-26979. doi:10.1074/jbc.M103213200
- Lee, S., Chen, T.T., Barber, C.L., Jordan, M.C., et al. (2007) Autocrine VEGF signaling is required for vascular homeostasis. Cell, 30, 691-703. doi:10.1016/j.cell.2007.06.054
- Smadja, D.M., Bieche, I., Helley, D., Laurendeau, I., et al. (2007) Increased VEGFR2 expression during human late endothelial progenitor cells expansion enhances in vitro angiogenesis with up-regulation of integrin 6. Journal of Cellular and Molecular Medicine, 11, 1149-1161. doi:10.1111/j.1582-4934.2007.00090.x
- Rickham, P.P. (1964) Human experimentations. Code of ethics of the World Medical Association. Declaration of Helsinki. British Medical Journal, 2, 177.
- De Simone, G., Devereux, R.B., Roman, M.J., et al. (1994) Assessment of left ventricular function by the midwall fractional shortening-end-systolic stress relation in human hypertension. Journal American College of Cardiology, 23, 1444-1451. doi:10.1016/0735-1097(94)90390-5
- Devereux, R.B. and Reichek, N. (1977) Echocardiographic determination of left ventricular mass in man: Anatomic validation of method. Circulation, 5, 613-618. doi:10.1161/01.CIR.55.4.613
NOTES
*This work was supported by grant from MIUR-project PRIN 2009, which is fully acknowledged.
#Corresponding author.