Open Journal of Biophysics
Vol.2 No.2(2012), Article ID:18918,10 pages DOI:10.4236/ojbiphy.2012.22004
Levinthal’s Paradox Revisited, and Dismissed
Department of Physical Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel
Email: arieh@fh.huji.ac.il
Received November 2, 2011; revised January 6, 2012; accepted January 14, 2012
Keywords: Protein Folding; Levinthal Paradox; Hydrophilic Forces; Markov Process
ABSTRACT
This article is concerned with the so-called Levinthal’s paradox. It will be argued that many have sought a “solution” to Levinthal’s paradox, where in fact, the “solution” already appeared in Levinthal’s original articles. Most of the subsequent suggested “solutions” were inadequate solutions to a non-paradox. It is shown that the discovery of strong hydrophilic forces not only dismisses the Levintal paradox, but also provides a solution to the general problem of protein folding. A simple model based on the Markov process is presented to demonstrate how a strong biased-force can dramatically reduce the number of steps required to reach the stable native 3-D structure of the protein.
1. Introduction
Ever since it was realized that the process of denaturation of proteins can be reversed without any auxiliary agent, the protein folding problem became one of the major unsolved problems in molecular biology [1-10].
In a recent editorial of Science, the editors listed 125 unsolved—“What Don’t We Know?” questions in science [7]. One of this is:
“Can we predict how protein will fold? Out of a near infinitude of possible ways to fold, a protein picks one in just tens of microseconds. The same task takes 30 years of computer time.”
There are essentially two problems associated with the process of protein folding. The first is concerned with the questions of how and why protein folds to its native 3-D structure in a very short time. The second is concerned with the factors that confer stability to the native structure of the protein.
These questions had presented formidable challenges to chemists, biochemists and physicists. In this paper we focus only on the first question, the one referred to in the quotation from Science editorial, which is also known as the Levinthal paradox [11]. The second question, as well as its answers, has been discussed in a recent article [12] and a monograph [9].
In the author’s opinion, the main hindrance to finding a solution to the protein folding problem has been the adherence to the hydrophobic (HØO) dogma, which states that various HØO effects (both solvation and interaction) are the “dominant forces” in protein folding [1,13]. The origin of this idea is contained in Kauzmann’s suggestion that: “The hydrophobic bond is probably one of the more important factors involved in stabilizing the folded configuration…” [1] This, quite modestly formulated hypothesis has been enormously exaggerated by many authors [9].
Note that Kauzmann’s hypothesis does not say anything about the role of the hydrophobic effect on the dynamics of the process of protein folding. Yet surprisingly the literature abounds with many claims that the dominant forces in protein folding are the HØO effects.
An exhaustive analysis of all the solvent induced effects on protein folding reveals that the hydrophilic (HØI) effects are much more important than the corresponding HØO effects [8,9]. The discovery of the strong HØI effects—both interaction and forces—has removed the crux out of the mystery of the protein folding problem (as well as other related problems such as self assembly and molecular recognition).
In this article we discuss the role of the HØI forces in answering the question of why protein folds along a narrow range of pathways and in a relatively short period of time.
We start in the Section 2 with a few quotations from Levinthal who has most eloquently formulated the problem of protein folding [11]. This is sometimes known as the Levinthal paradox. We shall see in Levinthal’s writings that there is nary a hint of a paradox. Instead one finds a well formulated problem, as well as a hint of possible solution of that problem.
In Section 3, we discuss some attempts to “solve” the so-called Levinthal’s paradox. In Sections 4 and 5, we suggest a way of implementing the HØI forces to achieving an answer to the protein folding problem. Some concluding remarks are presented in Section 6.
2. The Origin of the So-Called Levinthal’s Paradox
We begin with a few quotations from Levinthal’s articles [11], which are relevant to what is now referred to as the Levinthal Paradox.
a) “Let us ask ourselves how proteins fold to give such a unique structure. By going to a state of lowest free energy? Most people would say yes and indeed, this is a very logical assumption. On the other hand, let us consider the possibility that it isn’t so.”
b) “How accurately must we know the bond angles to be able to estimate these energies? Even if we knew these angles to better than a tenth of a radian, there would be 10300 possible configurations in our theoretical protein. In nature, proteins apparently do not sample all of these possible configurations since they fold in a few seconds, and even postulating a minimum time for going from one conformation to another, the proteins would have time to try the order of 108 different conformations at most before reaching the final state.”
c) “We feel that protein folding is speeded and guided by the rapid formation of local interactions which then determine the further folding of the peptide. This suggests local amino acid sequences which form stable interactions and serve as nucleation points in the folding process.”
d) Then, is the final configuration necessarily the one of the lowest free energy? We do not feel that it has to be. It obviously must be a metastable state which is in a sufficiently deep energy well to survive possible perturbations in a biological system. If it is the lowest energy state, we feel it must be the result of biological evolution, i.e. the first deep metastable trough reached during evolution happened to be the lowest energy state. You may then ask the question, “Is it a unique folding necessary for any random 150-amino acid sequence?” and I would answer, “Probably not.”
The propositions quoted above are cast in a form which is reminiscent of a mathematical proof by contradiction; let us assume that X is true, reach an absurd result, then conclude that our assumption cannot be true. In fact, we have here all the elements of such a proof.
Statement a) raises two questions: 1) How does a protein fold to give a unique structure? and 2) Is this unique structure the state of lowest free energy? We shall refer to the first question as the Levinthal question. The second question, as well as its answer will not be discussed here. It is related to Anfinsen’s thermodynamic hypothesis, and it is discussed elsewhere [9].
Statement b) essentially concludes that if one assumes a random sampling of the configuration space of the protein, then one arrives at an absurd result. Statements c) and d) suggest possible answers to the two questions raised in a).
Clearly, there exists no paradox in obtaining an absurd result based on an unrealistic assumption. Levinthal immediately recognized that the absurd result he reached follows from the wrong assumption of a random search over the immense configurational space. Levinthal did not see that absurd result as a paradox, as so many others did. He immediately reached the (almost) correct solution, as stated in quotation c) above. Namely, that there must be preferential pathways of folding, “guided by rapid formation of local interactions.” Although Levinthal did not specify what these “guiding interactions” are, his solution to the absurd result (based on unrealistic assumption) is almost correct. Instead of “guiding interacttions,” one should use the term “guiding forces.” Though these forces are derived from the interactions, it is the magnitude of the force acting on the groups of the protein that determines the speed of the folding process. The main question left unanswered by Levinthal is: What are these strong forces that guide the protein to its native structure in a relatively short time? We now know that these forces originate from the water, more specifically the solvent-induced forces exerted on the hydrophilic groups along the backbone and the side chains of the protein [8,9].
3. Attempts to Solve the Levinthal Paradox
During the past 40 years many have sought for a solution to the (non-existent) Levinthal paradox.
Perhaps the most serious and much acclaimed attempt to “solve” the paradox was published by Zwanzig et al. [14]. In their introduction, Zwanzig et al. wrote:
“The main point of this paper is to show by mathematical analysis of a simple model that Levinthal’s paradox becomes irrelevant to protein folding when some of the interactions between amino acids are taken into account.”
This is exactly the answer given by Levinthal himself namely, that the interactions between different parts of the protein can guide the folding process. As we have pointed out above, the important guiding factors are the forces rather than the interactions. Zwanzig et al. do not offer any answer to the question regarding these forces, nor do they specify which are “some of the interactions between amino acids”. Furthermore, the model used by Zwanzig et al. is not a realistic one, and might even be misleading.
Zwanzig et al. drew from Dawkins’ brilliant ideas of explaining the mechanism of evolution [15-17]. Briefly, the protein is viewed as a sequence of N bonds, and the “connecting bond between two neighboring amino acids can be characterized as “correct” or “incorrect” (Correct means native in biology). Then they assume some rate constant (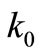 ) for the transition “correct” → “incorrect”, and another rate constant (
) for the transition “correct” → “incorrect”, and another rate constant (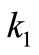 ) for the transition “incurrect → correct”. Assuming further that the ratio (
) for the transition “incurrect → correct”. Assuming further that the ratio (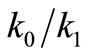 ) is small, they calculated the mean first-passage time to reach the fully “correct” conformation.
) is small, they calculated the mean first-passage time to reach the fully “correct” conformation.
It should be noted that the metaphor used by Dawkins, is barely suitable for explaining evolution to the layperson [15,17]. The mechanism arriving at the “correct” target, as proposed by Dawkins demonstrates the possibility of occurrence of an event which is perceived to be highly improbable. As such, Dawkins model achieves its goal of removing the mystery out of the evolutionary process. However, even in evolution, there exist no “correct” or “incorrect” results. In fact, Dawkins himself recognized that his explanation is not relevant to the actual process of evolution [9,15]. Evolution does not pose any goals or targets to reach. Nevertheless, one can simply define a “correct” outcome as one which has some evolutionary advantage. This is not the case for the protein folding process. Therefore, Dawkin’s metaphor is not adequate for the process of protein folding. The main objection to this model is that one cannot justify the preferential transition from “an incorrect bond” to “a correct bond” at each stage of the protein folding process.
In evolution, a transition from “incorrect” to “correct” is biased according to some selection criterion, i.e. the “correct” result has some advantages, and therefore that result survives. There exists no analog of the selection criterion in the process of protein folding.
Furthermore, Zwanzig et al. do not provide a plausible reason for the particular assignment of the values of the rate constants 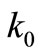 and
and 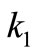 in terms of either molecular interactions or forces. Therefore, the model used by Zwanzig et al., as well as the specific solution of the model is not relevant to the protein folding problem.
in terms of either molecular interactions or forces. Therefore, the model used by Zwanzig et al., as well as the specific solution of the model is not relevant to the protein folding problem.
There are many statements in which evolution theory is invoked in connection with the problem of protein folding. In a recent article Wolynes writes: [18]
“Evolution solved the protein-folding problem. A major goal of bio-molecular science has been to understand how this was done.”
Of course, evolution does not solve any problem, nor was the protein folding problem posed to Nature. Evolution only evolves and a product which has some evolutionary advantages survives. It is clear that Wolynes means “solved” in his first sentence only in a figurative sense, in the same sense when people say today that some bacteria “developed” resistance to some drug. Of course, bacteria do not “develop” anything. In a given population there are many mutants of the same bacteria, some of which are resistant to a specific drug. When that drug is administered, only those mutants that are resistant to the drug would survive. To an outside observer, it looks as if the population as a whole has “developed” a resistance to the drug.
Thus, it is acceptable to use the word “develop” in the sense that this is how it seems to an observer who is not aware of the existence of resistant mutants in the original population. However, it is meaningless to try to “learn” from the bacteria (or from Evolution) how they “developed” the resistance to the drug.
Similarly, during evolution proteins, or rather polypeptides were synthesized. Some folded, while others did not. Of those that folded some reached a stable 3-D structure, some did not. Of those that reached a stable structure, some had a special advantage, while others did not, and so on.
Looking at the final outcome of a functional protein one can say figuratively that Nature or Evolution has “solved” the problem of folding a polypeptide into some useful 3-D structure. This is acceptable only if we understand that in the “population” of all the peptides which were synthesized during evolution, some have folded into a useful 3D-structure. Not because this structure was the target of Evolution, and not because Evolution had faced the problem of how to fold a specific protein. However, one cannot attempt an understanding of how Evolution has “solved” the protein folding problem, simply because Evolution did not solve any problem. Any attempt to “learn” from evolution must therefore lead to a dead-end.
4. Possible Approach to Solving the Protein Folding Problem
The more realistic mechanism for folding and the one alluded to by Levinthal, is the one based on the forces acting on each group at any given configuration of the protein. In this view there is no “correct” or “incorrect” bond, or a configuration. There is also no need to involve a folding “code”, or a “target” to be reached. At each stage of the process of folding there are many possible transitions, some are more probable and some less probable, or even improbable. This view leads to a range of pathways, which we may refer to as the preferential folding pathways, along which the protein folds with high probability, and with negligible probability along all other pathways. In other words, this view effectively reduces the immense number of pathways to a narrow “corridor” of protein folding pathways, within which there is some degree of randomness. However, randomness of this kind does not allow the protein to wander at any direction as the “drunken golfer,” seeking a single hole in a flat featureless landscape. Instead, the strong hydrophilic (HØI) forces, force and guide the protein to fold along a narrow range of pathways.
A suitable theoretical framework which is sufficiently general as well as realistic is to view the process of protein folding as a Markov chain [7]. In this view, the protein visits a finite number of states in the process of folding. The folded state (or states) may be viewed as an approximate absorbing state (or group of states). Thus, for each state i, there are transition probabilities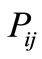 . These are the conditional probabilities of moving from state i to state j. The increments in time are assumed to be discrete. Thus, we have for any state i
. These are the conditional probabilities of moving from state i to state j. The increments in time are assumed to be discrete. Thus, we have for any state i
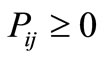 ,
,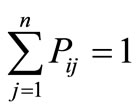 (1)
(1)
i.e. the transition probabilities are non-negative, and the probability to move from i to any other state j (including the state j = i) is one. The magnitude of the transition probabilities are determined by the forces acting on the protein, which in turn, are determined by the gradients in the Gibbs energy landscape. A reader of an earlier version of this manuscript commented that the “force” is derived from the energy landscape, and that the entropy contribution was neglected. This is not so. The “force” is derived from the potential of mean force, and includes contribution from both energy and entropy.
For simplicity we can choose the state n to be an approximate absorbing state, i.e. once the system reaches that state, it stays there for a sufficiently long time, so that it can fulfill its biological function and therefore it can be considered to be there “forever”. The folded state denoted here by the state n, is characterized by the transition probabilities
 (2)
(2)
within this Markov-chain view of the protein folding process, we can characterize two extreme cases as follows:
The random search of the configurational space is equivalent to the assumption that at each state i there is equal probability to move to each of the states j which are accessible from state i, i.e.
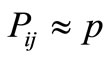 (3)
(3)
for each i and for each j accessible from i
The other extreme case is that from each i the protein can move to a single state with high probability. Let us denote this state by i + 1, i.e. at each step we move from i to i + 1 until we reach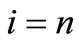 , hence
, hence
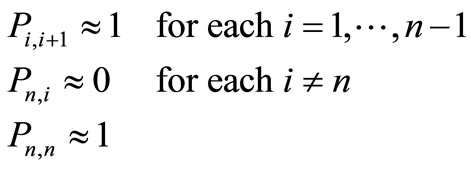 (4)
(4)
The more realistic view is that at each state i, there is a group of states which are accessible from i, but have a distribution of probabilities. For instance starting from![]() , the system can reach states
, the system can reach states . The transition probabilities to these
. The transition probabilities to these 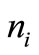 states, is such that some states are reached with relatively higher probability. These three probabilities are depicted in Figure 1.
states, is such that some states are reached with relatively higher probability. These three probabilities are depicted in Figure 1.
It is intuitively clear that if such preferential transition probabilities exist for each state i, then there will also be preferential pathways to go from state 1 to state n. These will not be the random searches in the configurational space, nor a single path implied by the second extreme model. Instead, the transitions will have some preference to go along a specific path, with some random deviations from the preferable paths. In such a Markov chain one can also compute the average number of steps to reach the absorbing state [9].
5. A Simple Model for “Folding” by Means of the Markovian Process
In this section we describe a simple process. Lest it will be misunderstood we emphasize that this is not a model for protein folding. It is raised here to demonstrate that a cause-biased process can drastically reduce the number of steps required to reach some stable state. This is to contrast the target-biased process.
We start with a small section of the protein and focus on one angle of internal rotation, Figure 2. We “freezein” the configurations of all other angles of internal rotation except the one on which we focus.
In the case of ethane, or hexamethyl ethane the internal potential energy of rotation would look like the one in Figure 3, i.e. a function with three minima and maxima. On the other hand, for a more complicated molecule, say six different groups substituting for the hydrogen atoms of ethane we might have a potential function of the form shown in Figure 4.
The details of the potential shape are not important here. What is important is that there might be some regions that are not accessible and within that region there might be minima that are even lower than the minima in the accessible region. For instance, suppose that every time we synthesize the molecule depicted on the lhs of Figure 4, the accessible region is always between angles say 120˚ - 350˚.
We now focus on the accessible region of angles. This region may be divided into two sub-regions. The first is referred to as transient (TR) states, includes all the points for which U(Ø) has a steep slope, say 120˚ - 160˚ and 250˚ - 350˚. The second, referred to as absorbing state, denoted AB, includes the points in the region 160˚ - 250˚, where the potential function is almost flat.
Clearly, whenever the configuration of the molecule is in the region TR there will be a strong force (or average
 (a) (b) (c)
(a) (b) (c)
Figure 1. Three possible schemes of transition probabilities. (a) Equal probabilities to move from state i to a group of nearby states; (b) Probability one to move from i to i + 1; and (c) High probability to move from i to i + 1, and relatively low probability to move to other states (the width of the arrows indicating the magnitude of the probabilities).
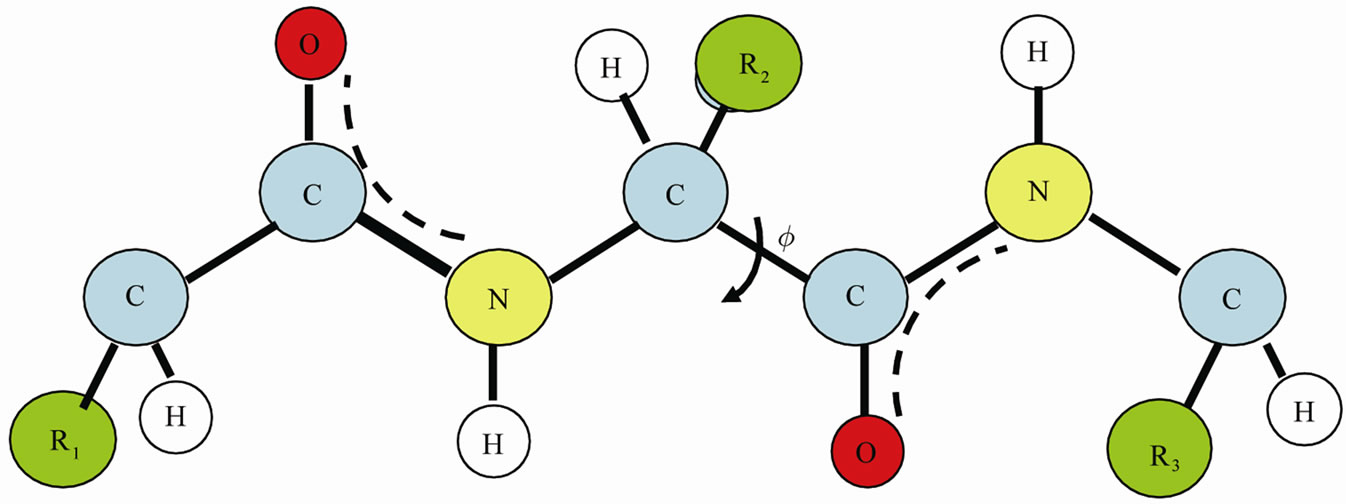
Figure 2. A small section of the protein. We focus on the internal rotation about the C-C bond, and “freeze-in” all other internal rotations.
force, in case that U(Ø) is the Gibbs energy landscape) towards region AB. On the other hand, in the region AB where U(Ø) is almost flat there will be no strong forces acting on the groups of the molecule that will cause the molecules to exit this region. For simplicity, suppose we always start with a transient configuration on the left hand side of AB. We now divide the region into a finite number of intervals, and without loss of generality we can enumerate all the configurations in TR by the numbers 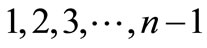 and refer to these configurations as transient states. The configurations in the region AB will be enumerated
and refer to these configurations as transient states. The configurations in the region AB will be enumerated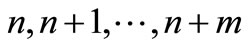 , and will be referred to as absorbing states. It is clear that starting with any point in the region TR, there will be preferential force to move that point, with high probability towards the region AB. On the other hand, once we arrive at the region AB there will be very small probability to get out from this region.
, and will be referred to as absorbing states. It is clear that starting with any point in the region TR, there will be preferential force to move that point, with high probability towards the region AB. On the other hand, once we arrive at the region AB there will be very small probability to get out from this region.
Again, without loss of generality we can collect all configurations in the region AB into one state denoted by the index n. Translating the forces into transition prob-
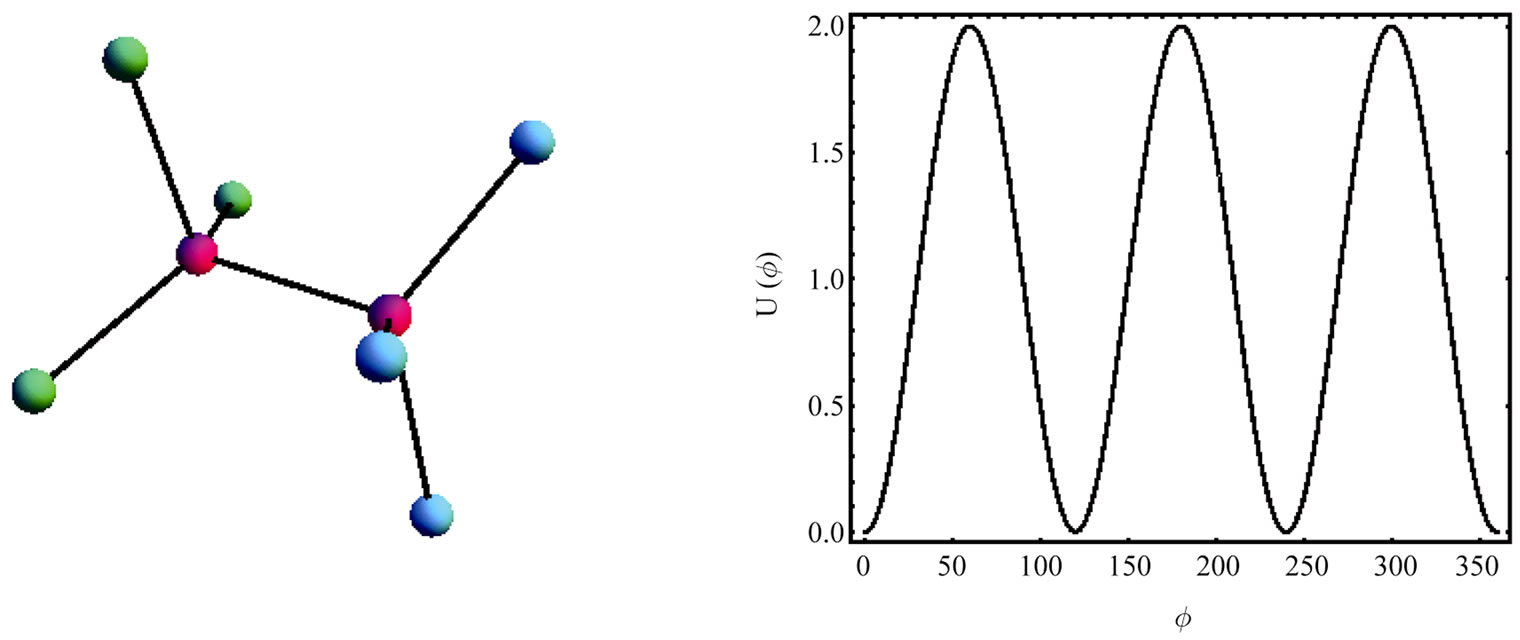
Figure 3. A simple rotational potential with three minima and three maxima.
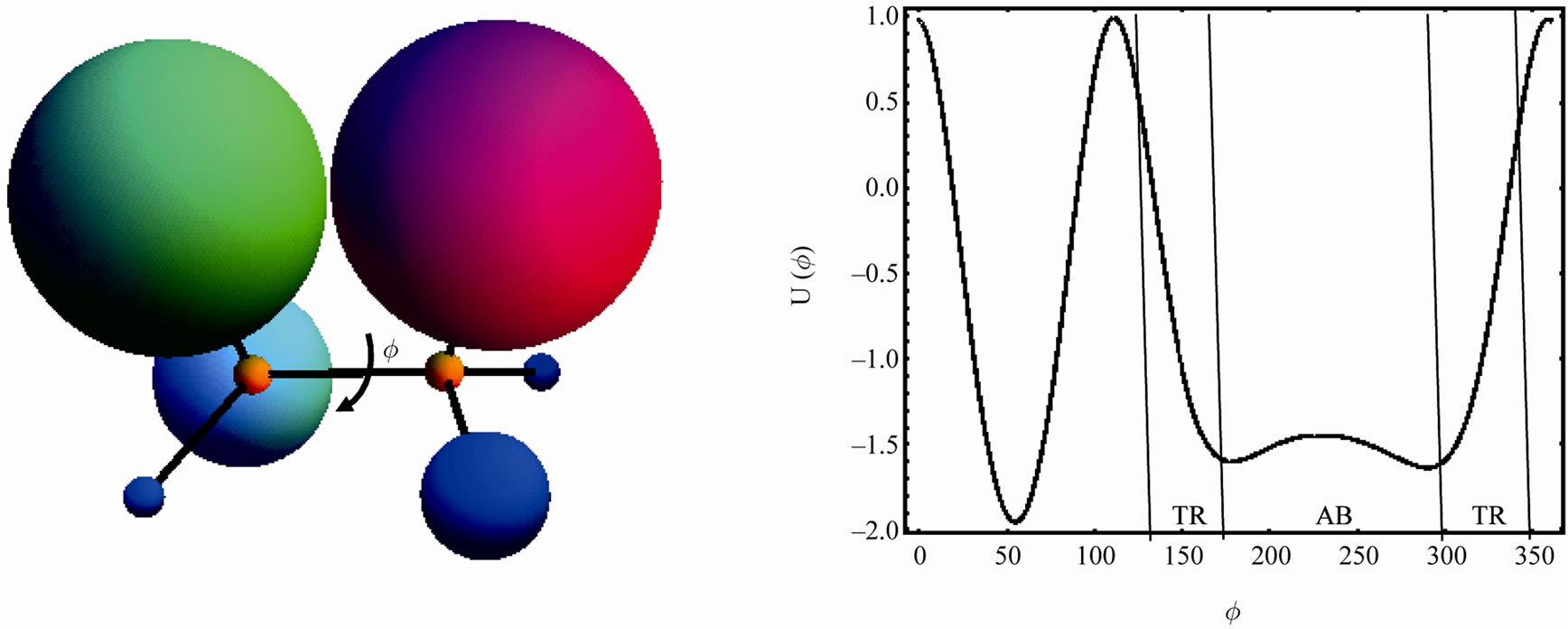
Figure 4. A more complex rotational potential. The region 350˚ and 120˚ is considered to be inaccessible. The accessible region (120˚ to 350˚) is divided into regions which are transients (TR) and a nearby absorbing (AB) state.
abilities, we write
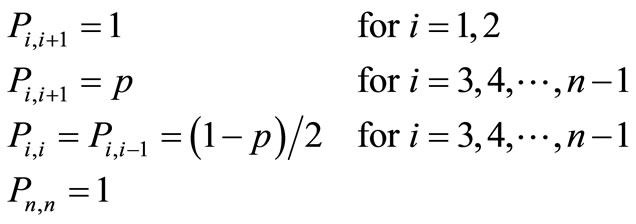 (5)
(5)
The choice of the transition probabilities for  was made for convenience of notation. This has no effect on the results when we consider very large number of configurations, say
was made for convenience of notation. This has no effect on the results when we consider very large number of configurations, say 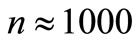 or more.
or more.
Clearly, if we chose  then starting from state
then starting from state ![]() we shall arrive at state n by exactly
we shall arrive at state n by exactly 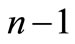 steps. On the other hand, if we chose
steps. On the other hand, if we chose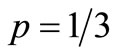 , the point will move either towards the left, towards the right or stay put with equal probability. As expected the number of steps required to arrive at AB will be much larger.
, the point will move either towards the left, towards the right or stay put with equal probability. As expected the number of steps required to arrive at AB will be much larger.
A typical transition probability matrix for 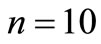 and
and 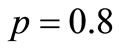 is
is
 (6)
(6)
with the transition probability matrix (6) we can calculate the average number of steps required to reach the state n, starting with any initial state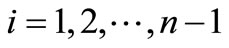 . To do this we write the matrix
. To do this we write the matrix 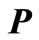 in the form.
in the form.
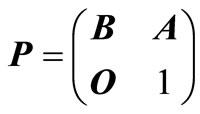 (7)
(7)
where 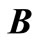 is a
is a 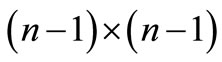 matrix,
matrix, 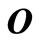 is a row vector of zeros
is a row vector of zeros 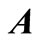 in a column vector, including all the transitions
in a column vector, including all the transitions 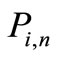 for
for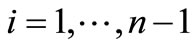 , and the element
, and the element  .
.
According to a theorem in Markov chain theory [19], the average number of steps required to reach the absorbing state n, from any initial state i, is obtained from the elements of the inverse matrix.
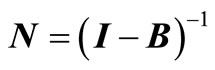 (8)
(8)
where 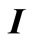 is a
is a 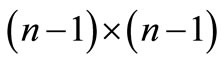 unit matrix. The required average quantities, denoted by
unit matrix. The required average quantities, denoted by 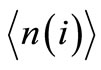 are obtained from the sum of the row elements of the matrix
are obtained from the sum of the row elements of the matrix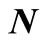 , i.e.
, i.e.
![]() (9)
(9)
The calculation of the vector 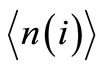 was done for the transition probabilities as given in (5).
was done for the transition probabilities as given in (5).
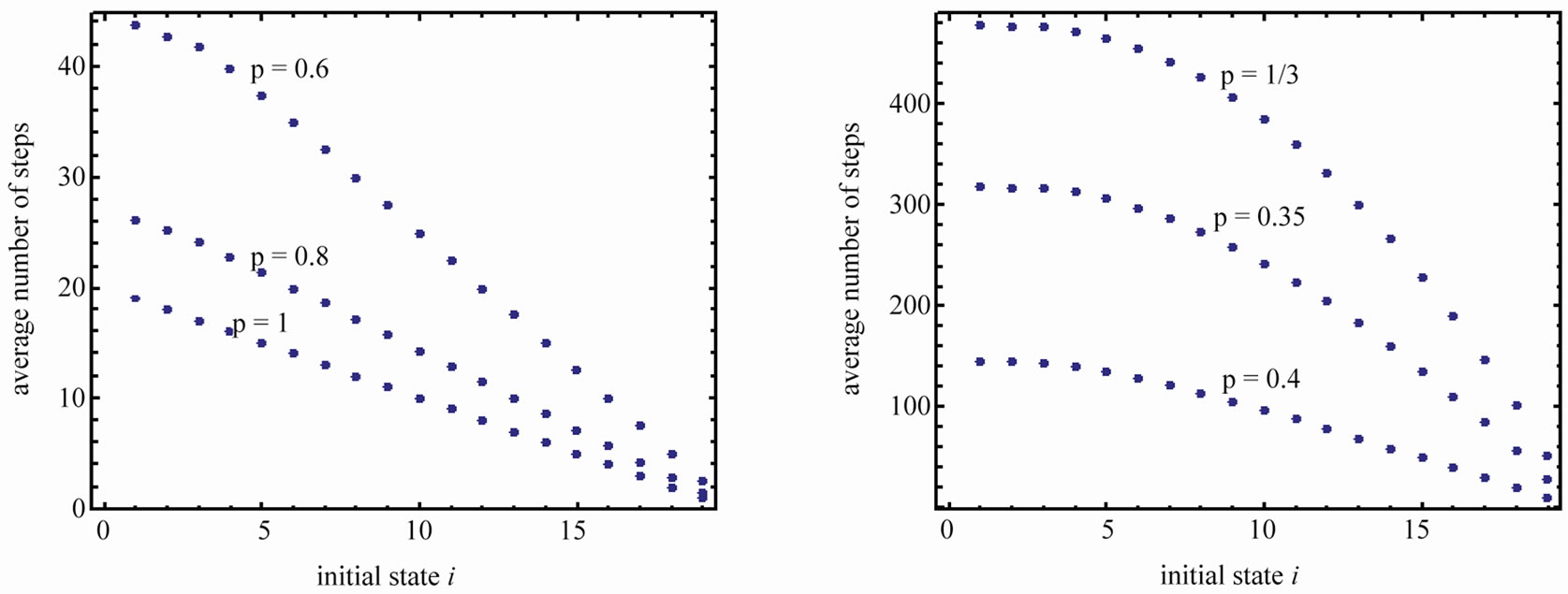
Figure 5 shows the average number of steps to reach the region AB for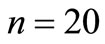 , and
, and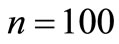 , and
, and
 . Note that the spacing between the lines increases dramatically when p approaches 1/3.
. Note that the spacing between the lines increases dramatically when p approaches 1/3.
Figure 6 shows the number of steps to reach AB from the initial state![]() , as a function of the number of states
, as a function of the number of states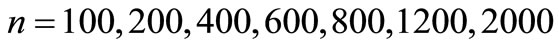 , and for the case
, and for the case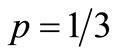 .
.
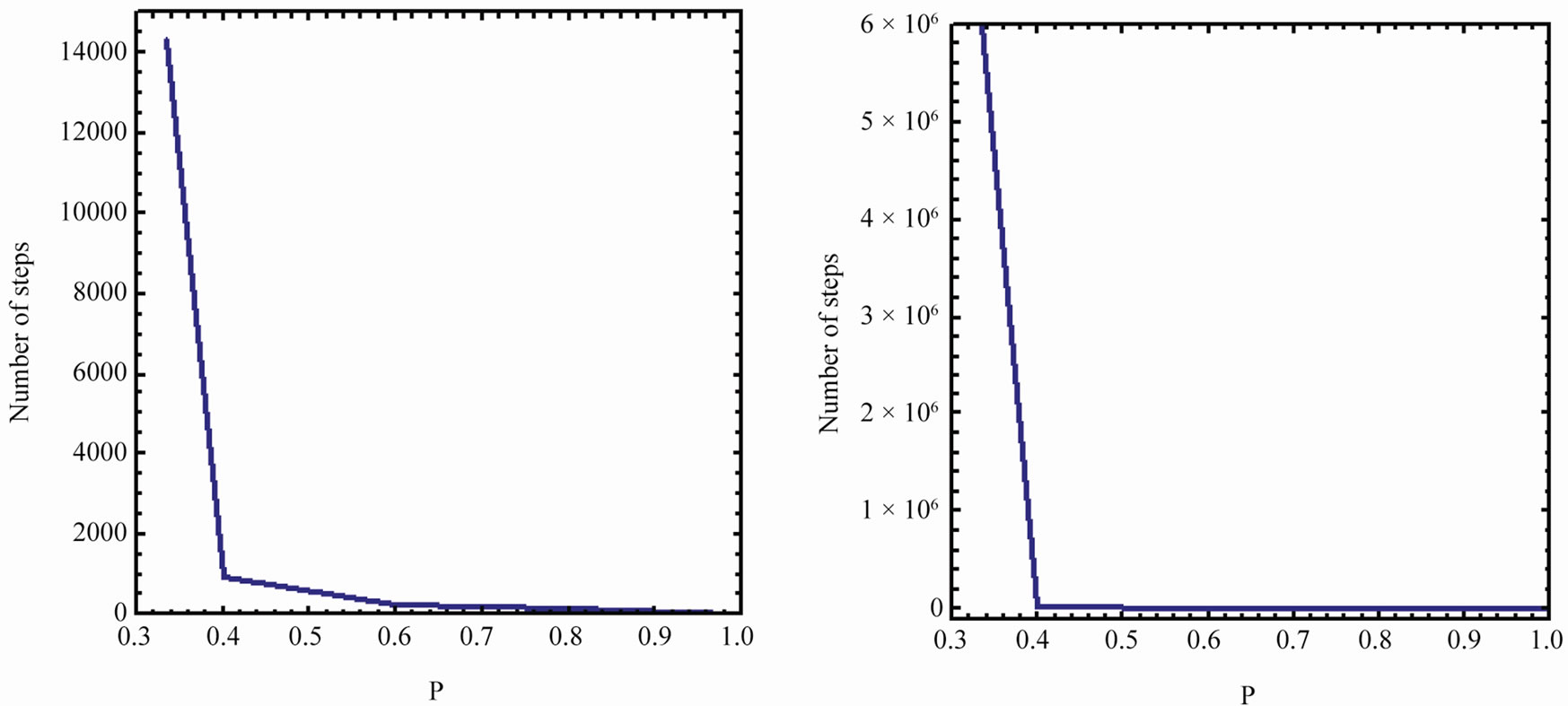
Perhaps, the most dramatic results of this model is shown in Figure 7 where we plot the average number of steps to reach AB, starting from![]() , for
, for , and varying p between 1/3 to 1. It is shown that for
, and varying p between 1/3 to 1. It is shown that for 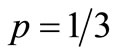 the number of steps is of the order of 106, and this number drops dramatically between
the number of steps is of the order of 106, and this number drops dramatically between 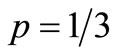 and
and 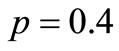 , then it stays relatively low around 2000, as expected for the extreme case of
, then it stays relatively low around 2000, as expected for the extreme case of  and
and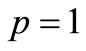 .
.
This is an important result. If we have a relatively
 (a)
(a)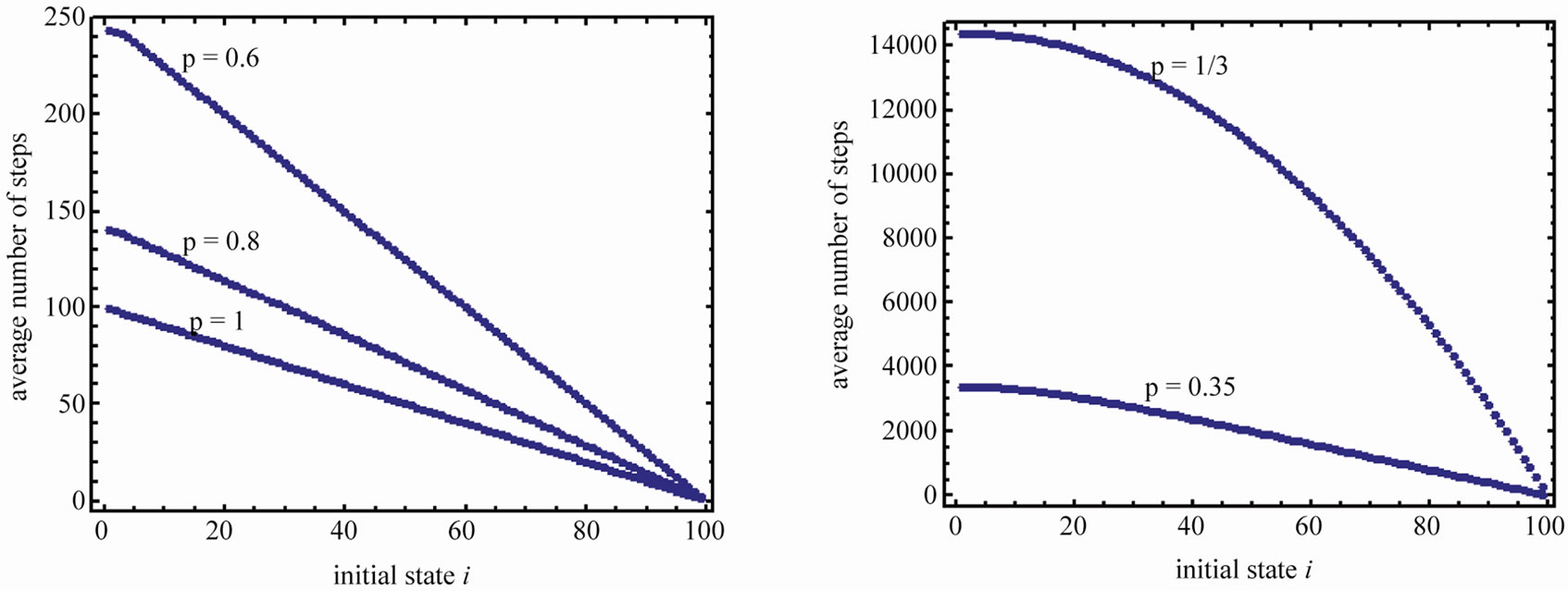 (b)
(b)
Figure 5. Average number of steps required to reach the absorbing state from any state i. (a) n = 20 and various p. (b) n = 100 and various p.
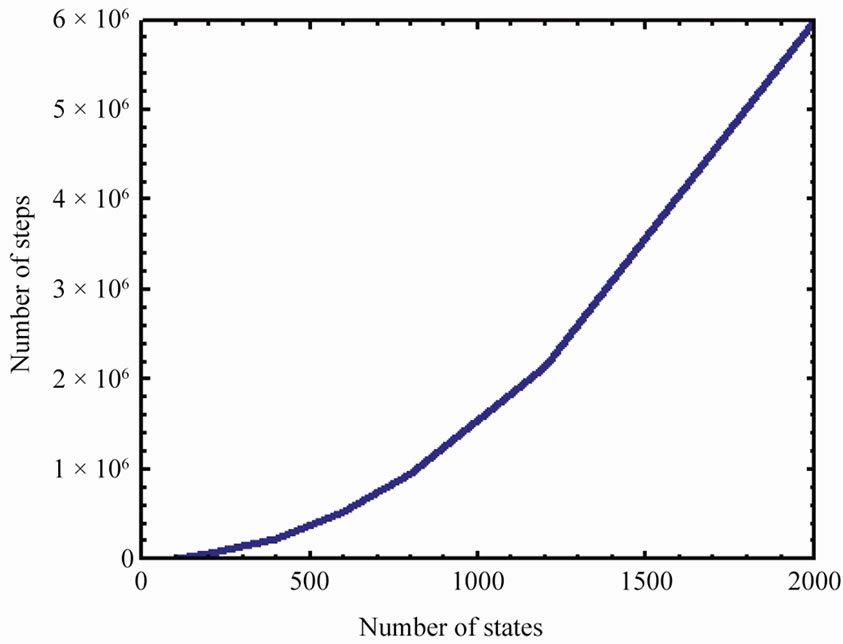
Figure 6. The average number of steps required to reach the state AB from i = 1, as a function of the number of states n (here p = 1/3).
strong force exerted on each group of the protein, at each stage of the folding process, then the average number of steps to reach the region AB dramatically decreases. This result in effect dismissed the so-called Levinthal paradox. It should be noted that this demonstration differs in a fundamental way from Zwanzig et al. approach. The transitions in our model are caused-biased, whereas in Zwanzig et al. they are target-biased. The cause, in our model are the forces, and the forces are obtained from the gradients in the Gibbs energy landscape. As such we do not claim anything whether these forces originate from the energy or the entropy. These are the total forces; both direct and solvent-induced forces acting on all the groups along the protein.
In real proteins we have many more degrees of freedom, and strong forces are exerted on many groups of the protein simultaneously. Also from each state i, there are many states which are accessible, not only two as in the example above, and the native state is not a single state, and certainly not an absolute absorbing state.
However, the extension from the simple model treated above to real protein should not pose any new difficulty. One can imagine that at each stage of the folding process, there are strong solvent-induced forces exerted on the various groups along the protein. These forces will force the protein to fold along a narrow range of pathways, not towards a pre-conceived target, and not towards the “correct” configuration for each bond. The overall process would be a speedy one, i.e. reaching the final relatively stable structure in a relatively short time. This procedure, in effect, answers Levinthal’s original questions quoted in sections II, and at the same time removes the crux of the difficulty from the general protein folding problem.
6. Discussion and Conclusion
In the previous section we studied a simple stochastic process involving a change in one parameter, the angle of rotation Ø. In real proteins, say M amino acids, we have at least 2 M internal rotational degrees of freedom. At each configuration of the protein there are forces exerted on each of the groups. These forces are derived from the potential of mean force, which is essentially the Gibbs energy landscape of the protein. It is almost universally
 (a) (b)
(a) (b)
Figure 7. The average number of steps required to reach AB from the initial state i = 1, as a function of p, for (a) n = 100 and (b) n = 2000.
recognized that the solvent-induced part of these forces are important. The main “unknown” in the protein folding process is not how and why the protein reaches its final 3-D structure in a short time, but what are the strong forces acting on the various groups of the protein, which force the protein to change its configuration within a narrow range of pathways. In my view once we have discovered the strong HØI forces exerted on the groups, we have not only dismissed the so-called Levinthal paradox, but we have also answered the question of how and why protein folds and reaches the final 3-D structure in a relatively short time.
Thus, the HØI forces explain the folding process and the HØI interactions also provide explanation for the stability of the protein. As we have discussed recently [9], the HØI interactions also provide answers to the questions of self-assembly among biomolecules, as well as molecular recognition.
This is as far as one can get in answering the general principle of protein folding. The question that remains is how to implement the hydrophilic (HØI) forces in studying specific proteins. In this regard, there is no need to study the entire Gibbs energy landscape. We have seen in Figure 4 that even in the simple model there could be regions which are un-accessible and therefore irrelevant to the process. In real proteins there are vast regions in the Gibbs energy landscape that are not accessible (either because of very strong repulsive forces between various groups along the chain, or because these configurations are very improbable, e.g. one or more knots in the chain of amino acids). There is also no need to speculate on the general shape of the Gibbs energy landscape. All we need is a tiny region in the Gibbs energy landscape which is relevant to the folding pathways of that specific protein.
In 1992 [20], upon recognizing the importance of the solvation part in the Gibbs energy landscape, I suggested to look at the solvation Gibbs energy landscape as a multi-dimensional space, each of its points representing one of the conformations of the protein. Gradients in this landscape correspond to lowering the solvation Gibbs energy of the protein. The protein will initially and preferentially move along one of these gradients until it reaches a space of lower dimensionality, where again there are gradients leading to another, yet a lower dimensionality space. This was described as a “tunnel within a tunnel,” until the protein is “drained” into the folded form [20]. In a two dimensional landscape we can imagine that starting from the top of the hills, the protein flows in the two dimensional space of x, y towards a “river”. Once a “river” is reached the flow continues along the gradient in the one dimensional path of the river. In a multi-dimensional case, at any stage the protein “flows” from a higher dimensional “river” (or tunnel) into a lower dimensional “river” (or tunnel), eventually reaching a one-dimensional river leading to the folded form. This final conformation does not have to be a global minimum in the Gibbs energy landscape as some have speculated following the so-called Anfinsen Dogma [21-23].
The rationale underlying this sequential reduction of the dimensionality of the space in which the protein “moves” is the following:
Suppose we start with the fully extended unfolded conformation. Initially, we would expect that the motion of the protein will be random until two or more hydrophilic groups are brought to such a distance that they exert strong hydrophilic force on each other. This force reflects the existence of a steep gradient in the original multi-dimensional landscape. Once the two hydrophilic groups are brought to a short distance such that they can form direct hydrogen bonds (as in an α-helix or β-sheet), some of the rotational angles ψ, Ø will be “locked” for a short time while the random motion about all other angles continues, but now in a lower dimensional space. Of course several direct HBs can occur simultaneously resulting in a further reduction in the dimensionality of the space in which the conformation of the protein moves. Some experimental evidence for the occurrence of waterbridges connecting hydrophilic groups of the backbone of proteins was reported [24,25].
As pointed out correctly by Dill and Chan [22], “Levinthal’s argument led to a search for folding pathways.” Indeed, Levinthal’s conclusion that the process of folding is “speeded and guided by rapid formation of local interactions” is basically correct. Also his doubts regarding the question of the existence of a conformation of “lowest free energy” (see quotations c) and d)) are correct too [9].
Unfortunately, the search for models of folding having a global minimum Gibbs energy conformation continues. In recent publications, one can find claims that the funnel model solves the Levinthal’s paradox [22]. Dill and Chan describe at least six different funnels, all of which have a common feature—the existence of a single global minimum in the Gibbs energy landscape. All of these models were inspired by the so-called Anfinsen’s Thermodynamic hypothesis. Unfortunately, neither Anfinsen’s hypothesis, nor Levinthal’s suggestion requires that the native structure be a global minimum in the Gibbs energy landscape [9,23].
A reader of an earlier version of the manuscript claimed that the distinction between the target-biased and the cause-biased motion of the protein is only semantic. I believe that the difference between the two approaches is profound and critical to the solution of the protein folding problem.
The motion of the protein is not “guided” or “speeded” towards a target. This is true in evolution theory and a fortiori true in protein folding. In the model described in this article, the motion is a result of the forces acting on the groups of the protein. The stronger the force is, the speedier the motion. Once the protein reaches a stable state, such that it stays there long enough to function, it will be in a local minimum in the Gibbs energy landscape. This minimum has nothing to do with the Second Law of thermodynamics, as so many have erroneously concluded [9,21].
REFERENCES
- W. Kauzmann, “Some Factors in the Interpretation of Protein Denaturation,” Advances in Protein Chemistry, Vol. 14, 1959, pp. 1-63. doi:10.1016/S0065-3233(08)60608-7
- T. E. Creighton, “The Problem of How and Why Proteins Adopt Folded Conformation,” Journal of Physical Chemistry, Vol. 89, No. 12, 1985, pp. 2452-2459. doi:10.1021/j100258a006
- E. Shakhnovich, “Protein Folding Thermodynamics and Dynamics: Where Physics, Chemistry and Biology Meet,” Chemistry Review, Vol. 106, No. 5, 2006, pp. 1559-1588. doi:10.1021/cr040425u
- L. Mirny and E. Shakhnovich, “Protein Folding Theory: From Lattice to All-Atom Models,” Annual Review of Biophysics and Biomolecular Structure, Vol. 30, 2001, pp. 361-396. doi:10.1146/annurev.biophys.30.1.361
- J. M. Shea and C. L Brooks, “From Folding Theories to Folding Proteins: A Review and Assessment of Simulation Studies of Protein Folding and Unfolding,” Annual Review of Physical Chemistry, Vol. 52, 2001, pp. 499-535. doi:10.1146/annurev.physchem.52.1.499
- K. A. Dill, “Polymer Principles and Protein Folding,” Protein Science, Vol. 8, No. 6, 1999, pp. 1166-1180. doi:10.1110/ps.8.6.1166
- D. Kennedy and C. Norman, “What Don’t We Know?” Science, Vol. 309, No. 5731, 2005, p. 75. doi:10.1126/science.309.5731.75
- A. Ben-Naim, “Molecular Theory of Water and Aqueous Solutions, Part I: Understanding Water,” World Scientific, Singapore, 2009. doi:10.1142/9789812837615
- A. Ben-Naim, “Molecular Theory of Water and Aqueous Solutions: Part II: The Role of Water in Protein Folding Self Assembly and Molecular Recognition,” World Scientific, Singapore, 2011.
- G. D. Rose, P. J. Fleming, J. R. Banavar and A. Maritan, “A Backbone Based Theory of Protein Folding,” Proceedings of the National Academy of Science, Vol. 103, No. 45, 2006, pp. 16623-16333. doi:10.1073/pnas.0606843103
- C. Levinthal, “Are There Pathways for Protein Folding,” Journal de Chimie Physique, Vol. 65, No. 1, 1968, pp. 44-45.
- A. Ben-Naim, “Some Aspects of the Protein Folding Examined in One-Dimensional Systems,” Journal of Chemical Physics, Vol. 135, No. 8, 2011, pp. 104-115. doi:10.1063/1.3626859
- K. Dill, “Dominant Forces in Protein Folding,” Biochemistry, Vol. 29, 1990, pp. 7133-7155. doi:10.1021/bi00483a001
- R. Zwanzig, A. Szabo and B. Bagchi, “Levinthal’s Paradox,” Proceedings National Academy of Science USA, Vol. 89, No. 1, 1992, pp. 20-22.
- R. Dawkins, “The Blind Watchmaker,” Norton, New York, 1987.
- K. A. Dill, “Polymer Principles and Protein Folding,” Protein Science, Vol. 8, No. 6, 1999, pp. 1166-1180. doi:10.1110/ps.8.6.1166
- R. Srinivasan and G. D. Rose, “Methinks It Is Like a Folding Curve,” Biophysical Chemistry, Vol. 101-102, 2002, pp. 167-171. doi:10.1016/S0301-4622(02)00147-3
- P. G. Wolynes, “Energy Landscapes and Solved ProteinFolding Problems,” Philosophical Transactions of the Royal Society A, Vol. 363, No. 1827, 2005, pp. 453-464.
- J. G. Kemeny and J. L. Snell, “Finite Markov Chains,” Van Nostrand Comp. Int., New York, 1960.
- A. Ben-Naim, “Statistical Thermodynamics for Chemists and Biochemists,” Plenum Press, New York, 1992.
- C. B. Anfinsen, “Principles that Govern the Folding of Protein Chains,” Science, Vol. 181, No. 4096, 1973, pp. 223-230. doi:10.1126/science.181.4096.223
- K. A. Dill and H. S. Chan, “From Levinthal to Pathways to Funnels,” Nature Structural Biology, Vol. 4, 1997, pp. 10-19. doi:10.1038/nsb0197-10
- A. Ben-Naim, “Pitfalls in Anfinsen’s Thermodynamic Hypothesis,” Chemical Physics Letters, Vol. 511, No. 1-3, 2011, pp. 126-128. doi:10.1016/j.cplett.2011.05.049
- M. Sundaralingam and Y. C. Sekharudu, “Water-Inserted Alpha-Helical Segments Implicate Reverse Turns as Folding Intermediates,” Science, Vol. 244, No. 4910, 1989, pp. 1333-1337. doi:10.1126/science.2734612
- M. Sundaralingam and Y. C. Sekharudu, “A Study of Alpha-Helix Hydration in the Polypeptides,” In: R. S. Sarma and M. H. Sarma, Eds., Structure and Methods, Vol. 2, Adenine Press, 1990, pp. 115-127.