Open Journal of Organic Polymer Materials
Vol.06 No.02(2016), Article ID:65485,13 pages
10.4236/ojopm.2016.62007
Synthesis and Properties of Polyurethane Elastomers with Trehalose Units
Kazunori Kizuka*, Shin-Ichi Inoue
Applied Chemistry Department, Aichi Institute of Technology, Toyota, Japan

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 29 December 2015; accepted 11 April 2016; published 14 April 2016
ABSTRACT
In recent years, sugar-derived polymer materials have been actively investigated. In research of polyurethane (PU), sugar has been used as a raw material because it has properties similar to polyol. However, the elastic property of the obtained PU is substantially lost. Hence, the introduction of a sugar unit to PU while maintaining the elastic property remains a challenge in polymer chemistry. Here, we report the synthesis of a polyurethane elastomer (PUE) with a trehalose unit using raw materials such as an aromatic diisocyanate (4,4’-diphenylmethane diisocyanate), polyols including a polyether polyol (polytetramethylene glycol), two polyester polyols (polycaprolactone and polycarbonate diol), and trehalose. Novel PUEs with trehalose units are synthesized by a one-shot method. Trehalose, which has non-reducing properties, is used as sugar. The use of trehalose, which has been scarcely applied to PUE, is essential to obtain the desired PUEs with sugar units.
Keywords:
Polyaddition Reaction, Polyurethane Elastomer, Trehalose, Cross-Linker

1. Introduction
Polymer materials derived from sugar have been actively investigated in recent years in the field of polyurethane (PU) [1] - [20] , because polymer materials using sugar are expected to replace materials derived from petroleum and because they are biodegradable. However, most investigations have used sugar as a raw material as it has similar properties to polyol. PUs are used in a surprising array of commercial applications, which, for convenience, are classified into seven major product types [21] - [27] : 1) flexible slab; 2) flexible molded foam; 3) rigid foam; 4) solid elastomers; 5) reaction injection molded materials; 6) carpet backing; and 7) one- and two-com- ponent formulations for coatings, adhesives, and sealants. Building on this background, we are interested in solid elastomers widely used for commercial products. Therefore, we are attempting to develop polymer materials with new functionalities [28] . Polyurethane elastomers (PUEs) containing sugar may be used for many applications such as heat insulation, tremor insulation, or as protective cases for commercial or industrial instruments. Typically, sugar is reduced in organic solutions, producing aldehyde and ketone groups. Thus, non-reducing sugar, which retains hydroxyl groups in solution, is ideal to introduce sugar units in the main chain. We select trehalose, which is scarcely reported on studies of PUEs containing trehalose units [29] , as a representative non-reducing sugar which is similar to sucrose. Trehalose has two primary and six secondary hydroxyl groups. Hence, we studied the synthesis of PUEs containing trehalose units and expected that trehalose acted as a crosslinker. Trehalose was a rare sugar naturally found in crustaceans, insects, and mushrooms. However, a synthetic method of producing trehalose was developed by Hayashibara Co., Ltd. in 1994 [30] . Thus, mass production of trehalose was now possible. Presently, trehalose was used as a raw material for food adjunct, cosmetics, and medical drugs in industrial applications. Here, we reported the synthesis of PUEs containing trehalose units using raw materials such as aromatic diisocyanate (4,4’-diphenylmethane diisocyanate (MDI)), polyols including a polyether polyol (polytetramethylene glycol (PTMG)) and two polyester polyols (polycaprolactone (PCL) and polycarbonate diol (PCD)), and trehalose by a one-shot method to form the corresponding PUEs containing trehalose units in the main chain.
2. Experimental
2.1. Materials
PTMG (molecular weight = 2000) (PTMG2000) (TERATHANE 2000) was supplied by Invista Industry, Texas, USA. PCL (molecular weight = 2000) (PCL2000) (PLACCEL 2000) was supplied by Daicel Industry, Osaka, Japan. MDI (MILLIONATE MT) and PCD (molecular weight = 2000) (PCD2000) (NIPPOLLAN 980N) were supplied by Tosoh Industry, Tokyo, Japan (Tosoh). MDI was purified by distillation under reduced pressure (267 - 400 Pa) at 100˚C before use. Trehalose was purchased from NacalaiTesque, Inc., Kyoto, Japan (Nacalai) and used without further purification. Tetrahydrofuran (THF) and benzene were purchased from Nacalai and distilled over calcium hydride under an Ar atmosphere. N,N-Dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were purchased from Nacalai and stored over 4 Å molecular sieves before use. The following compounds were purchased from commercial suppliers and used as received: DMSO (Euriso Top, Saint-Aubin, France), hexane (Nacalai), and acetone (Nacalai).
2.2. Synthesis of Polyurethane Elastomers Containing Sucrose
PUEs with trehalose units were prepared from MDI, one of the three polyols (PTMG2000, PCL2000, and/or PCD2000)), and trehalose by a one-shot method (Scheme 1). The recipe and trehalose content for each of the PUEs are listed in Table 1. The NCO/OH molar ratio was 2 for all PUEs containing trehalose units.
For example, the synthesis of PUE-PTMG-T1 was performed as follows. A trehalose/DMF solution was prepared from trehalose (0.34 g, 0.10 × 10−3 mol) and DMF (10 mL) at 100˚C for 15 min under an Ar atmosphere.

Table 1. Recipe and appearance of the polyurethane elastomers containing a trehalose unit.
aPolyols: polyoxytetramethylene glycol (molecular weight = 2000; PTMG2000), polycaprolactone diol (molecular weight = 2000; PCL2000), and polycarbonate diol (molecular weight = 2000; PCD2000).
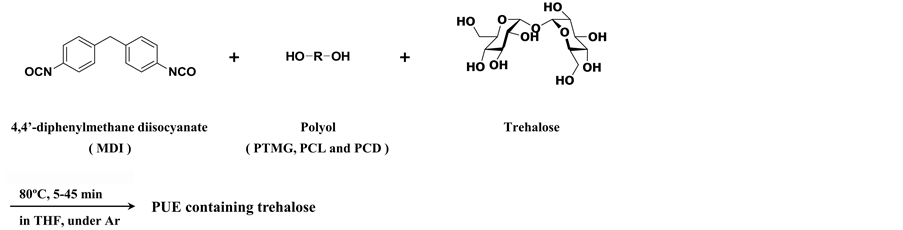
Scheme 1. Synthesis of polyurethane elastomers containing trehalose via the one-shot method.
MDI (5.0 g, 2.0 × 10−2 mol), PTMG2000 (18 g, 0.90 × 10−2 mol), THF (20 mL), and trehalose/DMF solution (10 mL) were added to a 100 mL four-necked separable reaction flask equipped with a mechanical stirrer, a gas inlet tube, and a reflux condenser. This mixture was stirred at 80˚C for 20 min under an Ar atmosphere. The thin polymer sheets (~0.5 mm) were obtained by casting the resulting solution (20 g) using a disposable container at room temperature (23 ± 2˚C) for 15 h. The obtained sheet was cured at 80˚C for 6 h in vacuo.
2.3. Characterization
All analyses and tests were performed at room temperature (23 ± 2˚C) unless otherwise indicated.
2.3.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectra were recorded on a Varian (California, USA) Unity Plus-300 (1H, 300 MHz; 13C, 75.4 MHz) NMR spectrometer. Chemical shift values for protons were referenced to the resonance of tetramethylsilane as the internal standard and values of carbon were referenced to the carbon resonance of DMSO-d6 (δ49.5).
2.3.2. Fourier Transform Infrared (FTIR) Spectroscopy
Fourier transform infrared (FTIR) spectra were recorded on a JASCO (Tokyo, Japan) FTIR-5300 spectrometer equipped with an attenuated total reflection (ATR) system, which used an ATR500/M with an ATR prism KRS-5.
2.3.3. Gel Permeation Chromatography (GPC)
The average molecular weight and molecular distributions were investigated using a Tosoh (Tokyo, Japan) gel permeation chromatograph (GPC) equipped with SD-8022, CCPD, CO-8020, and RI-8020. The measurement conditions for GPC were as follows: sample, 0.1 wt% (DMF/DMSO = 1/1 solution); solvent, DMF; column, TSK gels α-M and TSK GUARDCOLUMNα; flow rate, 500 µL/min at 40˚C; quantum, polystyrene transformation method.
2.3.4. Chemical Properties
Solubility tests were performed using 15 × 15 mm test pieces. Each test piece was soaked in a solvent (benzene, hexane, acetone, THF, DMF, or DMSO; 8.0 mL) at room temperature or 100˚C (for DMF and DMSO) for 24 h.
Swelling tests were performed using 15 × 15 mm test pieces. The degree of swelling (Rs) was calculated using the formula Rs(%) = W’/W × 100, where W’ is the weight of the test piece soaked in benzene for 24 h, and W is the weight of the test piece after drying at 30˚C for 24 h in vacuo.
2.3.5. Mechanical Properties
Hardness tests were performed using a Kobunshi Keiki (Kyoto, Japan) Asker Durometer (JIS A type) with test pieces stacked to achieve a thickness of 6 mm.
Tensile tests were performed on an Orientec (Tokyo, Japan) RTC-1225A with a model-U-4300 using a JIS 3-dumbbell as the standard sample and a crosshead speed of 100 mm/min.
2.3.6. Thermal Properties
Dynamic mechanical analyses (DMA) were performed on a Seiko Instruments (Chiba, Japan) DMS 6100 at 5˚C/min over −100˚C to 300˚C at 20 Hz under N2 atmosphere.
Differential scanning calorimetry (DSC) measurements were performed on a Rigaku (Tokyo, Japan) Thermo-Plus DSC-8230 at 10˚C/min from −120˚C to 200˚C under an Ar atmosphere. Approximately 9.5 mg of each PUE was weighed and sealed in an aluminum pan. The samples were rapidly cooled to −120˚C and then heated to 200˚C at 10˚C/min.
Thermogravimetric analyses (TGA) were performed on a Seiko Instruments (Chiba, Japan) TG/DTA6200 at 10˚C/min from 30˚C to 500˚C under N2 atmosphere.
2.3.7. Surface Analysis
Atomic force microscopy (AFM) analyses were performed on dried sheets at room temperature (23˚C ± 2˚C) in air using an Olympus NV2000. Most of the images were obtained in tapping mode (ACAFM) with a silicon nitride cantilever (OMMCL-AC 240TS-C2, Olympus optical) using a spring constant of 15 N/m and a resonating frequency of 20 KHz. The scanning rates were varied from 1 to 2 Hz. All the images presented here were reproduced from images obtained from at least three points on each sample surface.
3. Results and Discussion
3.1. NMR Spectroscopy
Analyses by 1H NMR (Figure 1) and 13C NMR (Figure 2) spectroscopy revealed that the polymers obtained were undoubtedly PUEs containing trehalose and that these polymers were composed from a urethane segment and trehalose. The NMR analyses indicated that trehalose was attached to the main PU chain as a cross-linker. For example, 1H NMR (DMSO-d6, 300 MHz) of PUE-PTMG-T1; δ8.54 (1H, CO-NH), 7.31 and 7.08 (1H, -C6H4-), 5.10 - 4.27 (1H, CH(OH)), 4.05 and 3.27 (2H, O-CH2), 3.78 (2H, -C6H4-CH2-C6H4-), and 1.49 (2H, -CH2-CH2-, -CH(R)-) and 13C NMR (DMSO-d6, 75.4 MHz) of PUE-PTMG-T1; δ154 - 147 (Urethane: C=O), δ138 - 135 (Urea: C=O), 129 (C=C), 118(C=C), 115(C=C), 70 (O-CH2), and 26 (CH2) (Figure 1).

Figure 1. 1H NMR spectra of PUE-PTMG-T1.

Figure 2. 13C NMR spectra of PUE-PTMG-T1.
3.2. FTIR Spectroscopy
The representative IR spectra of PUE-PTMG-T1-5 are shown in Figure 3. The IR spectroscopy analyses of PUE-PTMG-T1-5 were used to check the end of polyaddition reaction. The absence of the characteristic NCO band at 2265 cm−1, appearance of N-H stretching band at 3307 cm−1, N-H bending band and C-N stretching band of amide II at 1597 and 1535cm−1, and C=O stretching band at 1709 cm−1 confirmed the end of the polyaddition reaction and formation of PU linkages. The characteristic absorption bands at 2940 - 2850 cm−1 indicated that the -CH2- asymmetric stretching mode is available in the synthesized PUE-PTMG-T1. The band at 1647 cm−1 was attributed to the amide II stretching mode of PU. The band at 1093 cm−1 is due to the asymmetric stretching of C-O-C linkage.
Infrared (ATR, cm−1) ν3307 (N-H), 2937, 2851, and 2795 (C-H), 1709 (C=O), 1597 and 1535 (N-H).
3.3. GPC
The GPC of PUEs containing trehalose units is reported in Table 2. For example, PUE-PTMG-T1; Mw 340000; Mw/Mn 7.4.
3.4. Chemical Properties
PUEs with trehalose units yielded quantitatively in 10 - 25 min, and the actual trehalose contents in each synthesized PUE agreed with the theoretical values. Interestingly, the reaction solutions showed different inherent viscosities, and the viscosities of PUEs increased with the trehalose contents. In addition, all PUEs containing trehalose units were transparent. The solvent resistances of PUEs containing trehalose units were tested by immersing each PUE sheet in various solvents including hexane, benzene, toluene, acetone, THF, DMF, and DMSO. The results are presented in Table 3. All PUEs containing trehalose units were resistant to hexane and acetone and swelled in benzene, THF, DMF, and DMSO at room temperature (23 ± 2˚C). Notably, the corresponding PUEs without trehalose dissolved completely in DMSO and DMF at 100˚C, whereas the PUEs containing trehalose contents dissolved only slightly. In general, the PUEs with trehalose units exhibited good solvent resistance. These results suggested that the PUEs containing trehalose units form interpenetrating polymer networks [29] .

Figure 3. FTIR (ATR method) spectra of PUE-PTMG-T1-5.
Table 2. Physical Properties of the polyurethane elastomers with trehalose unit.
aMeasurement conditions: JIS A type, total thickness = 6 mm, room temperature (23˚C ± 2˚C). bMeasurement conditions: benzene solvent at room temperature (23˚C ± 2˚C) for 24 h. cDifferential scanning calorimetry was performed at a heating rate of 10˚C/min from −120˚C to 200˚C under an Ar atmosphere. dThermogravimetric analysis was performed at a heating rate of 10˚C/min from 30˚C to 500˚C under an N2 atmosphere. eMeasurements conditions: solvent = N,N-dimethylformamide, sample = 0.1 wt% (N,N-dimethylformamide/dimethyl sulfoxide = 1/1 solution), flow rate 500 L/min, measurement temperature = 40˚C.
Table 3. Solubility test of the polyurethane elastomers with trehalose unit.
○: dissolved, Δ: swelled, ×: undissolved; aMeasurement conditions: benzene, hexane, acetone, THF, DMF, or DMSO as the solvent at room temperature (23 ± 2˚C) or 100˚C (for DMF and DMSO) for 24 h. bRoom temperature (23˚C ± 2˚C); cRoom temperature (23˚C ± 2˚C) and 100˚C.
3.5. Mechanical Properties
The mechanical behavior of the crosslinked PUEs is dependent on the structural differences between PUEs containing trehalose units which were caused by changing the hard segment content, crosslinking density and intermolecular interactions between their hard segments. The stress versus strain curves for the trehalose-con- taining PUE sheets with different hard segment molar ratios are illustrated in Figure 4, and the tensile strengths and elongation at breaking points of the PUEs with trehalose units are shown in Figure 5. The mixture of the trehalose and polyurethane chain in the soft amorphous phases reduced the mobility of macromolecular chains, thereby generating stiff PUEs when trehalose was in the matrix phase. Trehalose is a rigid, whereas polyurethane is a ductile elastomer. The tensile strength and elongation at breaking points for PUE-(PTMG/PCD)-T1 were same as that for PUE-(PTMG/PCD), and the elongation at breaking point of PUE-PCL-T1 was lower than that for PUE-PCL However, the tensile strength and elongation at breaking points of PUE-(PTMG/PCD)-T2?T5 for polymers decreased with the trehalose contents. The tensile strengths of PUE-PCL-T2?T5 were same as that for PUE-PCL, and the elongation at breaking points of PUE-PCL-T2?T5 decreased with the trehalose contents. As a result, PUE-(PTMG/PCL/PCD)-T1 exhibited the best elastic behavior. These results suggested that the effect of trehalose on the higher order conformation expanded to the overall structure as the trehalose contents increased.
The effect of the various polyurethane microstructures on their macroscopic behavior was reflected in their hardness. Table 2 shows hardness of the trehalose-containig PUE sheets with different amount of hard segment content. Hardness of PUE-PTMG-T1-T4 were lower than that for PUE-PTMG, hardness of PUE-PCL-T1-T3 were lower than that for PUE-PCL, and hardness of PUE-PCD-T1-T2 were lower than that for PUE-PCD, respectively. These results show that hydrogen bonding, phase segregation, crosslink density and the plasticizer effect of trehalose dangling chains affect the hardness. A higher number of crosslinked hard segments leads to an increase in the hardness of the polyurethane materials.
3.6. Thermal Properties
DSC analyses of the PUEs containing trehalose units were performed from −120˚C to 200˚C under an Ar atmosphere. From Table 2, it is evident that one main transition occurred for the PUEs containing trehalose units. The values for the glass transition temperatures (Tg) of the corresponding PUEs without the trehalose content were −67.0˚C, −45.0˚C, and −54.0˚C for PUE-PTMG, PUE-PCL, and PUE-PCD, respectively. The Tg values of PUE-PTMG-T1-T5 were lower than that for PUE-PTMG and the Tg values of PUE-PCL-T1-T5 were higher than that for PUE-PCL. For PUE-PCD-T1-T5, The Tg values of T1-T3 were lower than that for PUE-PCD and theTg values of T4 and T5 were higher than that for PUE-PCD. Tg of PUEs with trehalose units increases with the concentration of hard segments and with of the number of crosslinks added. The increase in Tg of PUEs with trehalose units is due to the hindrance local motion of the polymer segments throughout the formation of physical and chemical crosslinks between molecular chains. Thus, the glass transition temperature of the PUEs with trehalose units is influenced by its crosslinking density and chemical structure. The difference in Tg values arise
 (a) (b) (c)
(a) (b) (c)
Figure 4. Tensile properties of the polyurethane elastomers containing a various amounts of trehalose. (a) PUEs using MDI, PTMG2000, and trehalose; (b) PUEs using MDI, PCL2000, and trehalose; (c) PUEs using MDI, PCD2000, and trehalose. Red: 1.4 wt% trehalose content, deep blue: 3.1 wt% trehalose content, green: 5.0 wt% trehalose content, orange: 7.6 wt% trehalose content, light blue: 10wt% trehalose content, black: PUEs without trehalose content.

Figure 5. Tensile strengths and elongation at the breaking points for PUEs with containing of the trehalose. Red: PUEs using MDI, PTMG2000, and trehalose; deep blue: PUEs using MDI, PCL2000, and trehalose; green: PUEs using MDI, PCD2000, and trehalose.
from several factors including the crosslinking density of the trehalose-based network and higher content of MDI in crosslinked polyurethane.
The thermal stabilities of the PUEs containing trehalose units were examined via TGA under an N2 atmosphere. The TG curves of the PUEs without trehalose and the PUEs with different trehalose component ratios are displayed in Figure 6 to analyze the effect of trehalose on stabilization further. Degradation temperature (T10) decreased evidently for the PUEs with trehalose. This result is attributed to the complicated process of PUE decomposition given that PUE is a copolymer composed of microphase-separated hard and soft segments.

Figure 6. Thermo gravimetric analyses of the polyurethane elastomers containing a various amounts of trehalose. (a) PUEs using MDI, PTMG2000, and trehalose; (b) PUEs using MDI, PCL2000, and trehalose; (c) PUEs using MDI, PCD2000, and trehalose. Red: 1.4 wt% trehalose content, deep blue: 3.1 wt% trehalose content, green: 5.0 wt% trehalose content, orange: 7.6 wt% trehalose content, light blue: 10 wt% trehalose content, black: PUEs without trehalose content.
DMA measurements of the PUEs with trehalose units were performed from −100˚C to 300˚C. The results are displayed (Figure 7). Rubbery flat regions were not observed in PUE-(PTMG/PCL/PCD)-T3?T5 but in PUE- (PTMG/PCL/PCD)-T1 and T2 at approximately 50˚C - 180˚C. In addition, the rubbery flat regions for PUE- (PTMG/PCL/PCD)-T1 and T2 declined significantly in comparison with those observed for PUE-(PTMG/PCL/ PCD). These results suggested that the molecular chain lengths between crosslinked polymers were reduced upon addition of a small amount of trehalose. Regarding PUE-(PTMG/PCL/PCD)-T3-T5, the characteristic S- shaped curves were not observed and the E’ values increased. These results suggested that the trehalose-con- taining PUEs calcified as the trehalose contents increased. The tanδ values additionally shifted to the high-tem- perature area as the trehalose contents and values of the peak top simultaneously increased. Furthermore, the two different peaks were present as small broad peaks in PUE-(PTMG/PCL/PCD)-T4 and T5. The peak in the low-temperature area may be attributed to the hard segment, whereas that on the high-temperature area may be attributed to the PU chains.
3.7. Morphology Evolution
The surface topography of the PUEs containing trehalose units was examined by AFM (Figure 8). AFM was

Figure 7. Dynamic mechanical analyses of the polyurethane elastomers containing a various amounts of trehalose. (a) PUEs using MDI, PTMG2000, and trehalose; (b) PUEs using MDI, PCL2000, and trehalose; (c) PUEs using MDI, PCD2000, and trehalose. Red: 1.4 wt% trehalose content, deep blue: 3.1 wt% trehalose content, green: 5.0 wt% trehalose content, orange: 7.6 wt% trehalose content, light blue: 10 wt% trehalose content, black: PUEs without trehalose content.
 (a) (b)
(a) (b) (c) (d)
(c) (d)
Figure 8. Atomic force microscopy images of the polyurethane elastomer without a trehalose unit (PUE-PTMG) and the polyurethane elastomer with a trehalose unit (PUE-PTMG-T5). (a) and (b): two dimensions; (c) and (d): three dimensions.
utilized to study the phase-segregated morphology ofPUEs with trehalose units. AFM investigations were conducted on the surface of the polymer, with a scanning area of 1000 × 1000 nm. Topographical heterogeneity is observed in the images of PUE containing trehalose units, which may reflect the existence of ordering tendencies in the polymer structure. In these images, by convention, the hard and soft microphases appear as bright and dark regions, respectively. By increasing the hard segment concentration, changes were observed in the surface morphology. The presence of bright and dark regions indicates the presence of microphase morphology. AFM images of the trehalose-containig PUE sheets have an extended smoother surface area compared to the AFM image observed in the polyurethane sheet without trehalose. The light-colored spots represent hard domains. These are dispersed all over the matrix which is formed by the soft domains while in the case of polyurethane without trehalose and appear as thick regions. Inclusions of hard segments can been in some limited areas. This could be explained by the possibility that those areas benefitted from a better ordering.
3.8. Discussion for Structure of Polyurethane Elastomers with Trehalose Unit
3D structures of the PUEs containing trehalose units were predicted using the characteristic structural and chemical features of trehalose as a reference. Specifically, the trehalose composed of two glucoses has two primary alcohols that are of the highest reactivity and six secondary alcohols which are of high reactivity [31] . Their alcohols should preferentially react with isocyanate, and the resulting materials form a sequential interpenetrating polymer network [32] . In addition, the initial chain polymerization reaction is expected to proceed gradually to form a simultaneous interpenetrating polymer network [32] . Hence, both networks are mixed in the PUE structure. Three types of interpenetrating polymer networks are networks I, II, and III (Figure 9). The network I is molecular model which react with isocyanate by six alcohols of high reactivity. The network II is molecular model which react with isocyanate by four alcohols of higher reactivity. The network III is molecular model which react with isocyanate by two primary alcohols that are of the highest reactivity. The network III partially generate in PUE containing many trehalose units. Therefore, the main networks in PUE-(PTMG/PCL/ PCD)-T1-T5 are networks I and II. These networks are mixed at random in each PUE. Consequently, as the trehalose contents in the PUEs increases, the molecular densities of the PUEs with trehalose units increase.
4. Conclusion
In summary, we achieved the synthesis of PUs that maintained the elastic property using the one-shot method. The use of trehalose for additive compound was essential to obtain the desired PUEs. This study demonstrated the

Figure 9. Interpenetrating polymer network in the PUE containing trehalose.
synthesis of PUEs by introducing trehalose units, which acted as a crosslinker in the main chain. The properties of PUE sheets were mainly governed by the stoichiometric balance of the components in the reaction and the degree of crosslinking.
Cite this paper
Kazunori Kizuka,Shin-Ichi Inoue, (2016) Synthesis and Properties of Polyurethane Elastomers with Trehalose Units. Open Journal of Organic Polymer Materials,06,63-75. doi: 10.4236/ojopm.2016.62007
References
- 1. Jovanovic, S., Dzunuzovic, J.V. and Stojanovic, Z. (2013) Polymers Based on Renewable Raw Materials—Part I. Kemija u Industriji, 62, 307-314.
- 2. Jovanovic, S., Dzunuzovic, J.V. and Stojanovic, Z. (2013) Polymers Based on Renewable Raw Materials—Part I. Kemija u Industriji, 62, 315-326.
- 3. Walter, R.F. (1969) Sucrose-Ethylene Diamine Polyols and Process for Preparing Same. U.S. Patent No. 3640997.
- 4. Neil, H.N. (1978) Sucrose Based Polyether Polyols. U.S. Patent No. 4230824.
- 5. Hatakeyama, H., Hirose, S. and Hatakeyama, T. (1995) Biodegradable Polyurethanes from Plant Components. Journal of Macromolecular Science, Part A: Pure and Applied Chemistry, A32, 743-750.
http://dx.doi.org/10.1080/10601329508010285 - 6. Asano, Y. and Hatakeyama, H. (2003) Rigid Type Polyurethane Foams Containing Saccharide and Lignin Structures in the Molecular Chain. Memoirs of Fukui University of Technology, 33, 275-282.
- 7. Hatakeyama, H. and Hatakeyama, T. (2005) Environmentally Compatible Hybrid Type Polyurethane Foams Containing Saccharide and Lignin Components. Macromolecular Symposia, 224, 219-226.
http://dx.doi.org/10.1002/masy.200550619 - 8. Chen, Q., Li, R., Sun, K., Li, J. and Liu, C. (2011) Preparation of Bio-Degradable Polyurethane Foams from Liquefied Wheat Straw. Advanced Materials Research, 217, 1239-1244.
http://dx.doi.org/10.4028/www.scientific.net/amr.217-218.1239 - 9. Wang, G. and Zhou, A. (2011) Soy Protein Based Biodegradable Flexible Polyurethane Foam. Advanced Materials Research, 152, 1862-1865. http://dx.doi.org/10.4028/www.scientific.net/AMR.264-265.1862
- 10. Garcon, R., Clerk, C., Gesson, J.-P., Bordado, J., Nunes, T., Caroco, S., Gomes, P.T., Minas da Piedade, M.E. and Rauter, A.P. (2001) Synthesis of Novel Polyurethanes from Sugars and 1,6-Hexamethylenediisocyanate. Carbohydrate Polymers, 45, 123-127. http://dx.doi.org/10.1016/S0144-8617(00)00323-4
- 11. Lim, H., Kim, E.Y. and Kim, B.K. (2010) Polyurethane Foams Blown with Various Types of Environmentally Friendly Blowing Agents. Plastics, Rubber and Composites, 39, 364-369.
http://dx.doi.org/10.1179/174328910X12691245469835 - 12. Ionescu, M., Mihalache, I., Zugravu, V. and Mihai, S. (1994) Inherently Flame Retardant Rigid Polyurethane Foams Based on New Triazinic Polyether Polyols. Cellular Polymers, 13, 57-68.
- 13. Guo, A., Demydov, D., Zhang, W., Zoran, W. and Petrovie, S. (2002) Polyols and Polyurethanes from Hydroformylation of Soybean Oil. Journal of Polymers and the Environment, 10, 49-52. http://dx.doi.org/10.1023/A:1021022123733
- 14. Mir, G., Sadeghi, M., Shamsi, R. and Sayaf, M. (2011) Fromaminolysis Product of PET Waste to Novel Biodegradable Polyuretuhanes. Journal of Polymers and the Environment, 19, 522-534.
- 15. Kim, S.H., Lim, H. and Kim, B.K. (2008) Effects of Initiator Type in Rigid Polyurethane Foams. Polymer Engineering & Science, 48, 1518-1523. http://dx.doi.org/10.1002/pen.21122
- 16. Kim, B.K. and Paik, S.H. (1999) UV-Curable Poly(ethylene glycol)-Based Polyurethane Acrylate Hydrogel. Journal of Polymer Science Part A, 37, 2703-2709.
http://dx.doi.org/10.1002/(SICI)1099-0518(19990801)37:15<2703::AID-POLA3>3.0.CO;2-A - 17. Zheng, L., Du, B. and He, Z. (2013) Treatment of Wool Scouring Wastewater by Immobilized Chitosan Bio-Mem- brane. Journal of Engineered Fibers and Fabrics, 8, 1-5.
- 18. Pan, X. and Webster, D.C. (2012) New Biobased High Functionality Polyols and Their Use in Polyurethane Coatings. ChemSusChem, 5, 419-429. http://dx.doi.org/10.1002/cssc.201100415
- 19. Deka, H. and Karak, N. (2009) Bio-Based Hyperbranched Polyurethanes for Surface Coating Applications. Progress in Organic Coatings, 66, 192-198. http://dx.doi.org/10.1016/j.porgcoat.2009.07.005
- 20. Frisch, K.C. and Kresta, J.E. (1977) An Overview of Sugars in Urethanes. Hickson, J.L., Ed., Sucrochemistry, ACS Symposium Series, Vol. 41, American Chemical Society, Washington DC, 238-256.
http://dx.doi.org/10.1021/bk-1977-0041.ch017 - 21. Szycher, M. (1999) Szycher’s Handbook of Polyurethanes, 3.4-9. CRC Press, Boca Raton.
- 22. Wirtz, H. and Schulte, K. (1973) Processing of Polyurethane Foam Systems. Kunststoffe, 63, 726-730.
- 23. Avar, G., Meier, W.U., Casselmann, H. and Achten, D. (2012) Polymer Science: A Comprehensive Reference. Polymer Science: A Comprehensive Reference, 10, 411-441.
- 24. Kuehn, A.F. (1986) Polyurethane—A Promising Prospect in Roll Coverings. PIMA, 68, 27-28.
- 25. Gagro, D. (2010) Polyurethanes Overview of the PUR Market. European Coatings Journal, 10, 9-11.
- 26. Bez, W. and Quack, G. (1983) Polyurethane and Latex Foam Carpet Backing Today. Cellular Polymers, 2, 31-53.
- 27. Hare, C.H. (2000) A Review of Polyurethanes: Formulation Variables and Their Effects on Performance. Journal of Protective Coatings & Linings, 17, 34-44.
- 28. Kizuka, K. and Inoue, S. (2015) Synthesis and Properties of Polyurethane Elastomers Containing Sucrose as a Cross-Linker. Open Journal of Organic Polymer Materials, 5, 103-112. http://dx.doi.org/10.4236/ojopm.2015.54011
- 29. Keisuke, K., Nobuhiko, H., Hiroyasu, M. and Yosio, I. (1979) Synthetic Polymers Containing Sugar Residues, 6. Novel Polyurethanes by Direct Addition Polymerization of α,α-Trehalose with Diisocyanates. Die Makromolekulare Chemie, 180, 2769-2773. http://dx.doi.org/10.1002/macp.1979.021801120
- 30. Maruta, J., Nakada, T., Kubota, M., Chaen, H., Sugimoto, T., Kurimoto, M. and Tsujisaka, Y. (1995) Existence of a Novel Enzyme Converting Maltose into Trehalose. Bioscience, Biotechnology, and Biochemistry, 59, 2189-2190.
http://dx.doi.org/10.1271/bbb.59.2189 - 31. Vila Verde, A. and Campen, R.K. (2011) Disaccharide Topology Induces Slowdown in Local Water Dynamics. The Journal of Physical Chemistry B, 115, 7069-7084. http://dx.doi.org/10.1021/jp112178c
- 32. Daniel, K., Sperling, L.H. and Utracki, L.A. (1991) Interpenetrating Polymer Networks, ACS.
NOTES

*Corresponding author.