Open Journal of Organic Polymer Materials
Vol. 2 No. 4 (2012) , Article ID: 23975 , 10 pages DOI:10.4236/ojopm.2012.24008
Synthesis and Characterization of Triarylamine-Based Block Copolymers by Combination of C-N Coupling and ATRP for Photorefractive Applications
Graduate School of Bio-Applications and Systems Engineering, Tokyo University of Agriculture and Technology, Tokyo, Japan
Email: *Kogino@cc.tuat.ac.jp
Received July 18, 2012; revised August 20, 2012; accepted August 31, 2012
Keywords: C-N Coupling; ATRP; Triarylamine; Acrylate Ester; Photorefractivity
ABSTRACT
Poly(4-butyltriarylamine)s with t-butyldimethylsilyl terminal protecting group (PBTPA-TBS) with various molecular weights were prepared by C-N coupling polymerization. The resulting precursors were postfunctionalized and subsequently used as macroinitiators for atom transfer radial polymerization (ATRP) of n-butyl acrylate (n-BA) and ethyl acrylate (EA). Both the polymerization processes were controlled and the polymers were characterized by 1H NMR, gel permeation chromatography (GPC) and thermal properties, which confirmed the successful synthesis of all the polymers. The microphase separated behaviors of the poly (4-butyltriarylamine)-block-poly (butyl acrylate) (PBTPA-b-PBA) were examined by AFM in the film showing phase separation structures for all the polymers. The photorefractive property of the composite based on PBTPA-b-PBA block copolymer was evaluated by two-beam coupling experiment. A relative high gain coefficient of 42.7 cm−1 was obtained at the electric field of 31 V/mm.
1. Introduction
Because of the excellent charge transporting properties, triarylamine (TAA) derivatives have been intensively studied and been widely used in thin layer electrooptical devices such as electroluminescent, solar cell and photorefractive holograph [1-4]. Especially, the polymeric TAA derivatives exhibit extensive application prospects due to the potential advantages such as possibility of extended p-conjugation and excellent film-forming property. The common reactions for the synthesis of poly(triarylamine) (PTAA) where TAA units are linked together in the main chain are Ullman reaction [5], palladium or nickel catalyzed coupling reactions [6], and simple oxidative polymerization using ferric chloride as an oxidizing agent [7,8]. Recently, our group have. reported a new approach to synthesize PTAA from A-B type monomer by C-N coupling polymerization using palladium based system as a catalyst [9-11]. A serious of PTAA-based block copolymers have been successfully prepared by adding terminal modifying macroinitiators during the polymerizations. The physical and electrical properties were significantly influenced not only by the chemical structure of polymers, but by their morphologies, which suggests the possibility of designing new TAA-based materials to meet the requirements of applications.
Generally, photorefractive (PR) materials possess both the photoconductivity and electro-optical (EO) activity. It is known that the high mobility of EO active component plays an important role in the high performance PR materials [12]. Therefore it is important that the material has low glass transition temperature (Tg). Nevertheless better photoconductive nature of PTAA, high Tgs of PTAA derivatives prevent the utilization as PR materials. Moreover, the alkyl substituted PTAA show poor miscibility with EO chromophore, which could lead to the poor stability of the host-guest PR systems. In order to solve these problems, it is proposed to utilize block copolymers. It is possible that introduction of soft block with high polarity to PTAA improves the miscibility of EO chromophore. Furthermore, the Tgs for both domains could be controlled independently in a microphase sepatated sample [13].
The process of ATRP can provide well-defined polymers from numerous monomers [14-16]. Therefore polymers prepared by other methods such as cationic, ROMP polymerization and GRIM polymerization have been reported to be employed as macroinitiators for ATRP [17- 19]. However, the application of maroinitiator synthesized by C-N coupling to ATRP has not yet been reported. In this paper, the block copolymers containing PTAA segment and poly (n-butyl acrylate) (PBA) or poly (ethyl acrylate) segment were prepared by combination of C-N coupling and ATRP for the first time. The PR properties of composites consisting of synthesized block copolymers, EO chromophore, and photosensitizer were preliminarily investigated. The unique structures of these block copolymers are expected to improve the performance of PR materials as described above.
2. Experimental
2.1. Materials
4-(4'-Bromophenyl)-4"-butyldiphenylamine, 3 and 2-(4-bromophenoxyl) ethanol, 1 were synthesized according to reported procedures [9,20]. n-Butyl acrylate (n-BA) and ethyl acrylate (EA) were distilled from calcium hydride under reduced pressure and stored in freezer. Tetrahydrofunan (THF) was purified by distillation under nitrogen from sodium benzophenone ketyl. Toluene, anisole and N-methylpyrrolidone (NMP) were distilled from calcium hydride and stored under nitrogen at room temperature. The other reagents and solvents were used as purchased without any further purification.
2.2. Characterization
1H NMR spectra were recorded with a JEOL ECX300 spectrometer operating at 300 MHz. Molecular weight and its distribution were determined by gel permeation chromatography (GPC) with JASCO UV-970 and RI- 2031 detectors. Chloroform was used as an elute at a flow rate of 0.5 mL/min and the GPC system was calibrated with polystyrene standards. Differential scanning calorimetric (DSC) analyses were performed on a Rigaku DSC-8230 under nitrogen atmosphere at heating and cooling rates of 10˚C /min. Thermogravimetric analyses (TGA) were performed on Shimadzu DTA-60 under nitrogen atmosphere at heating rates of 10˚C/min. Atomic force microscopic (AFM) measurements were performed on JEOL JSPM 4200 system in trapping mode (phase and topographic modes) with an MPP-11100-10 silicon probe (resonant frequency: 300 kHz, force constant: 40 N/m). All thin film of polymers were spin-cast onto glass slide with a MIKASA 1H-D7 spin coater from THF solutions at 1500 rpm for 30 s.
2.3. Synthesis of 2-(4-Bromophenoxy)EthoxyTert-Butyldimethylsilane 2
To the mixture of 2-(4-bromophenoxyl)ethanol (5.38 g, 24.8 mmol), tert-butyldimethylchlorosilane (3.92 g, 26 mmol) and dichloromethane (30 mL), pyridine (2.23 mL, 28 mmol) and 4-dimethylaminopyridine (0.01 g, 0.08 mmol) were added slowly under nitrogen atmosphere at 0˚C. The solution was stirred at 0˚C for 30 min, and then the temperature was increased to 25˚C. After 24 h, solvents were evaporated, and the products were extracted with diethyl ester. The organic layer was washed by water and dried over MgSO4. The concentrated residue was purified by column chromatography (silica/toluene). Transparent liquid (5.62 g) was obtained. Yield: 62%. 1H NMR [300 MHz, CDCl3, δ(ppm)] δ7.39 (d, 2H), δ6.81 (d, 2H), 4.02 (d, 2H), 3.98 (d, 2H), 0.90 (s, 9H), 0.11 (s, 6H).
2.4. Synthesis of Tert-ButyldimethylsilylTerminated Poly (4-Butyltriarylamine) (PBTPA-TBS) 4
The example of synthetic details was as follows: a 20-mL flask equipped with a magnetic stirrer and a nitrogen inlet was charged with the mixture of 4-(4'-bromo-phenyl)-4"-n-butyldiphenylamine 3 (1.0 g, 2.7 mmol), sodium tert-butoxide (0.28 g, 3.0 mmol) and dry THF (5 mL). To this mixture, the solution of 2 (0.57 mg, 1.8 mmol), palladium (II) acetate (0.012 g, 0.054 mmol) and tri-tert-butyphosphine (0.044 g, 0.22 mmol) in THF (5 mL) was added under nitrogen atmosphere. After the mixture was stirred under reflux for 24 h, the solution of diphenylamine (0.34 mg, 2 mmol) in THF (0.5 mL) was added. The solution was continued stirring for 1 h under reflux, and then poured into methanol. The precipitate was dissolved in chloroform and then reprecipitated in acetone. After filtration and dried, 0.91 g of gray powder was obtained. Polymerization conditions and characteris tics of the resulting polymers were listed in Table 1. Yield: 60%. 1H NMR [300 MHz, CDCl3, d (ppm )] d7.43 (d, 50H), 7.15 - 7.03 (m, 104H), 6.84 (d, 2H), 4.02 (d, 2H), 3.97 (d, 2H), 2.58 (t, 26H), 1.61 (m, 26H), 1.38 (m, 26H), 0.94 - 0.90 (m, 48H), 0.11 (s, 6H).
2.5. Synthesis of Hydroxyl-Terminated Poly (4-Butyltriarylamine) (PBTPA-OH) 5
The mixture of PBTPA-TBS (0.7 g, 0.16 mmol), THF (40 mL) and hydrogen fluoride pyridine (pyridine/HF)
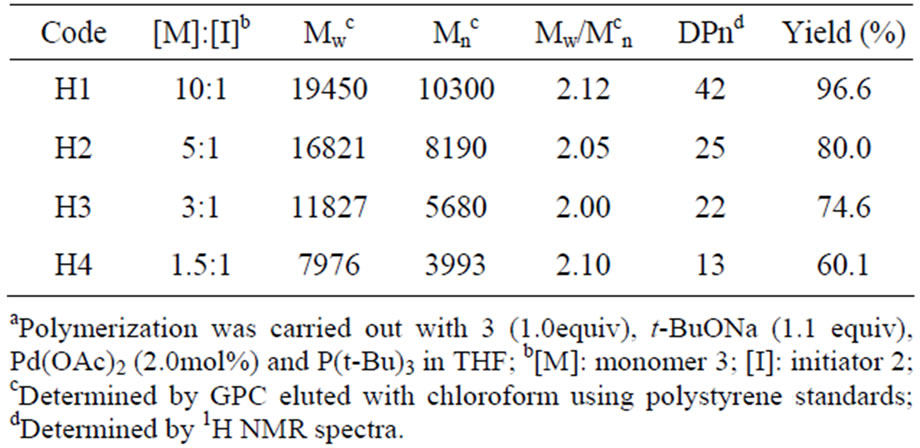
Table 1. Characteristics of PTPA-TBS prepared by C-N coupling under various reaction conditionsa.
(1.8 mL) [21] was placed into a 100-mL flask and stirred for 48 h at room temperature. After reaction, the mixture was poured into methanol. The precipitate was dissolved in chloroform, and then reprecipitated in acetone. Finally, 0.62 g of gray powder was obtained. Yield: 90%. 1H NMR [300 MHz, CDCl3, δ (ppm )] δ7.43 (d, 50H), 7.15 - 7.03 (m, 104H), 6.84 (d, 2H), 4.02 (d, 2H), 3.97 (d, 2H), 2.58 (t, 26H), 1.61 (m, 26H), 1.38 (m, 26H), 0.94 (t, 39H).
2.6. Synthesis of Bromo Ester Terminated Poly (4-Butyltriarylamine) (PBTPA-MI) 6
The reaction was carried out under nitrogen atmosphere. PBTPA-OH (0.5 g, 0.12 mmol) was dissolved in THF (62 mL), followed by addition of triethylamine (3.53 mL, 25.6 mmol) to the solution. Then 2-bromopropionyl bromide (2.33 mL, 23.3 mmol) was added slowly by a syringe at 0˚C. The solution was stirred at 0˚C for 30 min, and then the temperature was increased to 25˚C. After 48 h, the mixture was poured into methanol. The precipitate was dissolved in chloroform and then reprecipitated in acetone. Finally, 0.43 g of gray powder was obtained. Yield: 86%. 1H NMR [300 MHz, CDCl3, δ (ppm )] δ 7.43 (d, 50H), 7.15 - 7.03 (m, 104H), 6.80 (d, 2H), 4.45 (d, 2H), 4.35 (m, H), 4.10 (d, 2H), 2.58 (t, 26H), 1.78 (d, 3H), 1.61 (m, 26H), 1.38 (m, 26H), 0.94 (t, 39H).
2.7. Synthesis of PBTPA-Based Diblock Copolymers
The example of synthetic details was as follows: a 5-mL flask equipped with a magnetic stirrer and a nitrogen inlet was charged with PBTPA-MI (0.1 g, 0.023 mmol) and CuBr (72.9 mg, 0.51 mmol). After the evacuation followed with backfilling of nitrogen, the mixture of anisole/NMP (0.36 mL/0.36 mL), distilled n-BA (1.64 mL, 11.5 mmol) and N,N,N',N",N"-pentamethyldiethylenetriamine (PMDETA) (127 μL, 62 mmol) were added. Then the reaction mixture was stirred for 5 min. After five freeze-pump-thaw cycles, the flask was placed in a 95˚C oil bath for polymerization. After dilution with THF, the reaction mixture was filtered with alumina column in order to remove the copper catalyst. After concentration, the polymer was obtained by precipitation from methanol. Polymerization conditions and characteristics of block copolymers are listed in Table 2.
2.8. Photorefractive Measurement
PR devices were prepared as follows: THF solutions of PBTPA-b-PBA block copolymer, 2-[[4-(diethylamino) phenyl]methylene]propanedinitrile (DDCST) and 2,4,7- trinitro-9-fluorenone (TNF) were filtered through a 0.45 μm filter and cast onto indium tin oxide (ITO, 30 W)
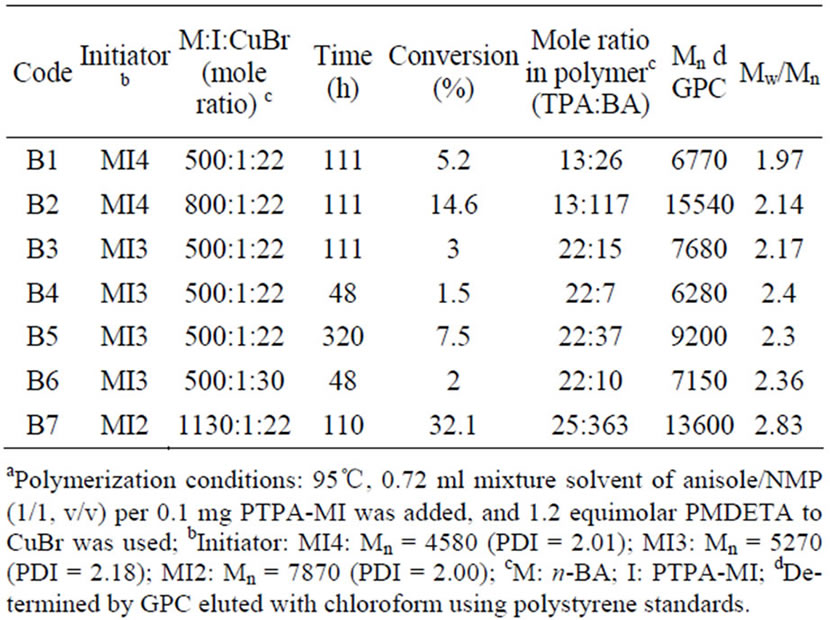
Table 2. Characteristics of PTPA-b-PBA prepared by ATRP under various reaction conditionsa.
covered glass substrate at room temperature. The viscous solutions were left to evaporate the residual solvent under vacuum for 6 h. Then the composite was sandwiched with another ITO-covered glass. The sample thickness was controlled to be approximately 80 μm by a Teflon spacer.
The photorefractive properties of the composites were preliminarily studied by the two beam coupling (2BC) experiment using a conventional experiment setup [22]. The coherent beam from an NEC GLS-5410 He-Ne laser operating at 633 nm was split into two writing beams with the equal intensity. The two p-polarized writing beams with an angle of 20˚ were overlapped on the sample, which was tilted at an angle of 50˚ with respect to the bisector of the two writing beams. Asymmetric energy transfer between the two beams can be obtained by the following experiment: Beam 1 was switched on/off, while the transmitted intensity of beam 2 was monitored with photodiodes (Hamamatsu Photomics, S2281), and the intensity of beam 1 was monitored as beam 2 was switched on/off. The 2BC gain is calculated according the Formula [23]:
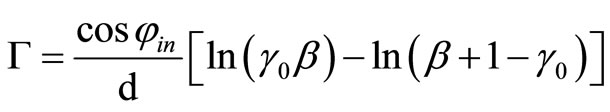 (1)
(1)
Where d is the thickness of the sample, is the tilting angle inside the sample. The experiment was carried out under room temperature, 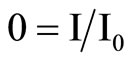 is the beam coupling ratio where I0 is the signal intensity without the pump beam and I is the signal intensity with the pump beam and is the light intensity ratio of the two incident beams.
is the beam coupling ratio where I0 is the signal intensity without the pump beam and I is the signal intensity with the pump beam and is the light intensity ratio of the two incident beams.
3. Results and Discussion
A new synthetic route was developed for the introduction of polyacrylate at PBTPA chain end using TBS terminated PBTPA as a precursor (shown in Scheme 1). The TBS end group was easily converted to hydroxyl, and then reacted with 2-bromopropionyl bromide to obtain PBTPA with bromo ester end group that was used as a macroinitiator for ATRP.
3.1. Synthesis of PBTPA-TBS Precursors
Our group has reported a self-condensing type monomer 3 with bromophenyl and diphenylamine moieties for building up well-defined polymer via C-N coupling reaction, which could be functionalized at both terminals separately by adding terminal modifying derivatives like an aryl bromide or arylamine derivatives as a terminator [9]. As a result, PBTPA-b-poly(ethylene oxide) [9] and PBTPA-b-polystyrene [11] were synthesized using polymer modified aryl bromides as terminal modifiers. However, preliminary studies revealed that the preparation of PBTPA-b-PBA via C-N coupling polymerization in the presence of a terminal modifier with PBA chain or ATRP initiating 2-bromopropionate moiety afforded PBTPA homopolymer without designed chain end in a relatively low yield. This is probably due to the prohibition of oxidative insertion of Pd in the terminator [9]. As a result, PBTPA-b-poly(ethylene modifier by ester groups. Therefore, different synthetic strategy is necessary, and new synthetic routes are explored. Here, aryl bromide with hydroxyl group and that protected with TBS group (1 and 2) were prepared (as shown in Scheme 1). The polymerization was carried out using palladium acetate and tri(tert-butyl)phosphine ligand as the catalyst, and tertbutoxide as base in THF for 24 h.
The 1H NMR spectrum of the product initiated by 1 was shown in Figure 1(A). The signals assignable to hydroxyl proton and methylene protons of the initiating end were almost negligible which indicated the expected product 5' was not obtained. While the signal derived from the diphenylamino group at 5.7 ppm was clearly observed. This suggested that aryl bromide with hydroxyl group hardly modified the diphenylamino terminal of 3, and the products were mainly the byproducts obtained via the self-polymerization of 3.
The structure of 2 was confirmed by 1H NMR in which the characteristic signals at 0.9 ppm and 0.11 ppm corresponding to the TBS group were observed, respecttively. The signals derived from methylene protons at 4.02 ppm and 3.98 ppm were also observed.
The structure of the product initiated from compound 2 (PBTPA-TBS) was confirmed by 1H NMR spectrum (Figure 1(B)). All the signals can be assigned, including the PBTPA chain and TBS end group. Moreover, no signal derived from the diphenylamino terminal at 5.7 ppm can be found, which indicated the complete modification of diphenylamino group by 2. Furthermore, unimodal and symmetric peaks were observed in the GPC traces of all the PBTPA-TBS products (Figure 2). In a previous study, it is reported that the GPC traces of the polymers modified by other aryl bromide in some cases exhibited a shoulder at a higher retention time region, which corresponds to the cyclic oligomers [9]. It is suggested that the aryl bromide derivate with TBS end group possesses afforded no cyclic oligomer because of high initiation efficiency of 3 by C-N coupling polymerization.
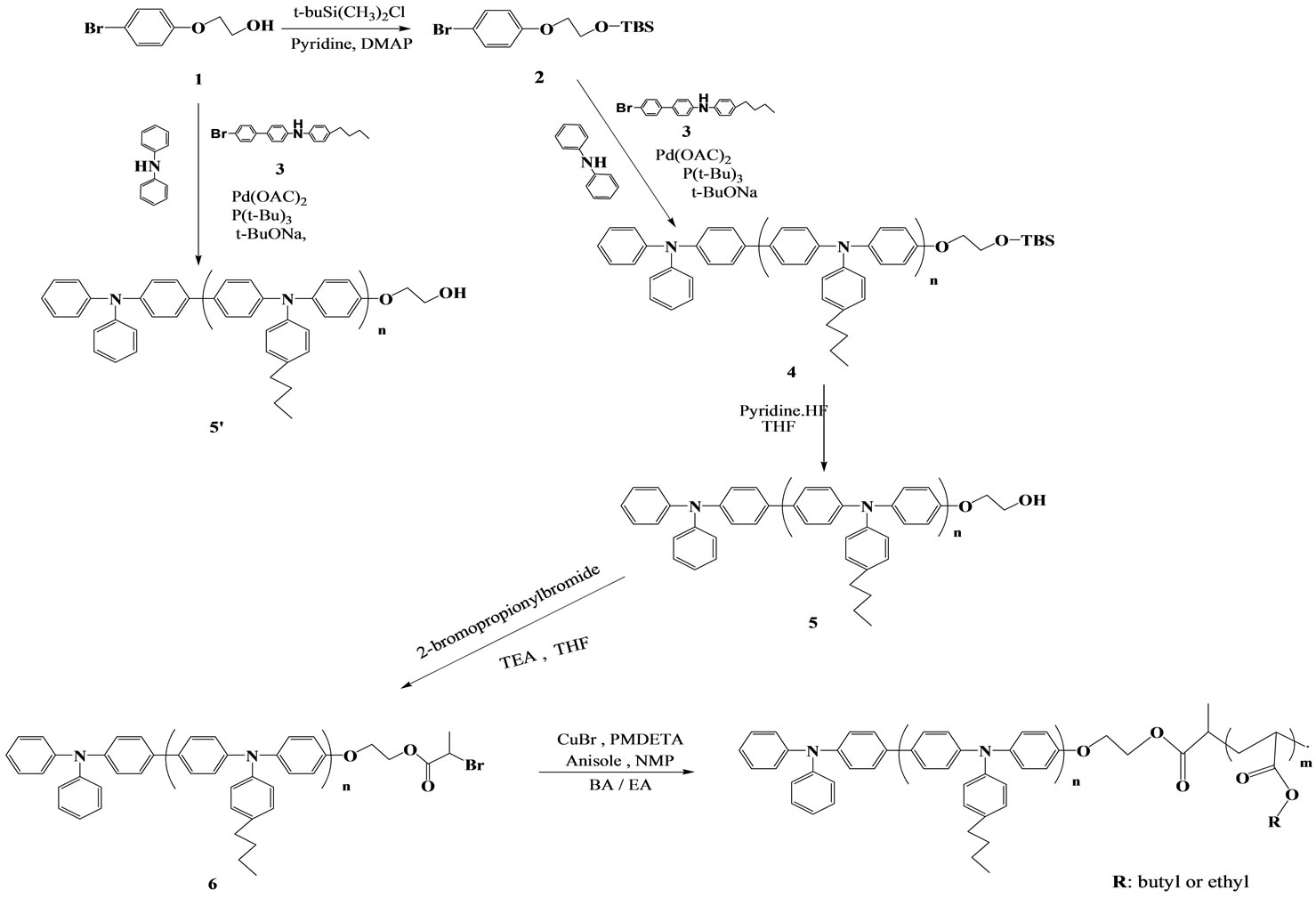
Scheme 1. Reaction scheme for synthesis of TPA-based macroinitiator and block copolymers.
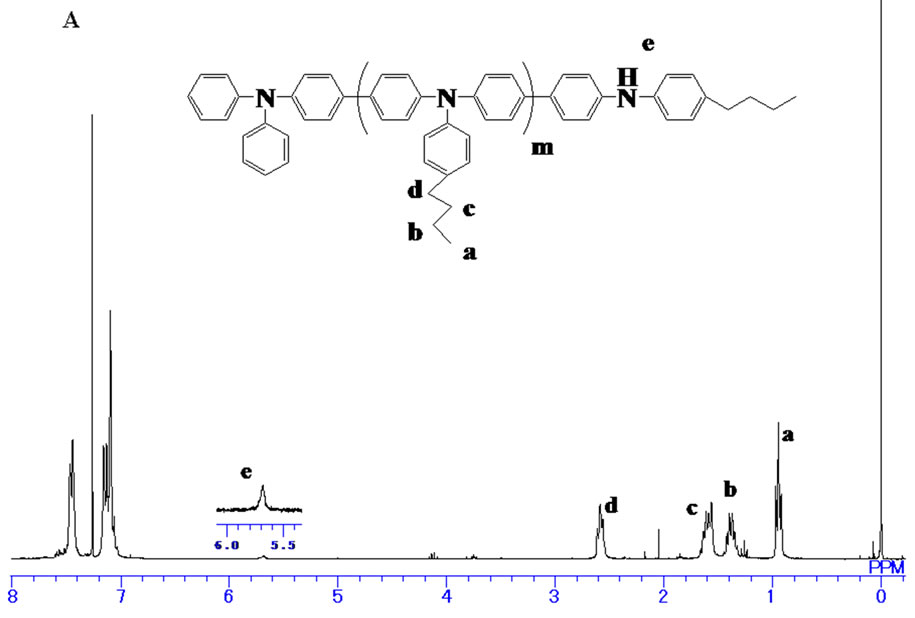
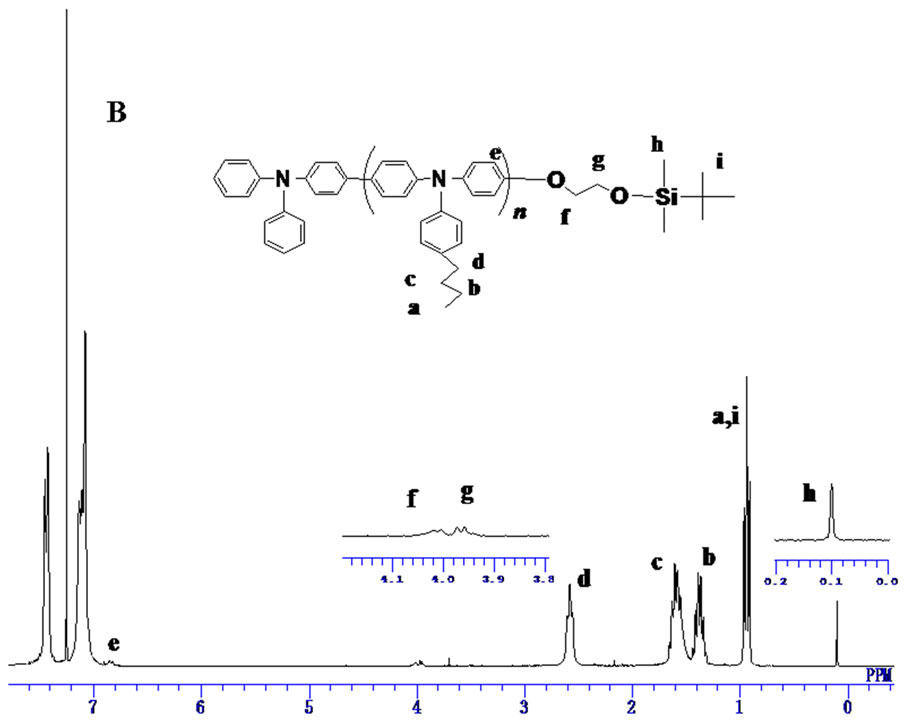
Figure 1. 1H NMR spectra of (A) PTPA initiated by 1, (B) PTPA initiated by 2.
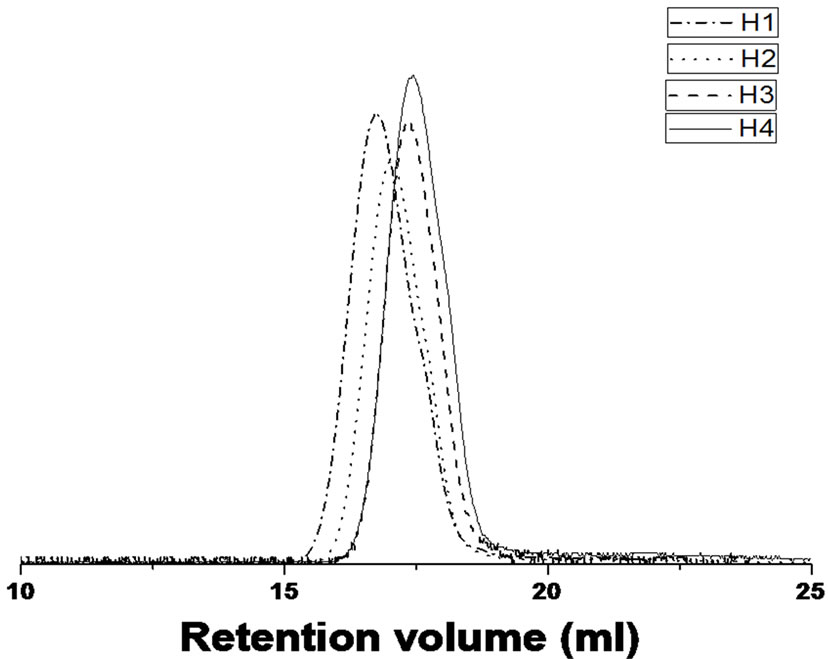
Figure 2. GPC traces of PTPA-TBS obtained under various conditions.
Table 1 shows the characteristics of PBTPA-TBS prepared with various reaction conditions. All the polymers showed high yields and narrow polydipersity in dexes around 2. The degrees of polymerization (DPn) were estimated from 1H NMR spectra by the comparing the integration of methylene protons from BTAA moiety (2.58 ppm) with that from end group (3.97 ppm). As ex pected, with the decrease of the ratio of monomer to initiator, lower polymerization degree was obtained. Poly mers with different polymerization degree ranging from 13 to 42 were obtained by adjusting the feed ratios. As suming that the reactivities in the oxidative insertion of modifier 2 and monomer 3 are comparable, the average degree of polymerization should be equal to the ratio of 3 to 2. It is noteworthy that the feed monomer ratios were obviously lower than the corresponding polymerization degree, which indicated the reactivity probability of monomer 3 is higher than modifier 2. It is possible that Pd migration after the reductive elimination is limited in the vicinity of reaction center, i.e., the subsequent oxidative insertion occurs preferentially at C-Br linkage in the same growing chain end. According to this mechanism, it is reasonable that higher reactivity of monomer com pared with the modifier. The detail about the reaction mechanism is now under investigation.
3.2. Synthesis of PBTPA Macroinitiators
Deprotection of TBS group of PBTPA-TBS was conducted using pyridine/HF in the THF solution. After the reaction, precipitations from methanol and acetone were carried out to get rid of the low molecular weight byproducts. The conversion of the TBS group to hydroxyl group was confirmed by the complete disappearance of signals for methyl protons at 0.11 ppm and 0.9 ppm in 1H-NMR (Figure 3(A)).
Finally, the PBTPA with hydroxyl end group (PBTPAOH) was esterified with 2-bromopropionyl bromide. The structure of the PBTPA with bromo ester end goup (PBTPA-MI) which used as a macroinitiator of ATPR was confirmed by 1H NMR. As shown in Figure 3(B) all signals were reasonably assigned. Besides the signals derived from PBTPA backbone, the characteristic signals assignable for bromo ester end group were observed at 4.45, 4.35, 4.10 and 1.78 ppm. The conversion was estimated to be nearly 100% due to the complete position shift of methylene protons belonged to end groups.
3.3. Synthesis of the PBTPA Based Diblock Copolymers by ATRP
Preparation of diblock copolymers containing both PBTPA segment and PBA or PEA segment was carried out by ATRP using CuBr as a catalyst, PMDETA as a ligand and bromoester terminated PBTPA as a macroinitiator. It is known that the typical ATRP is conducted in bulk. However, due to the macroinitiator PBTPA-MI is insoluble in acrylate monomers, the solvent is necessary. Nanda et al [24] have analyzed the effect of solvent polarity on the activation rate constants for Br– counterion with acrylate monomers. In this study, it was demonstra-
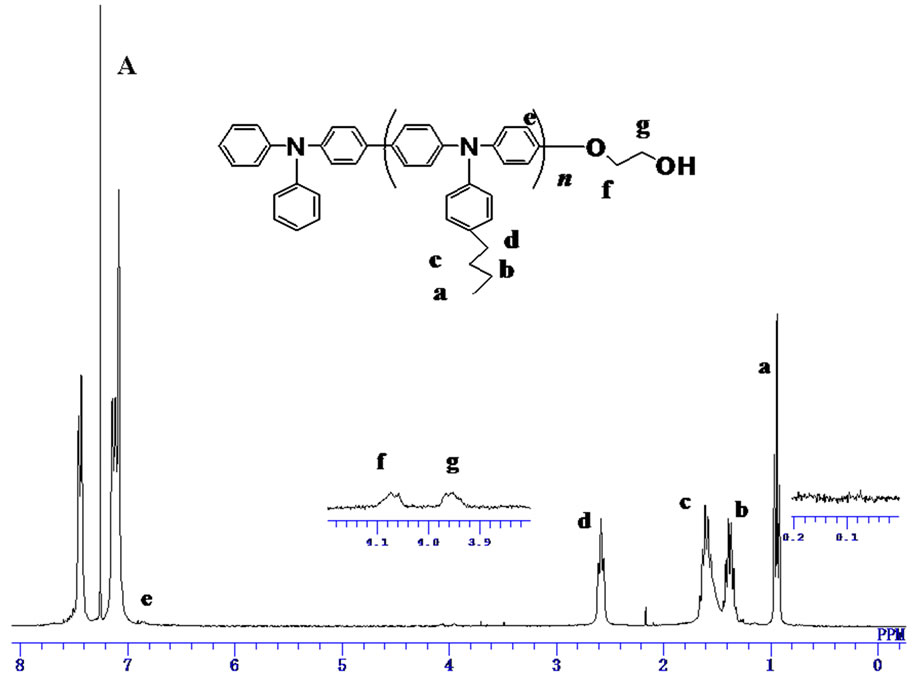

Figure 3. 1H NMR spectra of (A) PTPA-OH, (B) PTPAMI.
ted that the activation rate decreased with decreasing of the solvent polarity. Similar effect has been also reported in other systems [25-27]. Therefore the mixture solvent (1/1, v/v) of anisole, a good solvent for PBTPA-MI, and NMP, a solvent with relatively high polarity was used.
The structure of the PBTPA-b-PBA polymers was characterized by 1H NMR and all signals were reasonably assigned as shown in Figure 4(A). Besides the signals derived from PBTPA segment, the characteristic signals assignable to PBA segment were observed at 4.01, 2.25, 1.9 and 1.6 ppm respectively. The compositions of the copolymers were estimated by integrating methylene protons of PBTPA moiety 2.58 ppm versus that of PBA moiety 4.01 ppm.
As shown in Figure 5, the relative linear semilogarithmic kinetic plot ln([M]0/[M]t) versus time t indicates the living character of the polymerization and that the concentration of growing polymer radicals is constant.
From the data listed in Table 2, it could be found that the polymerization proceeded quite slowly, and the conversion of 32.1% was obtained after 110 h. The rate of polymerization in ATRP could be calculated using the following equation [28-30]:
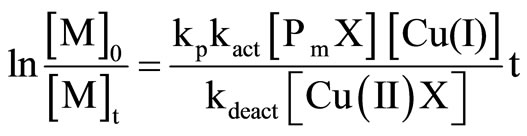 (2)
(2)
Where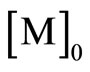 ,
,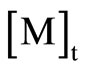 represent the concentration of monomer at time 0 and t, respectively; kp kact and kdeact are the rate constants of propagation, activation and deactivation, respectively; [Cu(I)], [PmX] are the concentration of copper in the lower oxidation state and the dormant species.
represent the concentration of monomer at time 0 and t, respectively; kp kact and kdeact are the rate constants of propagation, activation and deactivation, respectively; [Cu(I)], [PmX] are the concentration of copper in the lower oxidation state and the dormant species.
According the above equations, it was proposed that the lower conversion may result from the lower propagation rate, the lower concentration of the catalyst or the lower activation rate.
As expected, under the same conditions, the conversion is dramatically increased with increasing the feed ratio of monomer to macroinitiator, which can be attributed to the increase of the propagation rate [26]. In addition, when increasing the feed ratio of catalyst, the conversion increased (B4, B6). However, regardless of the reaction conditions, the polymerizations still proceeded more slowly than other systems reported.
It has been reported that the activation rate was significantly influenced by the structure of initiators [24, 31]. It is known the activation rates of dormant chains depend in two factors: 1) the stability of the formed radicals, 2) steric effects. The rigid structure of PBTPA-MI could results in high steric congestion. In order to investigate the effect of structures of PBTPA-MI on the activivation rate of ATRP, two kinds if macroinitiators with Mn = 4580 (PDI = 2.01) and Mn = 5270 (PDI = 2.18) were employed for the copolymerization. Under the same reaction conditions, the copolymer B3 initiated by PBTPAMI with longer PBTPA chain showed lower conversion and broader polydispersity compared with copolymer B1 with shorter PBTPA chain. Therefore, it is reasonable to deduce that the low activation of dormant PBTPA chain may be the main reason leading to low reaction rate.
GPC curves of PTPA-b-PBA block copolymers synthesized with different reaction times and the corresponding macroinitiator are given in Figure 6. The molecular weight increased with the increase of reaction time, which was agreed with that illustrated in Figure 5. In addition, the monomodel and symmetric shapes indicated the formation of block copolymers without ho mopolymerization.
PBTPA-b-PEA polymers were prepared under the similar conditions. The structures were confirmed by 1H NMR and all signals were reasonably assigned as shown in Figure 4(B). All the results were listed in Table 3. It could be found that polymerization also proceeded quite slowly. This may be also attributed to the low activation of dormant PBTPA chain end to a radical species.
3.4. Thermal Properties
The thermal properties of the block copolymers were examined by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA).
In the DSC profile of the diblock copolymer (Table 2, Code B1), the separated Tgs of both segments were clearly observed at –45˚C as well as 175˚C (Figure 7). This suggested the micro-phase separation occurs in block copolymer samples. The observed Tg values for each moiety are similar with each others, which indicated
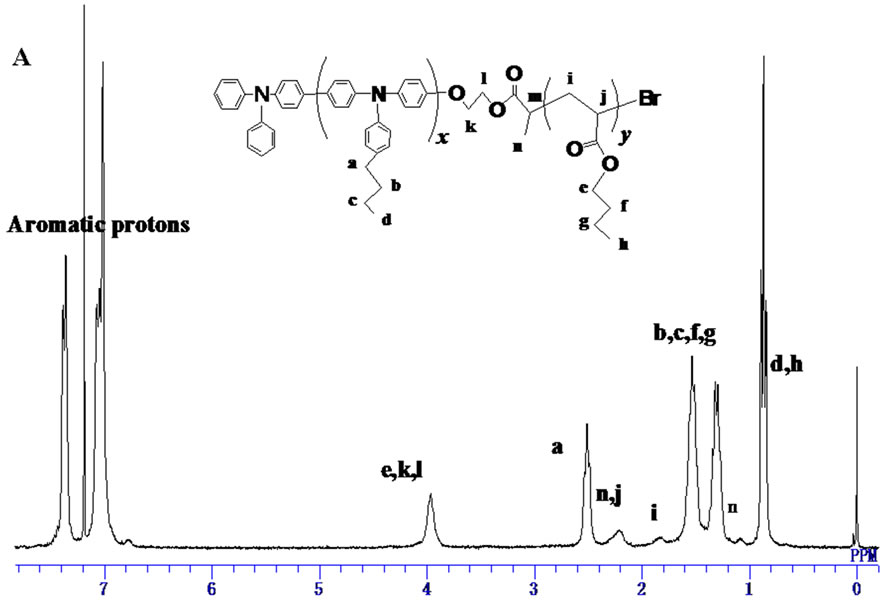
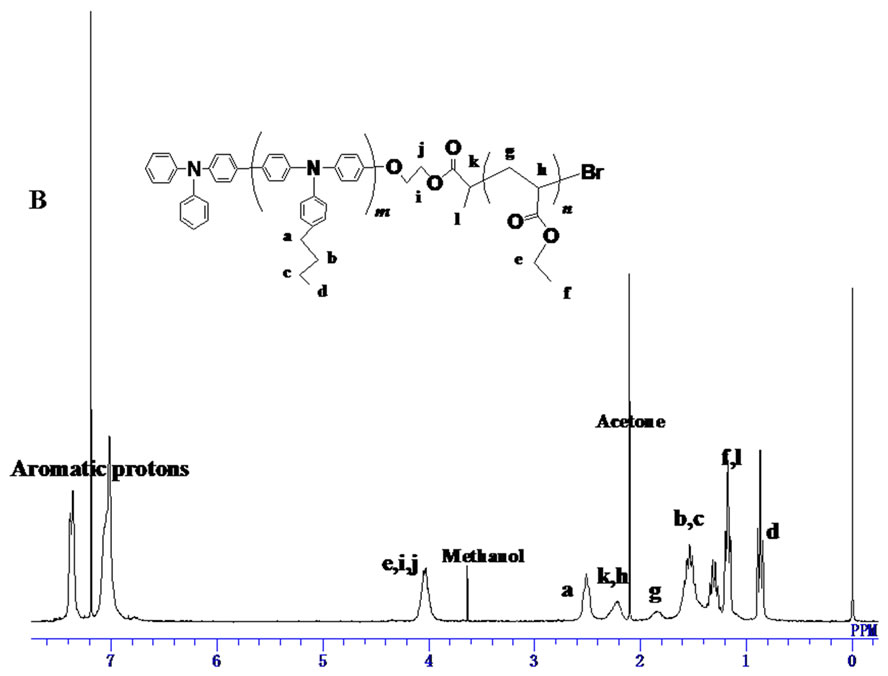
Figure 4. 1H NMR spectra of (A) PTPA-b-PBA, (B) PTPAb-PEA.
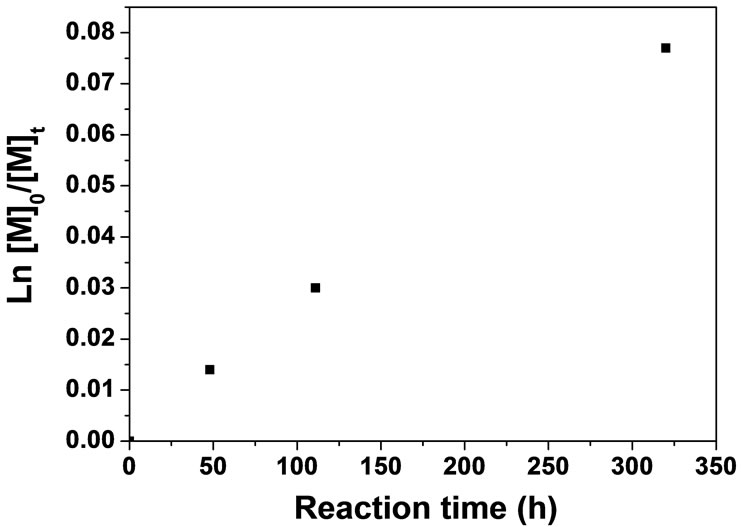
Figure 5. Semi-logarithmic kinetic plot for the ATRP of BA in anisole/NMP (50% by volume) using [M]:[I]:[CuBr]: [PMDETA] = 500:1:22:26. PTPA-MI with Mn = 5270 (PDI = 2.18) is used as initiator.
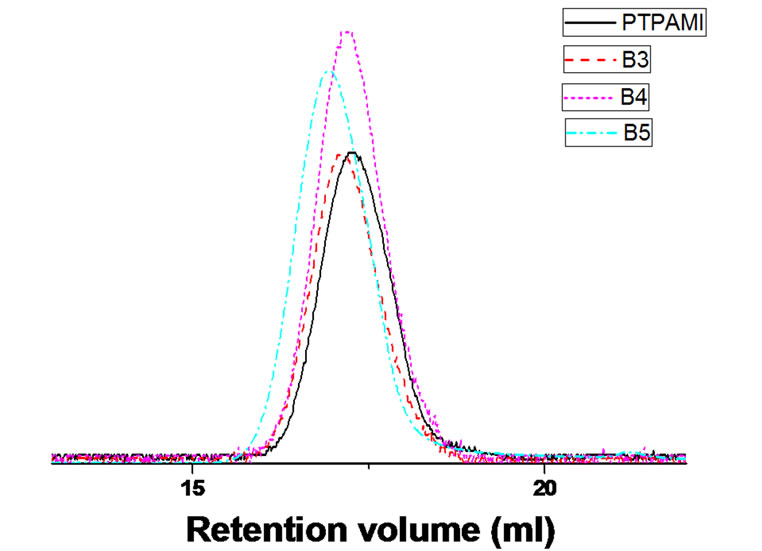
Figure 6. GPC traces of PTPA-b-PBA polymers and the corresponding macroinitiator.
the length of the BTAA and n-BA or EA segments does not influence Tg obviously under our experimental conditions.
As shown in Figure 8, the thermal decomposition of PBTPA-b-PBA (Table 3, B1) and PBTPA-b-PEA (Table 3, B9) mainly occurs in two steps. For PBTPA-bPBA, the first step of mass loss occurs between 350˚C to 440˚C, which is attributed to the decomposition of PBA segment. The second mass loss occurs from 440˚C which is assigned to the decomposition of PBTPA moiety. PBTPA-b-PEA (B9) showed the similar curve with that of PBTPA-b-PBA as shown in Figure 8.
3.5. Surface Morphology of the Copolymer Films
Atomic force microscopy (AFM) observation was conducted to investigate the surface morphologies of the PBTPA-b-PBA films. The films of the copolymers were prepared by spin coating from THF solution and then transformed their structures thermodynamically in equilibrium at 180˚C for 12 h. Figure 9(A) showed the phase image of B1 in which the weight ratio of PBTPA and PBA was about 1.2:1. Microphase separation with diameter 25 - 50 nm cylindrical domains of PBTPA moiety can be clearly observed and each cylinder is aligned perpendicular to the surface. In the film of B2 in which the weight ratio of PBTPA and PBA decreased to 1:4 (Figure 9(B)), it seems that sphere morphology of PBTPA domain exists on the surface. In B4 film (Figure 9(C)), clearly phase separated structure was not observed. This is probably due to weaker repulsion of each block resulting from the shorter PBA chain or the increase of stiffness with longer BTAA chain.
3.6. PR Property
2BC experiment was performed to verify the PR property of the prepared devices. Generally, a large amount of plasticizers are added to lower the Tg and to stabilize the systems in the polymeric PR composites [32,33]. How-
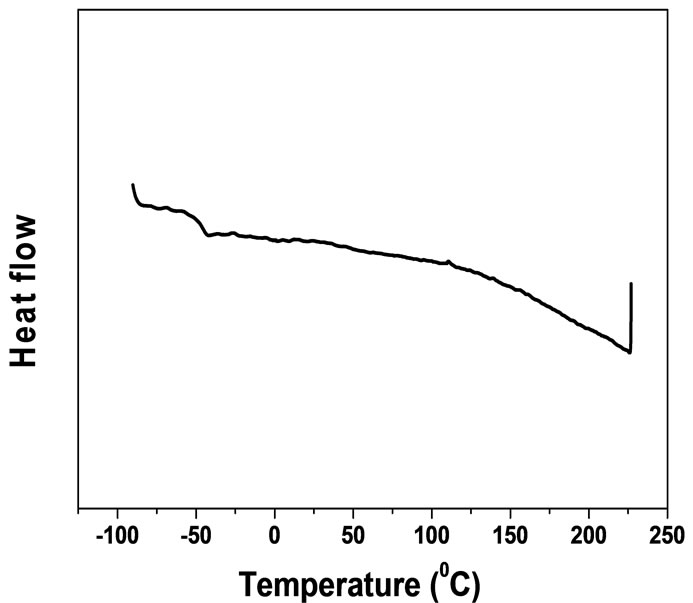
Figure 7. The DSC curve of B1 (Second heating).
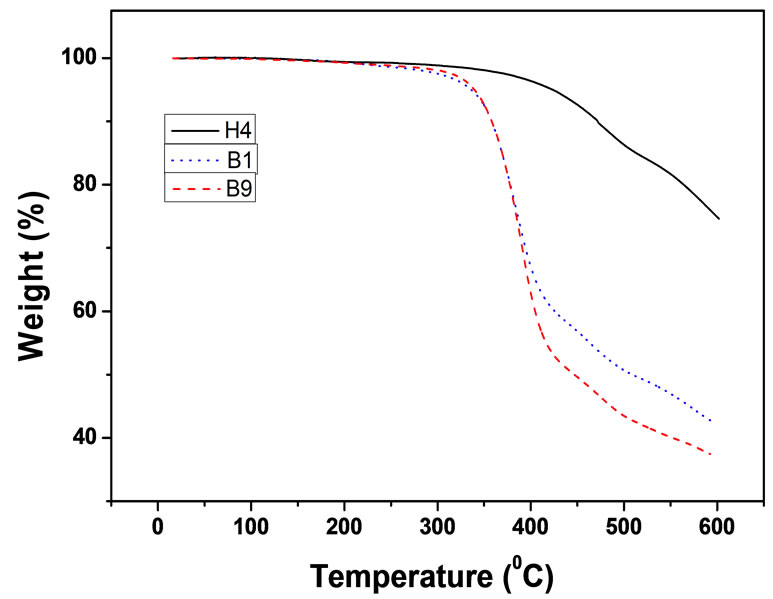
Figure 8. TGA thermograms of PTAP-based (co)polymers.
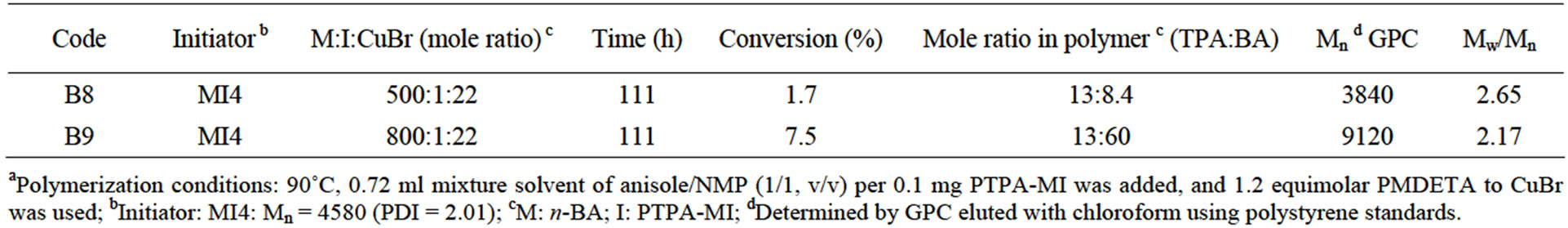
Table 3. Characteristics of PTPA-b-PEA prepared by ATRP under various reaction conditionsa

Figure 9. The phase mode AFM images of PTPA-b-PBA films prepared from 1wt% THF solution. (a) B1; (b) B2; (c) B4.
ever, the additional plasticizer could lower the proportion of effective PR components. Moreover, it has been reported plasticizer possesses its own influence on the PR effect [34]. Thus, the composites without adding plasticizers are preferable. Here two RP composites containing 88wt% of different PBTPA-b-PBA copolymers, B2 and B7 respectively, 11wt% DDCST as electro-optic chromophore and 1wt% TNF as sensitizer were prepared without adding any plasticizer, which possess the minimum components and are good candidate of understanding the interaction of different components during the photorefractive effect.
The PR devices were generally obtained by two sandwiched plates. The composite containing B7/DDCST/TNF could be easily sandwiched at 60˚C and form transparent films with thickness 80 mm. While the composite based on B2 which has the higher content of PBTPA segment could not be sandwiched even at 180˚C due to the stiff characteristics of the composite. This suggested the device fabrication was controlled by the weight ratio of the PBTPA moiety to PBA moiety in the block copolymers. Moreover, the composite film based on B2 seemed opaque due to PBTPA shows little compatibility with DDCST. This is understandable because of the phase separation and/or crystallization of DDCST.
In B7 based device, a relative high gain coefficient of 42.7 cm−1 was observed at the electric field of 31 V/mm. This observation and incompatibility of PBTPA with DDCST suggest that DDCST molecules exist predominantly in a PBA domain with lower Tg, and possess enough mobility to afford reorientation enhancement [35]. The detail studies about the effect of the unique multi-phased sytems on the photorefractive performance are under way.
4. Conclusion
Well-defined diblock copolymers based on PBTPA and polyacrylate were successfully synthesized by combination of C-N coupling and ATRP. The BTAA-based polymers were used as initiator of ATRP for the first time. The unique rigid structure of PBTPA leads to the slow polymerization rate of ATRP. Incorporation of polyacrylate segments into copolymers with PBTPA results in microphase separation of PBTPA domain with high Tg and PBA domain with low Tg, which was confirmed by AFM image. A relative high gain coefficient of 42.7 cm−1 was observed in the PR composite based on PBTPAb-PBA block copolymer at the electric field of 31 V/mm, which may be attributed to the unique design of micro-phase separated structure. The detail of PR property is under investigation.
REFERENCES
- H. Y. Lin, G. S. Liou, W. Y. Lee and W. C. Chen, “Poly (triarylamine): Its Synthesis, Properties, and Blend with Polyfluorene for White-Light Electroluminescence,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 45, No. 9, 2007, pp. 1727-1736. doi:10.1002/pola.21940
- Y. P. Zou, Y. Zhou, G. L. Wu, Y. F. Li and C. Y. Pan, “Electroluminescent Fluorene-Based Alternating Polymers Bearing Triarylamine or Carbazole Moieties in the Main Chain: Synthesis and Properties,” Journal of Applied Polymer Science, Vol. 111, No. 2, 2009, pp. 978-987. doi:10.1002/app.29156
- M. Thelakkat, J. Hagen, D. Haarer and H. W. Schmidt, “Poly(triarylamine)s-Synthesis and Application in Electroluminescent Devices and Photovoltaics,” Synthetic Metals, Vol. 102, No. 1-3, 1999, pp. 1125-1128. doi:10.1016/S0379-6779(98)01412-X
- D. Wright, U. Gubler, W. E. Moerner, M. S. Declue and J. S. Siegel, “Photorefractive Properties of Poly(siloxane) triarylamine-Based Composites for High-Speed Applications,” The Journal of Physical Chemistry B, Vol. 107, No. 20, 2003, pp. 4732-4737. doi:10.1021/jp027456i
- Y. Shirota, T. Kobata and N. Noma, “Starburst Molecules for Amorphous Molecular Materials. 4,4',4"-Tris (N,Ndiphenylamino) Triphenylamine and 4,4',4"-Tris [N-(3-methylphenyl)-N-phenylamino] Triphenylamine,” Chemistry Letters, Vol. 18, No. 7, 1989, pp. 1145-1148. doi:10.1246/cl.1989.1145
- M. Ishikawa, M. Kawai and Y. Ohsawa, “Synthesis and Properties of Electrically Conducting Polytriphenylamines,” Synthetic Metals, Vol. 40, No. 2, 1991, pp. 231-238. doi:10.1016/0379-6779(91)91778-9
- K. Ogino, A. Kanegate, R. Yamaguchi, H. Sato and J. Kurjata, “Oxidative Coupling Polymerization of 4-Methyltriphenylamine,” Macromolecular Rapid Communications, Vol. 20, No. 3, 1999, pp. 103-106. doi:10.1002/(SICI)1521-3927(19990301)20:3<103::AID-MARC103>3.0.CO;2-Q
- C. Takahashi, S. Moriya, N. Fugono, H. C. Lee and H. Sato, “Preparation and Characterization of Poly(4-alkyltriphenylamine) by Chemical Oxidative Polymerization,” Synthetic Metals, Vol. 129, No. 2, 2002, pp. 123-128. doi:10.1016/S0379-6779(02)00010-3
- K. Tsuchiya, T. Shimomura and K. Ogino, “Preparation of Diblock Copolymer Based on Poly(4-N-Butyl-Triphenylamine) via Palladium Coupling Polymerization,” Polymer, Vol. 50, No. 1, 2009, pp. 95-101. doi:10.1016/j.polymer.2008.10.057
- K. Tsuchiya, T. Sakakura and K. Ogino, “Synthesis of Triphenylamine Copolymers and Effect of Their Chemical Structures on Physical Properties,” Macromolecules, Vol. 44, No. 13, 2011, pp. 5200-5208. doi:10.1021/ma200940v
- K. Tsuchiya, T. Kikuchi, M. Songeun, T. Shimomura and K. Ogino, “Synthesis of Diblock Copolymer Consisting of Poly(4-butyltriphenylamine) and Morphological Control in Photovoltaic Application,” Polymers, Vol. 3, No. 3, 2011, pp. 1051-1064. doi:10.3390/polym3031051
- W. E. Moerner, S. M. Silence, F. Hache and G. C. Bjorklund, “Orientationally Enhanced Photorefractive Effect in Polymers,” Journal of the Optical Society of America B, Vol. 11, No. 2, 1994, pp. 320-330. doi:10.1364/JOSAB.11.000320
- L. M. Leung and J. T. Koberstein, “DSC Annealing Study of Microphase Separation and Multiple Endothermic Behavior in Polyether-Based Polyurethane Block Copolymers,” Macromolecules, Vol. 19, No. 3, 1986, pp. 706-713. doi:10.1021/ma00157a038
- T. E. Patten and K. Matyjaszewski, “Atom Transfer Radical Polymerization and the Synthesis of Polymeric Materials,” Advanced Materials, Vol. 10, No. 12, 1998, pp. 901-915. doi:10.1002/(SICI)1521-4095(199808)10:12<901::AID-ADMA901>3.0.CO;2-B
- K. Jankova, X. Y. Chen, J. Kops and W. Batsberg, “Synthesis of Amphiphilic PS-b-PEG-b-PS by Atom Transfer Radical Polymerization,” Macromolecules, Vol. 31, No. 2, 1998, pp. 538-541. doi:10.1021/ma9710186
- K. Matyjaszewski, B. Goebelt, H. J. Paik and C. P. Horwitz, “Tridentate Nitrogen-Based Ligands in Cu-Based ATRP: A Structure-Activity Study,” Macromolecules, Vol. 34, No. 3, 2001, pp. 430-440. doi:10.1021/ma001181s
- Q. Lin, M. Konas, R. M. Davis and J. S. Riffle, “Preparation and Properties of Poly(Alkyl-Vinyl-Ether-2-ethyl- 2-oxazoline)Diblock Copolymers,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 31, No. 7, 1993, pp. 1709-1717. doi:10.1002/pola.1993.080310709
- A. Kajiwara and K. Matyjaszewski, “Formation of Block Copolymers by Transformation of Cationic Ring-Opening Polymerization to Atom Transfer Radical Polymerization (ATRP),” Macromolecules, Vol. 31, No. 11, 1998, pp. 3489-3493. doi:10.1021/ma971445j
- Y. Lee, K. I. Fukukawa, J. Bang, C. J. Hawker and J. K. Kim, “A High Purity Approach to Poly (3-hexylthiophene) Diblock Copolymers,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 46, No. 24, 2008, pp. 8200-8205. doi:10.1002/pola.23091
- I. Seiichi, K. Koichi, F. Takashi and N. Jun, “Synthesis and Complexing Ability of Azacrownophanes: The Cyclodextrin Catalysis of the Photochemical Cyclization Reaction,” Journal of Inclusion Phenomena and Macrocyclic Chemistry, Vol. 39, No. 1-2, 2001, pp. 35-40.
- T. W. Greene and P. G. M. Wuts, “Protective Groups in Organic Synthesis,” 3rd Edition, John Wiley & Sons Ltd., New York, 1999. doi:10.1002/0471220574
- K. Okamoto, T. Nomura, S. H. Park, K. Ogino and H. Sato, “Synthesis and Characterization of Photorefractive Polymer Containing Electron Transport Material,” Chemistry Materials, Vol. 11, No. 11, 1999, pp. 3279-3284. doi:10.1021/cm990351g
- W. E. Moerner and S. M. Silence, “Polymeric Photorefractive Materials,” Chemical Reviews, Vol. 94, No. 1, 1994, pp. 127-155. doi:10.1021/cr00025a005
- A. K. Nanda and K. Matyjaszewski, “Effect of [PMDE TA]/[Cu(I)] Ratio, Monomer, Solvent, Counterion, Ligand, and Alkyl Bromide on the Activation Rate Constants in Atom Transfer Radical Polymerization,” Macromolecules, Vol. 36, No. 5, 2003, pp. 1487-1493. doi:10.1021/ma0340107
- G. Chambard, B. Klumperman and A. L. German, “Effect of Solvent on the Activation Rate Parameters for Polystyrene and Poly (Butyl Acrylate) Macroinitiators in Atom Transfer Radical Polymerization,” Macromolecules, Vol. 33, No. 12, 2000, pp. 4417-4421. doi:10.1021/ma992153g
- M. F. García, J. L. de la Fuente, M. Fernández-Sanz and E. L. Madruga, “The Importance of Solvent Polar Character on the Synthesis of PMMA-B-PBA Block Copolymers by Atom Transfer Radical Polymerization,” Polymer, Vol. 42, No. 23, 2001, pp. 9405-9412. doi:10.1016/S0032-3861(01)00514-6
- X. S. Wang, N. Luo and S. K. Ying, “Controlled Radical Polymerization of Methacrylates at Ambient Temperature and the Synthesis of Block Copolymers Containing Methacrylates,” Polymer, Vol. 40, No. 14, 1999, pp. 4157-4161. doi:10.1016/S0032-3861(98)00759-9
- K. Matyjaszewski and J. Xia, “Atom Transfer Radical Polymerization,” Chemical Reviews, Vol. 101, No. 9, 2001, pp. 2921-2990. doi:10.1021/cr940534g
- K. Matyjaszewski, “Mechanistic and Synthetic Aspects of Atom Transfer Radical Polymerization,” Journal of Macromolecular Science: Pure and Applied Chemistry, Vol. 34, No. 10, 1997, pp. 1785-1801. doi:10.1080/10601329708010308
- A. Goto and T. Fukuda, “Determination of the Activation Rate Constants of Alkyl Halide Initiators for Atom Transfer Radical Polymerization,” Macromolecular Rapid Communications, Vol. 20, No. 12, 1999, pp. 633-636. doi:10.1002/(SICI)1521-3927(19991201)20:12<633::AID-MARC633>3.0.CO;2-2
- K. Ibrahim, B. Löfgren and J. Seppälä, “Towards More Controlled Poly (n-butyl Methacrylate) by Atom Transfer Radical Polymerization,” European Polymer Journal, Vol. 39, No. 5, 2003, pp. 939-944. doi:10.1016/S0014-3057(02)00309-9
- I. K. Moon, C. S. Choi and N. Kim, “Synthesis and Characterization of a Low-Tg Photorefractive Composite,” Journal of Photochemistry and Photobiology A: Chemistry, Vol. 202, No. 1, 2009, pp. 57-62. doi:10.1016/j.jphotochem.2008.11.011
- H. N. Giang, K. Kinashi, W. Sakai and N. Tsutsumi, “Photorefractive Composite Based on a Monolithic Polymer,” Macromolecular Chemistry and Physics, Vol. 213, No. 9, 2012, pp. 982-988. doi:10.1002/macp.201100647
- J. Ostrauskaite, H. R. Karickal, A. Leopold, D. Haarer and M. Thelakkat, “Poly[bis(triphenylamine)Ether]s with Low Glass Transition Temperatures as Photoconductors in Fast Photorefractive Systems,” Journal of Materials Chemistry, Vol. 12, No. 1, 2002, pp. 58-64. doi:10.1039/b105930k
- O. Ostroverkhova and W. E. Moerner, “Oganic Photorefractives: Mechanisms, Materials, and Applications,” Chemical Reviews, Vol. 104, No. 7, 2004, pp. 3267-3314. doi:10.1021/cr960055c
NOTES
*Corresponding author.