World Journal of Cardiovascular Diseases
Vol. 2 No. 4 (2012) , Article ID: 23677 , 10 pages DOI:10.4236/wjcd.2012.24038
Mechanisms of C-reactive protein, interleukin-6 and soluble CD40L blood concentration changes after coronary stent implantation
![]()
1Institut Cardiologic, Clínica Rotger, Palma de Mallorca, Spain
2Fundació Banc de Sang i Teixits de Les Illes Balears, Palma de Mallorca, Spain
Email: *amerino@clinicarotger.es
Received 25 June 2012; revised 29 July 2012; accepted 23 August 2012
Keywords: Angioplasty; Inflammation; C-Reactive Protein; Interleukin-6; CD40 Ligand
ABSTRACT
Background: The pathway linking inflammation and thrombosis has been extensively studied. Experimental data support that arterial thrombosis also induces a detectable inflammatory response, which in turn, activates prothrombotic pathways closing a vicious circle that interconnects inflammation and thrombosis. Aim: We designed this study to investigate the causes of inflammatory markers increase after coronary angioplasty. Methods: We analyzed the interrelationship of thrombotic and inflammatory markers and the effect of blocking thrombus formation on the inflammatory response in 50 patients undergoing high thrombotic risk coronary angioplasty. The relationship of platelet number to soluble CD40 Ligand, Interleukin-6 and C-reactive protein blood levels was studied. Half of the study population was treated with standard antithrombotic drugs and the other half with the standard therapy plus platelet GP IIb-IIIa receptor inhibitor Eptifibatide. Results: There was a clear correlation between basal platelet count and sCD40L basal levels, post-angioplasty sCD40L increase and post-angioplasty IL-6 levels and post-angioplasty IL-6 levels with post-angioplasty CRP levels. Postangioplasty CRP, IL-6 and sCD40L blood levels were influenced by GP IIb-IIIa treatment in patients with angiographic thrombus. Conclusion: Platelet aggregation induces a pro-inflammatory response which is blocked by a GP IIb-IIIa inhibitor agent, particularly in patients with patent angiographic thrombus.
1. INTRODUCTION
Atherosclerosis is a chronic inflammatory disease [1]. It is generally accepted that the chronic and subtle increase in blood levels of several inflammatory markers, such as CRP or IL-6, reflects the underlying inflammatory process which affects the arterial wall [2]. Inflammatory markers show a sharp rise in the setting of ACS and after percutaneous coronary interventions, that lasts only for 48 - 72 hours, then returning to the previous levels [3-5]. Current opinion relates the sudden increase in CRP and IL-6 at the initiation of ACS to a “inflammatory burst”.
Intra-arterial thrombus formation, which is a pathophysiological landmark of ACS, can induce a pro-inflammatory reaction, and is a more plausible cause for the increase in inflammatory markers than the “systemic inflammatory burst” [6]. Many studies give support to the hypothesis that platelet activation/aggregation—by means of CD40L and sCD40L activity—may induce a pro-inflammatory cytokine response [7-11].
The CD40 ligand is a transmembrane protein that has cell recognition and interaction functions. Both CD40L and sCD40L interact with CD40 receptor, expressed in the membrane of many cells inducing a pro-inflammatory response [7-11]. Human platelets carry preformed CD40L molecules, which rapidly appear on the platelet surface following stimulation by thrombin [11]. Interaction of CD40 with its ligand CD40L induces a rapid downregulation of membrane-bound CD40L and the release of the soluble form of CD40L [10,11]. Therefore, the CD40L/sCD40L system appears to be a controlled pathway by which the organism produces a local thromboinflammatory reaction directed to prevent focal blood leaks without inducing an exaggerated systemic inflammatory reaction, which is: 1) Very rapid and limited in time, because it takes minutes to up to 6 hours to develop; 2) Limited in magnitude, because platelets are anucleated cells that release all the CD40L/sCD40L they carry but cannot produce more on demand like other types of cells such as lymphocytes or endothelial cells; and 3) Local, because sCD40L has a very unstable molecular structure and a short half-life.
Given the abundance of platelets in relation to cells with secretory ability in the vascular system, the CD40L/ sCD40L system has an enormous potential to induce a detectable inflammatory reaction in the bloodstream [10,11].
In this study, we try to address the interrelationship which happens between intra-arterial thrombus formation and the subsequent inflammatory response in vivo. We have analyzed the relationship of platelet number to sCD40L, IL-6 and CRP blood levels in a prothrombotic condition such as coronary stent implantation, and assessed the effects of treatment with a specific anti-platelet agent such as the platelet GP IIb-IIIa receptor inhibitor eptifibatide.
Aim. We designed this study to investigate the interrelationship between intraarterial thrombosis and the inflammatory response.
2. METHODS
2.1. Study Design and Inclusion Criteria
This study was designed as a prospective, non-randomized, controlled, open-label trial. FIfty patients in risk of a thrombotic complication were consecutively selected from those who underwent a coronary angioplasty procedure with stent implantation at the Catheterization Laboratory at Clinica Rotger, Palma de Mallorca, Spain. The study was approved by the Hospital Ethical Committee and in accord with the Helsinki Declaration of 1975 as revised in 1983. Inclusion criteria were clinical, angiographic or both. Clinical criteria were: Unstable angina with chest pain at rest and ECG changes in the previous 48 hours, or severe stable (Canadian Cardiovascular Society class ≥ 3 angina), and no serum creatine kinase or lactate dehydrogenase elevation on inclusion. Angiographic criteria were: Culprit lesions defined as angiographically complex by the Ambrose et al. classification [12-14] with or without “angiographic thrombus”. Patients with acute MI, primary angioplasty in acute MI, recent surgery (<3 months), recent stroke and metabolic, inflammatory, infectious and neoplastic diseases which could induce CRP elevation were excluded.
2.2. Angioplasty Procedure
Coronary angioplasty was performed in the first 48 hours after admission in patients with unstable angina, in the first 48 hours after post-MI angina, and, in all cases, immediately after the diagnostic angiography which showed the complex lesion that allowed the patient`s inclusion.
All patients underwent the standard angioplasty protocol of our Institution, which at the time of the study included treatment before the procedure with aspirin (100 mg/day), heparin (15.000 IU) and clopidogrel (75 mg/day; no pre-treatment, no loading dose).
Twenty-five consecutive patients received open-label treatment with the glycoprotein IIb-IIIa inhibitor Eptifibatide (180 mcg/Kg i.v. bolus plus 2 mcg/Kg/min infusion) immediately after the procedure and for the following 24 hours and the next 25 patients received standard antithrombotic therapy as described above.
2.3. Coronary Angiography and Quantitative Measurements
Coronary angiography was performed by femoral approach using 6F catheters, using orthogonal planes for each major coronary artery. The variables selected from diagnostic angiography are expressed in Table 1. The optimal single-plane projection was selected, which identified the stenosis in its greatest severity with minimal foreshortening or overlapping of branches, and enddiastolic frames were chosen for quantitative angiographic analysis using a validated automated edge detection algorithm (CAAS. Pie Medical Systems. Maastricht. Netherlands). All angiographic data were evaluated by an author blinded to randomization and clinical outcomes.
2.4. Blood samples
Clinical data and blood test results were prospectively gathered and included in our data base. Blood samples were collected immediately before the intervention, and at 24 and 48 hours after the procedure. Haemoglobin was measured by colorimetry (Hmx, Beckman Coulter), platelet number by impedance (Hmx, Beckman Coulter), fibrinogen by coagulometry (ACLTOP 500, Instrumentation Laboratory), creatinine by Jaffe rate method by Unicel DxC800 (Synchron Clinical System, Beckam Coulter), glucose by oxygen rate method by Unicel DxC800 (Synchron Clinical System, Beckman Coulter), Na and K by indirect potentiometry electrodes by Unicel DxC800 (Synchron Clinical System, Beckman Coulter), HDL by Direct measured by timed-end point method Unicel DxC800 (Synchron Clinical System, Beckman Coulter), LDL was calculated by Friedewald formula, triglycerides by timed-end point method by Unicel DxC800 (Synchron Clinical System, Beckman Coulter), and homocysteine by polarized immunofluorescence assay by Axym (Abbott).
CRP samples were collected in a separate test tube and analyzed immediately after the procedure. C-Reactive Protein levels were determined by Rate Turbidimetry (IMMAGE® Immunochemistry systems. Beckman Coulter). The IMMAGE® Immunochemistry Systems
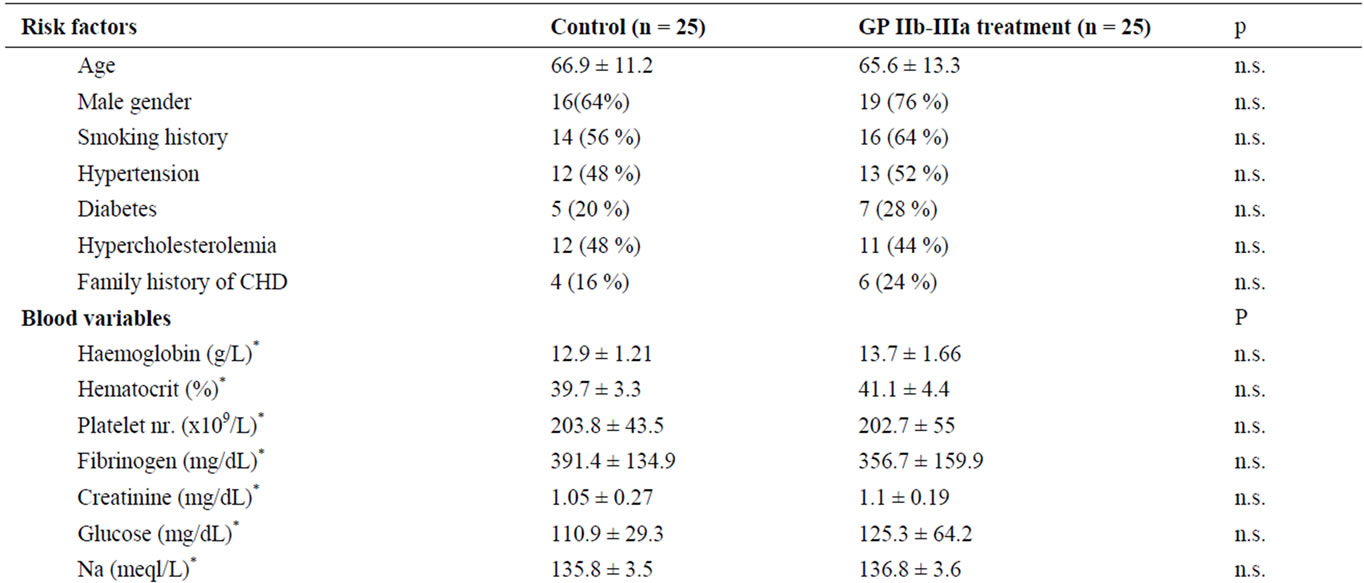
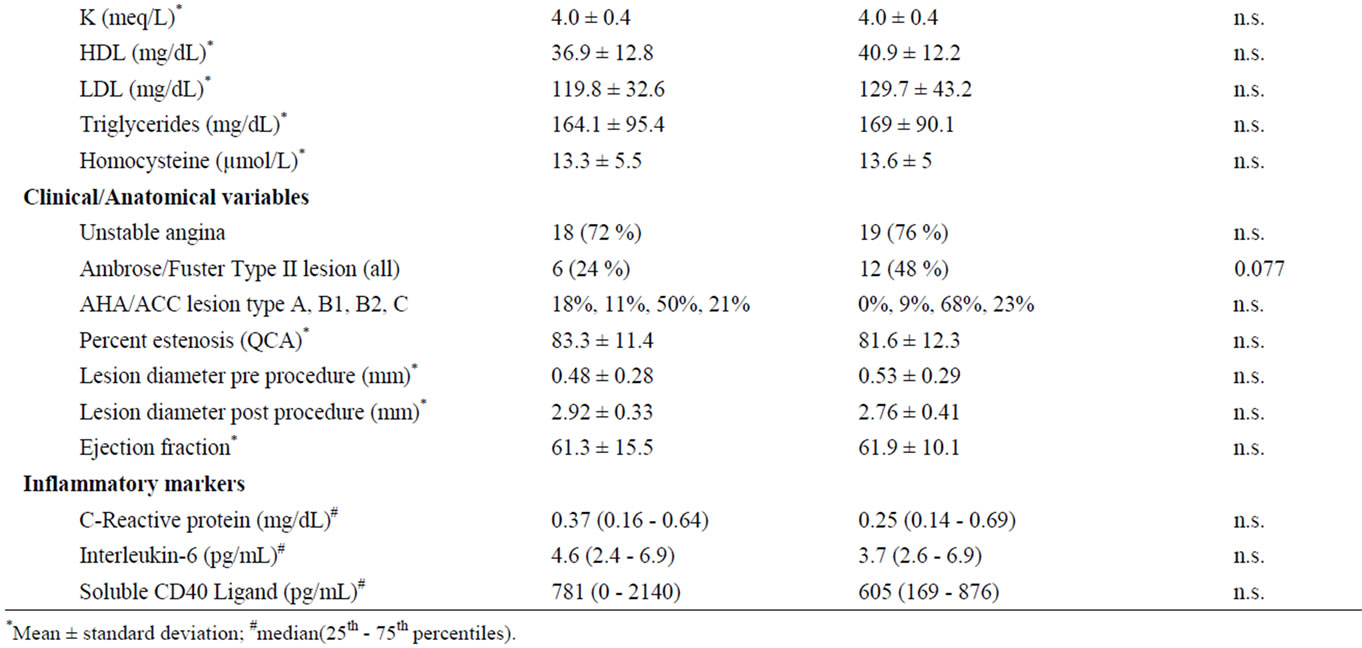
Table 1. Risk factors, except for age are expressed in percentage of cases. Blood variables and Clinical/Anatomical variables in percentage of cases an mean ± standard deviation. CHD: Coronary heart disease. Inflammatory markers in median (25th - 75th percentiles).
CRPH reagent is based on the highly sensitive Near Infrared Particle Immunoassay rate methodology. An antiCRP antibody-coated particle binds to CRP in the patient sample resulting in the formation of insoluble aggregates causing turbidity. The rate of aggregate formation is directly proportional to the concentration of CRP in the sample. The median normal value for CRP is 0.8 mg/L (0.08 mg/dL), with 90 percent of normal values <3 mg/L (0.3 mg/dL) and 99 percent <10 mg/L (1 mg/dL) [15]. Values over 10 mg/L (1 mg/dL) that suggest acute inflammation should be excluded [15] and were not found in our patients.
Blood samples for IL-6 and CD40L/sCD40L quantification were collected at room temperature in non-anticoagulated tubes. Next, samples were centrifugated at 2.000 rpm × 10 min, serum was extracted and then frozen at −80˚C until the end of the study, when the analysis were performed.
Interleukin-6 was analyzed by using the Human Inflammation Kit, BDTM Cytometric Bead Array (BD Biosciences) following the manufacturer’s instructions. Lower detection limit was 2.5 pg/ml. Intra-essay coefficient of variation was 8% and inter-essay was 10%.
2.5. Soluble CD40L Determination
Soluble and membrane bound CD40L levels were determined by using a commercial ELISA kit which recognizes the sCD40L C-terminal (Human sCD40L ELISA. Bender Med-systems. Vienna. Austria). Lower detection limit was 0.095 ng/ml. Intra-essay coefficient of variation was 4% and inter-essay was 6.8%. We analyzed the “absolute blood level of sCD40L” [16,17], which combines the freely circulating sCD40L and CD40L expressed in circulating cells, namely platelets. For that purpose, we collected blood samples at room temperature, and serum was separated after blood clotting in nonanticoagulated tubes. That means, we measured the sCD40L which was already released plus the membrane-bound CD40L which was expressed on activated platelets and cleaved to release additional sCD40L after blood clotting in the test tube.
2.6. IL-6 −174 GC Gene Promoter Polymorphism
Analysis of IL-6 polymorphism was done as described elsewhere [18]. DNA was amplified by polymerase chain reaction (PCR) by using the primers IL-6-Fw: 5’-GTCT ACAACAGCCGCTCACA-3’ and IL-6-Rv: 5’-ATCTTT GTTGGAGGGTGAGG-3’. 10 ml of the PCR product were digested with 2 units of Nla III, in a final volume of 20 μl. Digested products were analyzed by electrophoresis on a 2% agarose gel stained with ethidium bromide (10 mg/ml). Nla III digestion of the IL-6 500 bp amplicon shows 3 bands of 264, 207 and 29 bp in the −174 G allele, the −174 C allele shows 4 bands of 264, 122, 85 and 29 bp.
2.7. Statistical Analysis
Chi-square test was applied to compare qualitative variables. Quantitative variables were compared by linear regression equations. Continuous variables were summarized by median and interquartile ranges. Changes in levels of serum inflammatory markers from baseline to 24 hours were assessed relative to baseline (before drug administration), and changes from 24 to 48 hours (after the drug’s effect was over). Differences between control and eptifibatide groups were compared using the nonparametric Kruskall-Wallis test, because data were not normally distributed. A sample size of 50 was calculated to produce an 80% power to detect a 20% difference between treatment groups with regard to the rise in inflammatory markers with a significance level of 0.05.
3. RESULTS
3.1. Patients
Unstable angina was the clinical presentation in 37 patients (76%), and there was a nonsignificant trend towards more unstable patients in the GP IIb-IIIa treatment group (Table 1). The two groups were similar regarding the factors potentially affecting the objectives of the study. There were no differences in age, gender nor risk factor presentation among both, treatment an control groups. There were also no differences between groups in rheological, metabolic, atherothrombotic, anatomical or inflammatory variables which could have affected the objectives of the study (Table 1).
The percentage of patients with thrombus/complex lesions was high, but the type II lesions were equally distributed among both treatment groups (Table 1). Unstable angina patients presented as much as a 34.4% of type II lesions as opposed to only 8% of stable angina patients (p < 0.05).
3.2. The Thrombosis-Inflammation Link
We propose a sequence of events that links intra-arterial thrombosis and inflammation which is:
Platelet activation/aggregation→CD40L expression/ sCD40L release→IL-6 production→CRP production.
Following the proposed sequence of events, in all patients before treatment, basal platelet number was related to basal sCD40L/CD40L levels (r = 0.60; p < 0.001), (Figure 1). In non-treated patients, the 24 hour sCD40L/ CD40L increase was related to 24 hour post-angioplasty IL-6 concentration (r = 0.54; p < 0.01) (Figure 2). Finally, 24 hours post-angioplasty IL-6 and CRP levels in non-treated patients showed also a significant linear correlation (r = 0.49; p < 0.05) (Figure 3). There was no significant relationship between sCD40L/CD40L increase and post-angioplasty CRP levels, suggesting that the pro-inflammatory effect is mediated by IL-6 production. This sequence of events demonstrate a return pathway from thrombosis (platelet aggregation) to inflammation (CRP increase), closing the vicious circle between inflammation and thrombosis (see discussion).
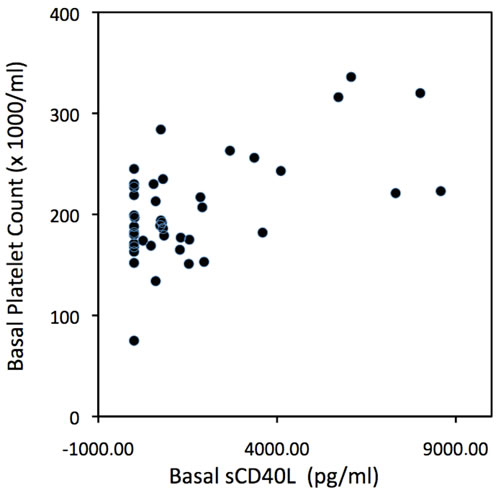
Figure 1. Scatter plot of basal (pre-treatment) platelet count vs basal sCD40L levels in all patients. (r = 0.60; p < 0.05).
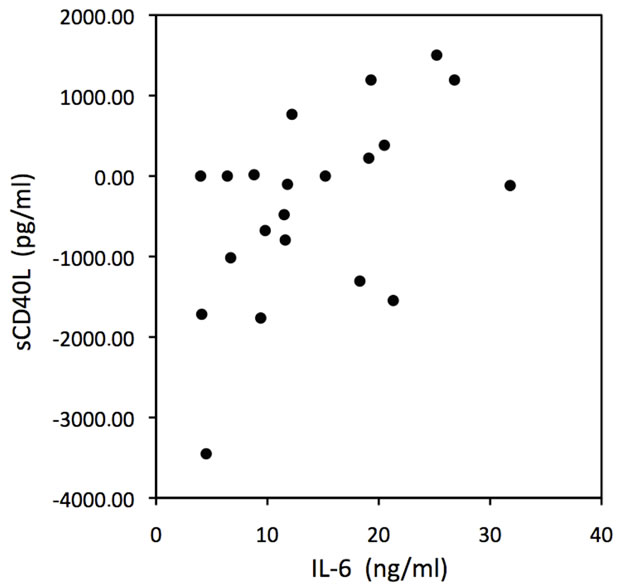
Figure 2. Scatter plot of 24 hours increase in sCD40L levels vs 24 hours IL-6 levels in non-treated patients. (r = 0.54; p < 0.01).
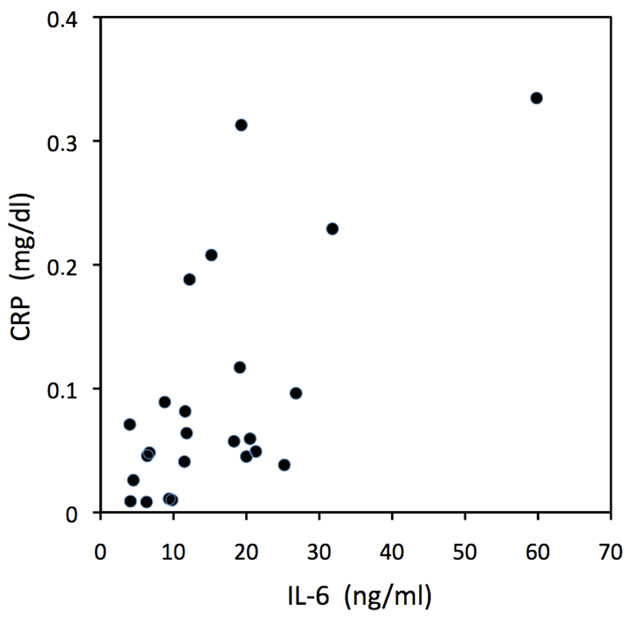
Figure 3. Scatter plot of 24 hours IL-6 levels vs 24 hours C-reactive protein levels in non-treated patients. (r = 0.49; p < 0.05).
3.3. Antithrombotic Treatment and Inflammation
3.3.1. C-Reactive Protein
After angioplasty CRP increased significantly in control patients. This rise was blocked by GP IIb-IIIa treatment (control basal median (25th, 75th percentiles): 0.37 (0.16, 0.64) vs. 24 h: 0.68 (0.45, 1.16) mg/dL; p < 0.05) (treated basal 0.25 (0.14, 0.69) vs 24 h: 0.37 (0.19, 0.79) mg/dL; p = ns). When we analyzed separatedly patients with and without thrombus (Figure 4), GP IIb-IIIa inhibition was effective in patients with angiographic thrombus.
3.3.2. Interleukin-6
IL-6 showed its typical 24 hour post-angioplasty peak (control basal: 4.6 (2.4, 6.9) vs. 24 h: 13.7 (8, 20.1) ng/dL; p < 0.001), which was not modified by GPIIb-IIIa treatment (treated basal 3.7 (2.6, 6.9) vs. 24 h: 8.6 (6.5, 19.6) ng/dL; p < 0.01). When we analyzed separatedly patients with and without thrombus (Figure 5), GP IIb-IIIa inhibition was effective in patients with angiographic thrombus.
3.3.3. sCD40L
Absolute sCD40L showed a significant decrease 24 hr after angioplasty which is consistent with the downregulation of the CD40L receptor observed after platelet aggregation in experimental studies. GP IIb-IIIa inhibittion reduced absolute sCD40L decrease in all patients by sparing sCD40L consumption induced by platelet aggregation (control basal: 781 (0, 2140) vs. 24h: 298(57, 816) pg/ml; p < 0.05)(treated basal 605(169, 876) vs. 24h: 722(273, 1998) pg/ml; p = ns). When we analyzed separatedly patients with and without thrombus (Figure 6), GP IIb-IIIa inhibition was effective only in patients without angiographic thrombus.
3.4. Thrombus Formation and the Inflammatory Response
Patients carrying the −174 GG (high IL-6 production) gene promoter polymorphism had more angiographic thrombus (65%) than GC heterozygotes (42%) or patients with the CC (low IL-6 production) polymorphism, who only 10% of them had angiographic thrombus (p = 0.016).
4. DISCUSSION
Although the link between inflammation and thrombosis has been extensively studied, both in “in-vitro” and “in-vivo” studies, the influence of thrombosis on inflammation has yield controversial findings in clinical studies. Lincoff et al. [19] and ourselves [20,21] found an inhibition of inflammatory markers rise after angioplasty in patients who had intensive antiplatelet treatment. James et al., in a GUSTO IV substudy, fail to show a relationship between antithrombotic treatment and inflammation inhibition [22]. That was probably due to the confounding effect of other variables such as myocardial damage which was observed in over 70% of the substudy population, and/or uncontrolled thrombus formation (they did not perform early coronary angiography). The population choosed for this particular study were unstable angina patients with elevated myocardial necrosis markers and recommended delayed coronary intervenetion until the end of the study. The patients selected for our experiment were unstable angina patients with no myocardial enzyme elevation at the time of angioplasty,
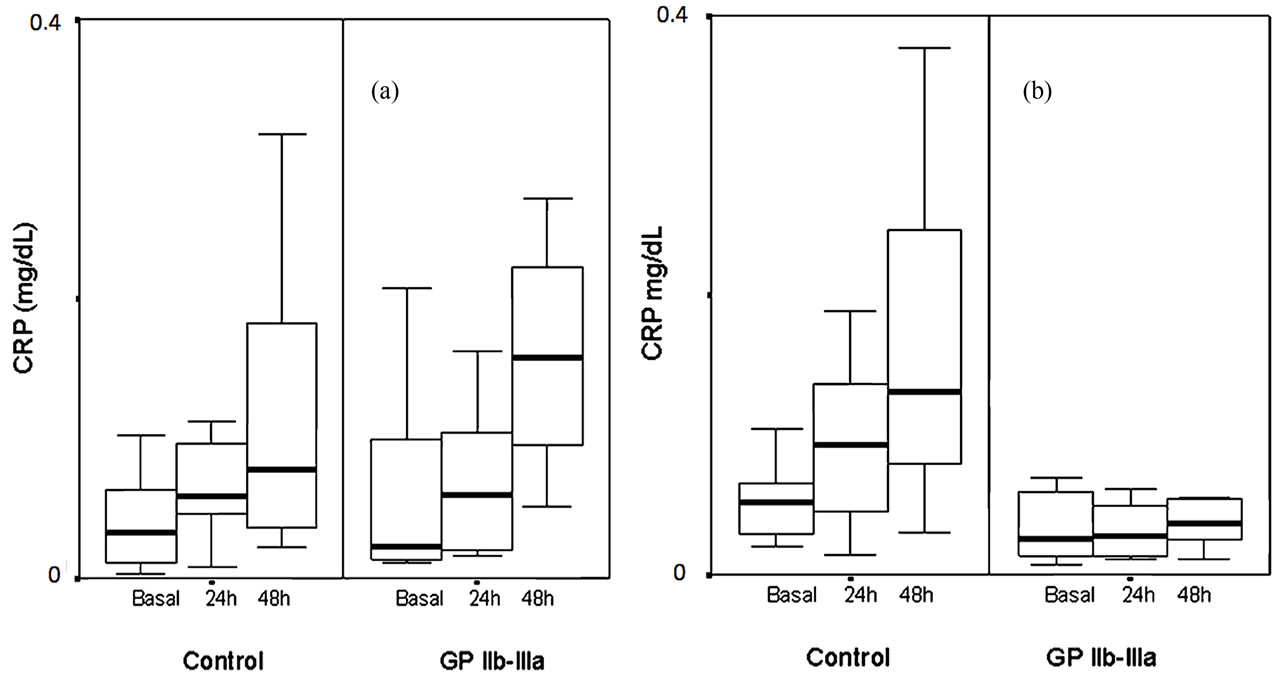
Figure 4. Box-and-whisker plot of changes in CRP levels from baseline to 24 hours, and from 24 to 48 hours postangioplasty in the treated and control groups. The box spans the interquartile range (25th to 75th percentiles), and the line within the box denotes the median. Whiskers extend from the 10th to the 90th percentiles. (a) Patients without angiographic thrombus: control basal median (25th, 75th percentiles): 0.33 (0.1, 0.65) vs. 24 h: 0.58 (0.41, 1.00) mg/dL; p < 0.01; treated basal 0.23 (0.13, 1.00) vs. 24 h: 0.59 (0.19, 1.19) mg/dL; p = ns. (b) Patients with angiographic thrombus: Control basal 0.52 (0.28, 0.71) vs. 24 h: 0.93 (0.44, 1.49) mg/dL; p < 0.05; treated basal 0.25 (0.13, 0.61) vs. 24 h: 0.27 (0.12, 0.54) mg/dL; p = ns.
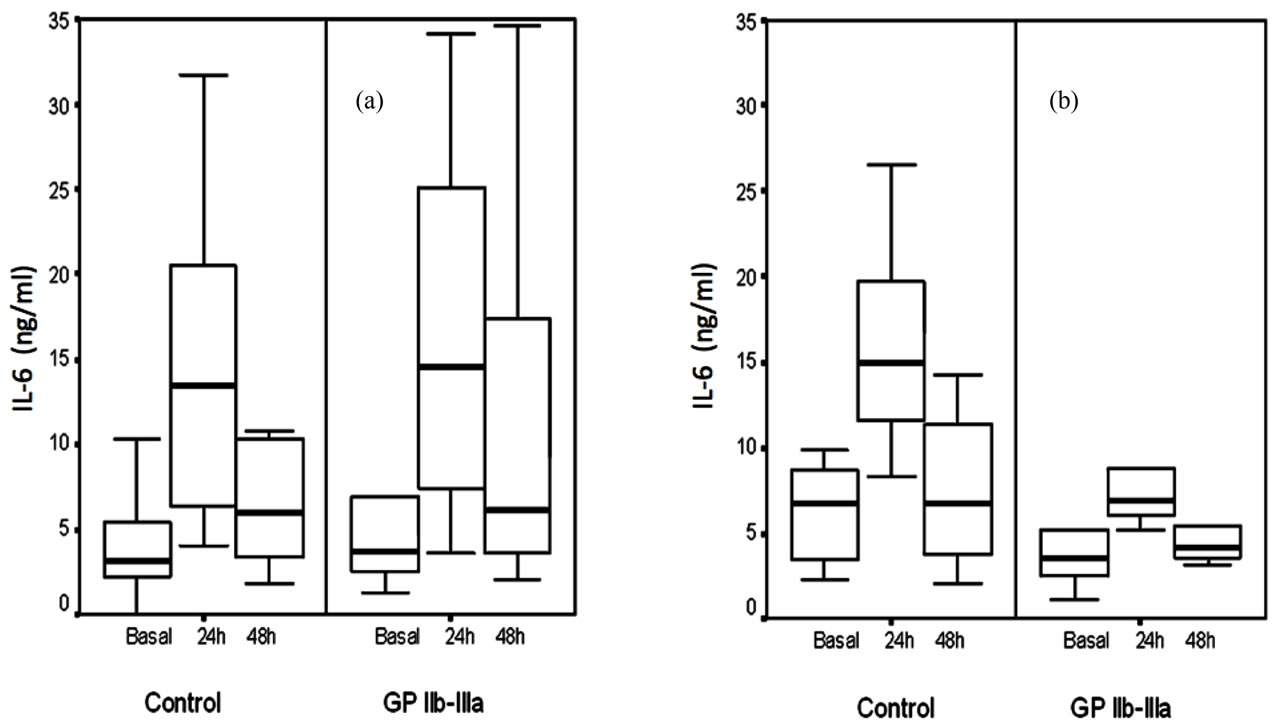
Figure 5. Box-and-whisker plot of changes in IL-6 levels from baseline to 24 hours, and from 24 to 48 hours postangioplasty in the treated and control groups. (a) Patients without angiographic thrombus: control basal median (25th, 75th percentiles): 3.1 (2.2, 5.6) vs. 24 h: 13.5 (6.4, 20.7) ng/ml; p < 0.01); treated basal 3.7 (2.5, 7) vs. 24 h: 14.6 (7.1, 26.1) ng/ml; p < 0.001. (b) Patients with angiographic thrombus: Control basal 6.7 (3, 9.3) vs. 24 h: 14.9 (11.5, 19.8) ng/ml; p < 0.01; treated basal 3.6 (2.2, 5.0) vs. 24 h: 7 (5.8, 9.5) ng/ml; p = ns.
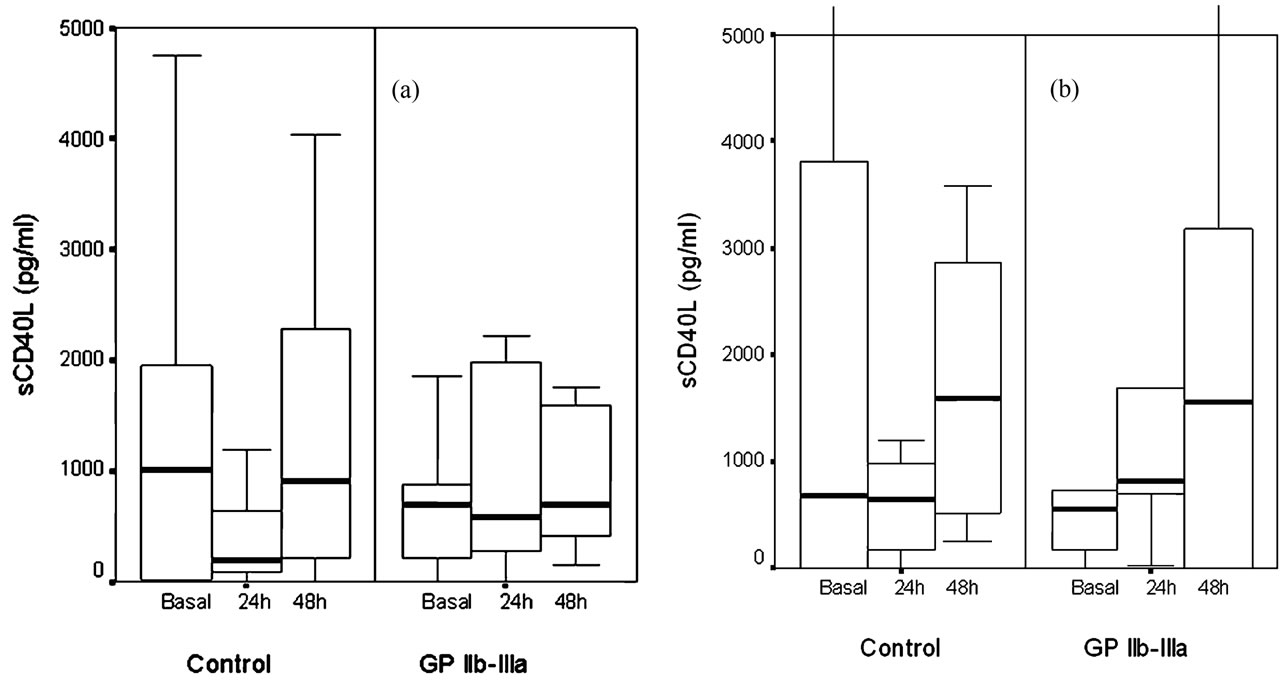
Figure 6. Box-and-whisker plot of changes in sCD40L levels from baseline to 24 hours, and from 24 to 48 hours postangioplasty in the treated and control groups. (a) Patients without angiographic thrombus: Control basal median (25th, 75th percentiles): 1017 (12, 2140) vs. 24 h: 198 (16, 728) pg/ml; p < 0.05; treated basal 693 (196, 895) vs. 24 h: 582 (255, 2040) pg/ml; p = ns. (b) Patients with angiographic thrombus: Control basal 686 (0, 3936) vs. 24 h: 649 (86, 986) pg/ml; p = ns; treated basal 546 (80, 680) vs. 24 h: 811 (658, 1577) pg/ml; p = ns.
so that the effect of stent implantation was not confounded by even subtle myocardial necrosis. Thus, we excluded patients with complicated procedures, acute myocardial infarction and any with periprocedural enzyme elevation.
Platelets are a key step in the arterial thrombotic complications of atherosclerosis and are known to carry many pro-inflammatory substances. So platelets can well be the link between thrombosis and inflammation. More than 300 proteins have been identified in human platelets [23]. Among them, some data point to the CD40L/ sCD40L system as playing the principal role between intra-arterial thrombosis and the increase in inflammatory markers [7-11].
We designed this experiment to assess if there is a relationship between intra-arterial thrombosis and inflamematory markers elevation after a pro-thrombotic event such as coronary stent implantation. We found that in non-treated patients: 1) There is a linear relationship between platelet number and sCD40L concentration, between sCD40L post-angioplasty increase and IL-6 postangioplasty concentration, and between IL-6 post-angioplasty concentration and CRP post-angioplasty concentration; 2) A GP IIb-IIIa inhibitor blocked CRP and IL-6 increase particularly in patients with angiographic thrombus, and preserved sCD40L consumption in all patients.
There are several issues to be discussed in relation to methodology, the findings themselves and their pathophysiological implications.
4.1. Methodological Issues
We did not perform randomization of patients in this study, although patients were consecutively assigned to treated and control groups. We controlled all the variables which may have altered the results of the experiment: Cardiovascular risk factors, metabolic indexes, and rheological and anatomical features. Second, the study of the pathophysiology of acute coronary syndromes is limited by the lack of an exact relationship between the actual intracoronary events and the patient’s clinical picture. We have used coronary angioplasty as a model of acute coronary syndrome. Both conditions share a common pathophysiological sequence after plaque rupture, and in the case of angioplasty, balloon inflation determines the exact time of plaque rupture. Other authors and ourselves have demonstrated an increase in CRP and IL-6 after angioplasty-induced plaque rupture, which peaks 24 - 48 hours after the procedure [19-21]. Percutaneous coronary intervention with stent implantation increases intra-arterial thrombosis in such a magnitude that requires intense antithrombotic treatment [24]. Some will argue that coronary intervention with stent implantation is not a pro-thrombotic condition, but experiments performed in primates clearly show that platelets form a mural thrombus inside the stent immediately after implantation, producing a thin layer which covers the stent’s luminal surface [25]. Abciximab, another GP IIbIIIa platelet receptor blocker, inhibits the formation of this platelet mural thrombus [25]. Third, there have been reports of the anti-inflammatory effects of Eptifibatide, but Eptifibatide has no direct anti-inflammatory effects. It is a cyclic heptapeptide which contains a KGD sequence which competitively blocks the platelet GP IIb-IIIa receptor and has no other known effects [26]. Forth, we classified patients having “angiographic thrombus” those who presented the features of Ambrose/ Fuster type II lesions as described in the Methods section. Angiographically complex lesions were described two decades ago and reflect the platelet rich thrombus image [12-14]. Finally, samples were collected at room temperature because we decided to analyze “absolute” sCD40L as explained in the Methods section. The rationale for “absolute” sCD40L determination is based on a practical as well as a physiological ground. First, it is much easier to achieve reproducible results because the samples must not be immediately frozen after blood collection, and the protocol used for sample freezing and storing may alter the results. Second, it seems more pathophysiologically suited to analyze the absolute and future potential of sCD40L release, than only the past sCD40L released which is responsible for only part of the pro-inflammatory reaction after a pro-thrombotic situation such as coronary stent implantation.
4.2. Questions Related To the Findings
Our study addresses for the first time the issue of how arterial thrombosis may induce a pro-inflammatory reaction in patients. The potential role of CD40L/sCD40L as a link between inflammation, atherosclerosis and thrombosis has been postulated for at least a decade [6,8,27,28]. However, it has not been established a relationship between platelet number and the inflammatory response. Platelets express CD40L within seconds of activation in vitro and in the process of thrombus formation in vivo [10,11]. Soluble CD40L increase was related to postangioplasty IL-6 levels in non-treated patients, showing a link between thrombosis and an amplification of the inflammatory response by the “messenger cytokine” (IL-6). This finding supports the pro-inflammatory effect of the CD40L/sCD40L system on several cell types involved in atherosclerosis reported in experimental studies [9,11,23]. Finally, as expected, since IL-6 is the most potent inductor of CRP production, IL-6 levels were related to CRP levels 24 hours post-angioplasty. This relationship applied only to non-treated patients, since GP IIb-IIIa inhibition blocked the inflammatory response.
There is no clear explanation for the lack of inflammatory response blockade by GPIIb-IIIa inhibitors in patients without angiographic thrombus. It seems that there act other unknown mechanisms which could induce inflammation, but the fact that platelet GP IIb-IIIa receptor inhibition acts in patients with thrombus opens to some discussion.
First, patients carrying the high IL-6 production polymorphism had more angiographic thrombus than those with the low IL-6 production polymorphism. This particular polymorphism modulates the response of IL-6 to arterial thrombosis [20], and it could explain why the GP IIb-IIIa inhibitor blocked CRP, IL-6 and sCD40L increase only in patients with angiographic thrombus. But in second place, platelets release all the sCD40L they carry and cannot produce more. This fact may explain why CRP and IL-6 increase was blocked by the GP IIb-IIIa agent in patients with complex lesions. Patients without complex lesions could retain more sCD40L/ CD40L potential able to induce CRP and IL-6 production.
4.3. Questions Related to Pathophysiology
Patients with Acute Coronary Syndromes and after coronary angioplasty show an increase in CRP and IL-6 levels that peaks 24 - 72 hours after the procedure, and then return to previous levels [3-5,19-22]. The current explanation is that there is a pro-inflammatory effect of coronary interventions, but we have shown that a return pathway exists from thrombosis to inflammation and explain the rise in inflammatory markers in these patients Platelets are the main source of sCD40L in the vascular system. We found a direct relationship between platelet number and sCD40L levels.
Soluble CD40L release from the platelet surface is the sole mechanism for the down-regulation of CD40L. Transmembrane CD40L from platelets is fully released as sCD40L and it has a half-life of approximately six hours [10,11]. These experimental data are fully translated to our clinical data. We found a decrease of sCD40L blood levels after coronary angioplasty, which was spared by platelet GP IIb-IIIa inhibitor Eptifibatide (Figure 6(a)). The change in sCD40L levels after angioplasty was related to IL-6 levels, and in turn, IL-6 post-angioplasty levels were related to post-angioplasty CRP levels (Figures 1-3). This chain of events may explain the increase in IL-6 and CRP that are observed in patients with Acute Coronary Syndromes and after angioplasty. Thus, CRP could be a marker of the thrombotic status of a single patient in the setting of an ACS.
Not only platelets but other major cells involved in the atherosclerotic process, such as smooth muscle cells and leukocytes participate in this closed loop. Inflammation can induce mural thrombus formation, and formed thrombus may amplify inflammation. Inflammation and thrombosis are so deeply interrelated that we could consider antithrombotic drugs as anti-inflammatory agents and conversely [23,29-32]. Patients who have a pro-inflammatory status have an increased risk of thrombotic complications, and in turn, thrombosis increases their pro-inflammatory conditions, closing a vicious circle which induces thrombotic complications. Antithrombotic treatment can block the inflammatory response and break this systemic amplification loop.
5. CONCLUSION
Inflammatory response after angioplasty is produced by a thrombotic sequence which relates platelet aggregation to sCD40L, to IL-6 and, finally to CRP increase. Platelet GP IIb-IIIa receptor inhibitor Eptifibatide block this inflamematory burst, particularly when angiographic thrombus is present.
6. ACKNOWLEDGEMENTS
This study was funded by the Spanish Society of Cardiology.
REFERENCES
- Ross, R. (1999) Atherosclerosis—An inflammatory disease. The New England Journal of Medicine, 340, 115-126. doi:10.1056/NEJM199901143400207
- Libby, P., Ridker, P.M., and Maseri, A. (2002) Inflammation and atherosclerosis. Circulation, 105, 1135-1143. doi:10.1161/hc0902.104353
- Liuzzo, G., Biasucci, L.M., Gallimore, R., et al. (1994) The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina. The New England Journal of Medicine, 331, 417-424. doi:10.1056/NEJM199408183310701
- Biasucci, L.M., Liuzzo, G., Grillo, R.L., et al. (1999) Elevated levels of C-reactive protein at discharge in patients with unstable angina predicts recurrent instability. Circulation, 99, 855-860. doi:10.1161/01.CIR.99.7.855
- Liuzzo, G., Buffon, A., Biasucci, L.M., et al. (1998) Enhanced inflammatory response to coronary angioplasty in patients with severe unstable angina. Circulation, 98, 2370-2376. doi:10.1161/01.CIR.98.22.2370
- Levi, M., van der Poll, T. and Büller, H.R. (2004) Bidirectional relation between inflammation and coagulation. Circulation, 109, 2698-2704. doi:10.1161/01.CIR.0000131660.51520.9A
- Furman, M.I., Krueger, L.A., Linden, M.D., Barnard, M.R., Frelinger III, A.R. and Michelson, A.D. (2004) Release of soluble CD40L from platelets is regulated by glycoprotein IIb/IIIa and actin polymerization. Journal of the American College of Cardiology, 43, 2319-2325. doi:10.1016/j.jacc.2003.12.055
- Aukrust, P., Damas, J.K. and Solum, N.O. (2004) Soluble CD40L and platelets: Self-perpetuating pathogenic loop in thrombosis and inflammation. Journal of the American College of Cardiology, 43, 2326-2328. doi:10.1016/j.jacc.2004.03.023
- Aukrust, P., Müller, F., Ueland, T., et al. (1999) Enhanced levels of soluble and membrane bound CD40 ligand in patients with unstable angina: Possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation, 100, 614-620. doi:10.1161/01.CIR.100.6.614
- Henn, V., Steinbach, S., Büchner, K., Presek, P. and Kroczek, R. (2001) The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood, 98, 1047-1054. doi:10.1182/blood.V98.4.1047
- Henn, V., Slupsky, J.R. and Grafe, M. (1998) CD40 ligand in activated platelets triggers an inflammatory reaction of endothelial cells. Nature, 391, 591-594. doi:10.1038/35393
- Ambrose, J.A., Winters, S.L., Arora, R.R., et al. (1985) Coronary angiographic morphology in myocardial infarction: A link between the pathogenesis of unstable angina and myocardial infarction. Journal of the American College of Cardiology, 6, 1233-1238. doi:10.1016/S0735-1097(85)80207-2
- Ambrose, J.A., Hjemdahl-Monsen, C.E., Borrico, S., Gorlin, R. and Fuster, V. (1988) Angiographic demonstration of a common link between unstable angina pectoris and non-Q wave myocardial infarction. Journal of the American College of Cardiology, 61, 244-247. doi:10.1016/0002-9149(88)90924-1
- Cohen, M., Merino, A., Hawkins, L., Greenberg, S. and Fuster, V. (1991) Clinical and angiographic characteristics and outcome of patients with rest-unstable angina occurring during regular aspirin use. Journal of the American College of Cardiology, 18, 1458-1462. doi:10.1016/0735-1097(91)90675-Y
- Myers, G.L., Rifai, N., Tracy, R.P., et al. (2004) CDC; AHA. CDC/AHA workshop on markers of inflammation and cardiovascular disease: Application to clinical and public health practice: Report from the laboratory science discussion group. Circulation, 110, 545-549. doi:10.1161/01.CIR.0000148980.87579.5E
- Bereczki, C., Nagy, E., Pal, A., Magyar, M.T. and Balla, J. (2003) Should soluble CD40 ligand be measured from serum or plasma samples? Letter. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 1129-1131. doi:10.1161/01.ATV.0000072368.37740.8E
- Ahn, E.R., Lander, G., Jy, W., Bidot, C.J., Jimenez, J.J., Horstman, L.L. and Ahn, Y.S. (2004) Differences of soluble CD40L in sera and plasma: Implications on CD40L assay as a marker of thrombotic risk. Thrombosis Research, 114, 143-148. doi:10.1016/j.thromres.2004.06.005
- Calvo, J., Martínez, N., Etxagibel, A., et al. (2002) Allelic frequencies of polymorphic variants of cytokine genes (IL-1A, IL-1B, IL-1RN, IL-6, IL-10, IL-12P40, and IFNG ) in a Spanish population. Inmunología, 21, 76-86.
- Lincoff, A.M., Kereiakes, D.J., Mascelli, M.A., et al. (2001) Abciximab suppresses the rise in levels of circulating inflammatory markers after percutaneous coronary revascularization. Circulation, 104, 163-167. doi:10.1161/01.CIR.104.2.163
- Merino, A., Gayá, A., Segura, I., et al. (2004) Platelet aggregation inhibition blocks C-reactive protein and interleukin-6 (IL-6) elevation after the coronary angioplasty: Effect of the −174 G/C IL-6 gene polymorphism. Journal of the American College of Cardiology, 94, 1300-1303. doi:10.1016/j.amjcard.2004.07.119
- Merino, A., Artaiz, M., Bergadá, J., Riera, M., Vidal, B. and Rodriguez, A. (2002) El eptifibatide reduce los valores de Proteina C Reactiva tras angioplastia coronaria. Revista Española de Cardiología, 55, 186-189.
- James, S.K., Siegbahn, A., Armstrong, P.A., et al. (2004) Activation of the inflammation, coagulation and fibrinolysis Systems, without influence of abciximab infusión in patients with non-ST-elevation acute coronary síndromes treated with dalteparin: A GUSTO IV substudy. American Heart Journal, 147, 267-274. doi:10.1016/j.ahj.2003.09.014
- Gawaz, M., Langer, H. and May, A.E. (2005) Platelets in inflammation and atherogenesis. The Journal of Clinical Investigation, 115, 3378-3384. doi:10.1172/JCI27196
- Silber, S., Albertsson, P., Aviles, F.F., et al. (2005) Guidelines for percutaneous coronary interventions.The task force for percutaneous coronary interventions of the European Society of Cardiology. European Heart Journal, 26, 804-847. doi:10.1093/eurheartj/ehi138
- Palmerini, T., Nedelman, M.A., Scudder, L.E., et al. (2002) Effects of abciximab on the acute pathology of blood vessels after arterial stenting in nonhuman primates. Journal of the American College of Cardiology, 40, 360-366. doi:10.1016/S0735-1097(02)01951-4
- Curran, M.P. and Keating, G.M. (2005) Eptifibatide: A review of its use in patients with acute coronary syndromes and/or undergoing percutaneous coronary intervention. Drugs, 65, 2009-2035. doi:10.2165/00003495-200565140-00007
- Anand, S.X., Viles-Gonzalez, J.S., Badimon, J.J., Cavusoglu, E. and Marmur, J.D. (2003) Membrane-associated CD40L and sCD40L in atherothrombotic disease. Journal of Thrombosis and Haemostasis, 90, 377-384.
- Aukrust, P., Wæhre, T., Damas, J.K., Gullestad, L. and Solum, N.O. (2001) Inflammatory role of platelets in acute coronary syndromes. Heart, 86, 605-606. doi:10.1136/heart.86.6.605
- Antoniades, C., Bakogiannis, C., Tousoulis, D., Antonopoulos, A.S. and Stefanadis, C. (2009) The CD40/CD40 ligand system. Journal of the American College of Cardiology, 54, 669-677. doi:10.1016/j.jacc.2009.03.076
- Libby, P. and Simon, D.I. (2001) Inflammation and Thrombosis: The Clot Thickens. Circulation, 103, 1718-1720. doi:10.1161/01.CIR.103.13.1718
- Libby, P., Ridker, P.M. and Hansson, G. (2009) Inflammation in atherosclerosis: From pathophysiology to practice. Journal of the American College of Cardiology, 54, 2129-2138. doi:10.1016/j.jacc.2009.09.009
- Muhlestein, J.B. (2010) Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Journal of Thrombosis and Haemostasis, 103, 71-82. doi:10.1160/TH09-03-0177
NOTES
*Corresponding author.