Open Journal of Inorganic Non-metallic Materials
Vol.2 No.1(2012), Article ID:17043,10 pages DOI:10.4236/ojinm.2012.21001
Comparison of Hydrothermal and Sol-Gel Synthesis of Nano-Particulate Hydroxyapatite by Characterisation at the Bulk and Particle Level
Institute for Materials Research, SPEME, University of Leeds, Leeds, UK
Email: s.j.milne@leeds.ac.uk
Received October 27, 2011; revised December 2, 2011; accepted December 23, 2011
Keywords: Hydroxyapatite; Hydrothermal; Sol-Gel; TEM-EDX; XRD; XRF; FTIR
ABSTRACT
Hydrothermal and sol-gel synthesis methods have been used to prepare nano-particulate hydroxyapatite (HA) powders for detailed characterisation. Bulk elemental analysis data are compared from X-ray diffraction, X-ray fluorescence and Fourier transform infrared spectroscopy. These show the presence of secondary phases in the sol-gel powders which can be attributed to evaporative loss of precursor phosphite phases during specimen preparation and breakdown of the primary HA phase during calcination. Only the primary HA phase is detected in the hydrothermally prepared powder. In addition, Ca/P ratios of each powder are determined at the particle level using transmission electron microscopy with energy dispersive X-ray spectroscopy (TEM-EDX), having first established a threshold electron fluence below which significant electron-beam-induced alteration of the composition of HA does not occur. The TEM-EDX results show a greater compositional variability of particles from the sol-gel preparation route compared to the hydrothermal route. Overall it is the combination of the analysis techniques that shows the hydrothermal synthesis route produces nearstoichiometric, single phase, hydroxyapatite.
1. Introduction
Hydroxyapatite (HA) forms the main mineral component of bone and teeth. Stoichiometric HA has the composition Ca10(PO4)6(OH)2 with a Ca/P ratio of 2.15 by atomic weight and 1.67 by atomic ratio. Naturally occurring HA typically forms nano-metre sized crystallites with a number of cation and anion impurities, for example , F–, Na+ and Mg2+ [1,2]. Within a pH range of 4.2 - 8.0 HA is more stable, i.e. less soluble, in aqueous media than alternative calcium phosphate ceramics, such as tricalcium phosphate, TCP (Ca3(PO4)2) [2].
, F–, Na+ and Mg2+ [1,2]. Within a pH range of 4.2 - 8.0 HA is more stable, i.e. less soluble, in aqueous media than alternative calcium phosphate ceramics, such as tricalcium phosphate, TCP (Ca3(PO4)2) [2].
Synthetic HA nano-particles can be produced by a number of techniques and excellent biocompatibility makes it of interest for use in a wide range of biomedical applications [3-5]. These include tissue scaffolds [6,7], tubule infiltration in dentine [8,9], implant coatings [10] and as matrices for controlled drug release [11]. By chemically modifying synthetic HA to more closely match the composition of natural HA; it may be possible to gain further improvements in biocompatibility and osteointergration.
There are two principal methods for the synthesis of hydroxyapatite: solid-state synthesis [12] and wet chemical methods. The latter generally offer lower temperature synthesis than solid-state reactions, from component oxides/carbonates, and common wet chemical routes include: precipitation [13], hydrothermal [14-16]. And solgel synthesis [3,17].
This paper compares the physical and chemical properties of nano-particulate powders of HA prepared using a hydrothermal and a sol-gel method. Hydrothermal synthesis is nano-particle preparation route which utilises heat and pressure to initiate reactions of reagents dissolved in water, or organic solvent (solvothermal). This opens up reaction chemistry not accessible under ambient conditions and crystalline HA can be produced without the requirement for post reaction calcination. The method is attractive as it can produce HA nano-particles with minimal or no agglomeration [14]. Sol-gel synthesis offers the potential to achieve effective mixing of the Ca and P precursors in solution and can retain this level of mixing in the gelled sample [3]. The gel contains organic species which can be eliminated by heating the dried gel powder to form crystalline HA, but this calcination process also promotes agglomeration of the primary particles.
In order to obtain an accurate determination of composition of HA powders, and to permit a detailed comparison of different synthesis methods, there is a need to consider the most appropriate chemical analysis methods. This paper compares bulk (X-ray diffraction [XRD], Xray fluorescence [XRF] and Fourier transform infrared [FTIR] spectroscopy) and individual particle (energy dispersive X-ray spectroscopy in transmission electron microscopy [TEM-EDX]) characterisation techniques for HA powders produced by the hydrothermal and sol-gel synthesis methods and, for comparison, a powder obtained from a commercial supplier (Sigma-Aldrich). The aim is to determine the synthesis route that produces the most stoichiometric and least variable HA powder and in the process to establish robust characterisation protocols for HA powders (from different sources).
Individual particle characterisation by TEM-EDX enables the compositional variability to be assessed while bulk analysis gives averaged data. However, the composition and structure of HA alters under the high energy electron beam of a an electron fluence > 107 electrons nm–2 at 200 KeV induces a radiolytic damage process involving phosphorus and oxygen loss, amorphisation and eventual re-crystallisation to calcium oxide (CaO). [18,19] In this study we confirm this behaviour and undertake TEM-EDX analysis below this threshold to assess the compositional variability between particles of the HAs synthesised via hydrothermal, sol-gel, and commercial routes. These particle analyses will be compared to bulk compositions obtained from XRD, XRF and FTIR in order to identify the optimum synthesis route for stoichiometric HA nano-particles.
2. Experimental
2.1. Hydrothermal Synthesis
For the hydrothermal method [14,16], 1.00 M stock solutions of calcium nitrate tetrahydrate (Ca(NO3)2×4H2O) (Fisher Scientific, reagent grade) (11.807 g) and diammonium hydrogen phosphate ((NH4)2HPO4, Fisher Scientific, analytical reagent grade) (6.603 g) were prepared using distilled water (50 mL), and subsequently diluted further to create 0.10 M solutions. A precipitate was formed by the drop-wise addition of the di-ammonium hydrogen phosphate solution (30 mL) to the calcium nitrate tetrahydrate solution (50 mL), with continuous stirring, to obtain a Ca/P ratio of 1.67 in the mixed solution. The pH of the resulting suspension was ~5.1, however the drop-wise addition of ammonium hydroxide (NH4OH) (Sigma-Aldrich, ACS reagent grade) during the mixing phase, allowed the pH to be raised to pH = 11.0. The addition of ammonium hydroxide to the synthesis solution allows for the control of pH, but also has an added benefit of preventing the formation of carbonate during synthesis [1,20]. An increase of OH– ions in the solution is also known to speed up the transformation rate from amorphous calcium phosphate (ACP) to hydroxyapatite, via a secondary octacalcium phosphate (OCP) phase [21- 23].
Vigorous stirring of the resulting solution was carried out for a further 10 minutes. The solution was then transferred into a 125 mL Teflon-lined hydrothermal reactor (Model 4748, Parr Instruments) which was then heated at 200˚C for 24 hours. Once air-cooled to room temperature, the particulate product was collected by centrifugation and washed to remove unwanted co-products by re-suspending the powder in distilled water, using ultrasonic agitation, for 5 mins and then sedimenting the particles by means of centrifugation at 6000 rpm for 5 mins. This process was repeated 6 times with a final wash carried out using methanol so as to limit agglomeration in the final dried powder. Drying was carried out in an oven at ~50˚C for ~4 hours [14].
2.2. Sol-Gel Synthesis
Sol-gel synthesis of HA [3,17], involved the hydrolysis of triethyl phosphite (C2H5O)3P) (10 mL) (Aldrich, 98%) in distilled water (60 mL). The mixture is sealed immediately with parafilm in a glass beaker and stirred vigorously. After a few minutes, the cloudy solution turns clear to signal the completion of hydrolysis. A stoichiometric amount (Ca/P ratio = 1.67) of calcium nitrate tetrahydrate (Fisher Scientific, reagent grade) (19.683 g) was dissolved in 50 mL anhydrous ethanol, and subsequently added drop-wise into the hydrolyzed phosphite solution, followed by 10 minutes of more vigorous stirring. The resulting solution was clear and subsequently aged at room temperature for 16 - 24 hours before drying. The ageing process is crucial in that it allows for the solution system to stabilise such that a monophasic HA can be produced; the time extent can vary depending on the chemical nature of the precursors used [3]. Insufficient ageing can result in weight loss during pyrolysis and the potential for undesired phases, such as CaO, to be observed [3,24].
The aged sol was dried using a hot plate: the solvents were driven off at ~60˚C until a viscous liquid formed, which was further dried in an oven at 100˚C for 12 hours. The resulting white gel was ground into a powder with a pestle and mortar. Two samples were prepared by calcining the gel at 500˚C and 700˚C respectively, for 2 hours [3].
The properties of hydrothermal and sol-gel prepared powders were compared to a commercial hydroxyapatite powder (Sigma-Aldrich, reagent grade).
2.3. Characterisation
Bulk compositional and phase analysis for all samples has been performed by: 1) powder X-ray diffraction (XRD) using a Philips/PANalytical X’pert Diffractometer with Cu Kα radiation (λ = 1.5045 Å); 2) X-ray fluorescence (XRF) using a PANalytical Axios-Advanced XRF spectrometer (University of Leicester) with a 4 kW Rhodium anode end window with a super sharp ceramic technology X-Ray tube; 3) FTIR spectroscopy using a Thermo Scientific iS10 FTIR spectrometer with an attenuated total reflection (ATR) accessory, and 4) transmission electron microscopy with energy-dispersive Xray spectroscopy (TEM-EDX).
For the TEM-EDX, samples were examined in a FEI CM200 field emission gun TEM operating at 197 kV and fitted with an Oxford Instruments ultra thin window ISIS energy dispersive X-ray (EDX) spectrometer and a Gatan imaging filter (GIF 200). The Ca and P content (across 100 individual particles per sample) was measured in the TEM by quantification of spot-EDX spectra; the Ca/P ratio, as determined from the ISIS processing software using virtual standards for the Ca and P Kα X-ray peaks, was monitored, at a take-off angle of 20˚ and a specimen tilt angle of 15˚. Following the TEM conditions outlined by Eddisford (2007) [19], the beam current at a fixed illumination (spot size 8 and largest, 200 μm, condenser aperture) was estimated by using an FEI calibration curve based on the measured brightness (i.e. exposure time) on the (large) fluorescent viewing screen of the TEM: current (in nA) = 4.875/(Exposure time [in seconds] for an emulsion setting of 2.0). Beam currents were then converted to electron fluence rates (number of electrons per unit area per second) and fluence (total number of electrons per unit area) by measuring the area of illumination and the total exposure time respectively. Particle sizes, of 50 particles per sample, were measured using Gatan’s Digital Micrograph software.
3. Results & Discussion
Particle morphology for the four samples is shown by TEM micrographs in Figures 1(a)-(d). Particle sizes were obtained by measuring, from TEM images, the dimensions of 50 particles per sample and reporting the lengths (Figures 2(a)-(d)) and aspect ratios (length/width). Commercially supplied HA exhibited elongated particles ≤ 200 nm in length, with the greatest proportion in the 50 - 75 nm range; aspect ratios varied from 1.0 to 13.9, with an average of 3.6, Figures 1(a) and 2(a).
Hydrothermally produced HA particles exhibited a well-defined rod-like morphology with lengths ≤ 300 nm and aspect ratios ranging from 1.8 to 7.8, with an average of 4.0, Figures 1(b) and 2(b).
Sol-gel derived HA powders possessed a more equiaxed morphology than the other powders (average aspects ratios ≤ 2); sizes were mostly 25 - 100 nm with a maximum of ~250 nm. The gel calcined at 500˚C possessed the highest proportion of sub 50 nm particles of any powder, (Figures 1(c) and 2(c)), aspect ratios varied from 1.0 to 3.2, with an average of 1.7. Increasing the calcination temperature from 500˚C to 700˚C shifted the size distribution to higher values, with some particles reaching 225 - 250 nm in length (Figures 1(d) and 2(d)), aspect ratios varied from 1.0 to 4.0, with an average of 1.7.
Previous studies have led to the assumption that a high pH reduces particle growth due to the increased number of OH− ions in the solution creating surface defects in the preferred plane of growth [22,25]. An increase in pH, however, has the benefit of preventing the formation of unwanted calcium phosphate phases, such as dicalcium phosphate anhydrate, DCPA (CaHPO4) and dicalcium phosphate dihydrate, DCPD (CaHPO4·(H2O)2); these are known to be unstable at pH values greater than 6 - 7 and should subsequently convert to the more stable HA phase [26].
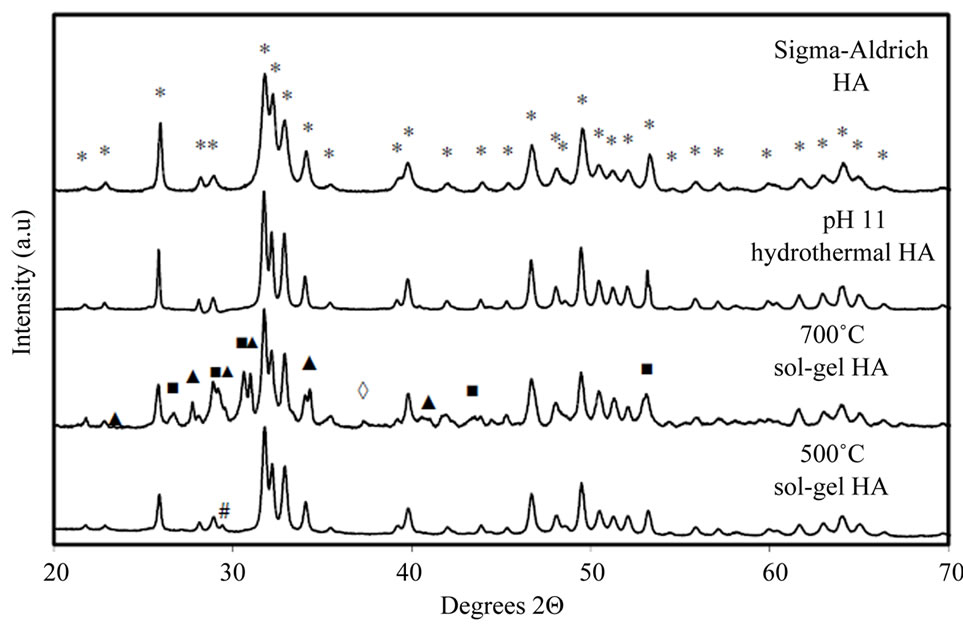
X-ray powder diffraction patterns for all four samples are shown in Figure 3(a). The commercial powder and the hydrothermal powder showed only peaks due to hydroxyapatite (JCPDS ref: 01-074-0566 [27]); peaks were sharper for the hydrothermal powder which is consistent with the larger particle size and good degree of crystallinity observed by TEM. The sol-gel derived HA, calcined at 500˚C, showed HA reflections plus a faint extra peak at 2Θ = 29.4˚, indicating a minor amount of secondphase calcium carbonate (CaCO3, JCPDS ref: 00-005- 0586 [28]). Increasing the calcination temperature to 700˚C resulted in significant levels of other secondary phases; β-tricalcium phosphate, β-TCP (Ca3(PO4)2, JCPDS ref: 01-070-2065 [29]), along with small amounts of dicalcium phosphate anhydrate, DCPA (CaHPO4, JCPDS ref: 01-070-0360 [30]) and calcium oxide (CaO, JCPDS ref: 01-077-2010 [31]). Whilst evaporation of un-reacted triethyl phosphite from the sol or gel could contribute to a net excess of Ca, leading to CaCO3 in the 500˚C sample for example, the other secondary phases observed after heating a gel to 700˚C may be a result of thermal decomposition of HA.
Previous studies suggest HA may decompose above temperatures as low as 600˚C. Increasing the calcination temperature to 700˚C and above, causes the decomposition of HA into β-TCP and CaO, following the proposed reaction [32,33]:
Ca10(PO4)6(OH)2 = 3Ca3(PO4)2 + CaO + H2O (1)
To confirm this, the commercial powder was subject to calcination for 1 hour at both 700˚C and 800˚C. X-ray powder diffraction patterns for these samples are shown in Figure 3(b); it is observed that the onset of decomposition, of HA to β-TCP, began at 700˚C, with a greater amount of β-TCP formed at 800˚C.
The decomposition temperature of HA may be strongly dependent on the particle characteristics and synthesis route for the HA powder [32,34]. In the present work, the
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 1. TEM images of (a) Sigma-Aldrich HA; (b) pH 11 hydrothermal HA; (c) 500˚C sol-gel HA and (d) 700˚C sol-gel HA. All of the preparations are nano-particulate and neither the hydrothermal nor sol-gel powders exhibit any significant porosity. The sol-gel powders are smaller and more equi-axed than the hydrothermal powders.
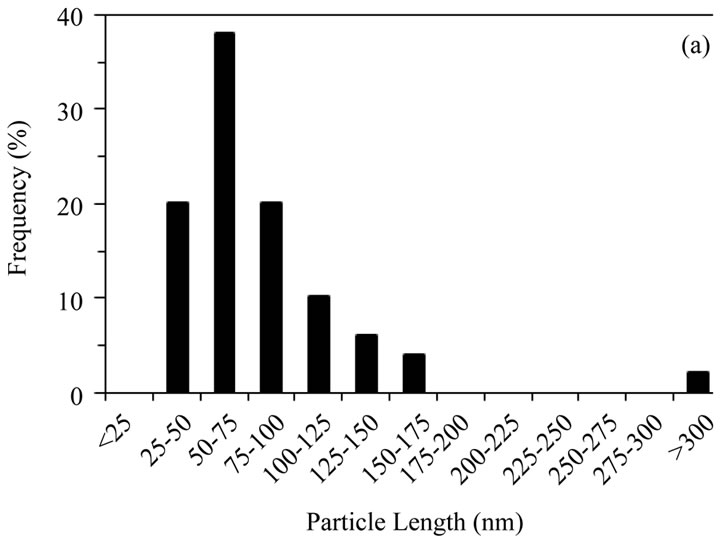
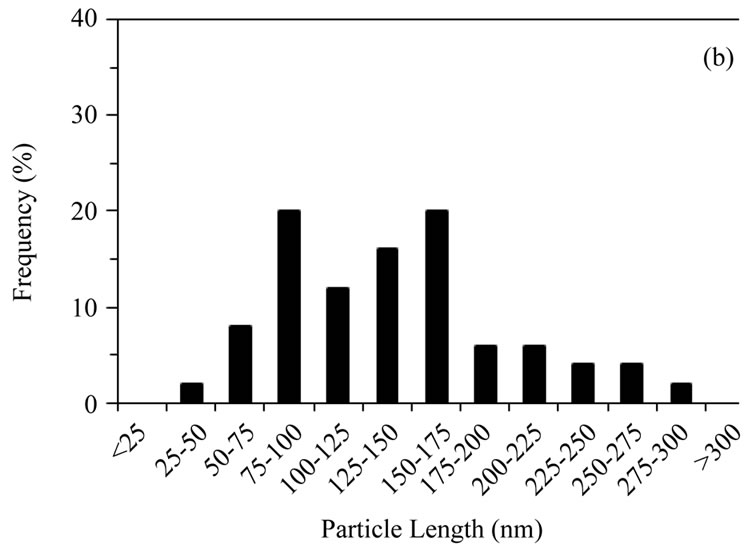
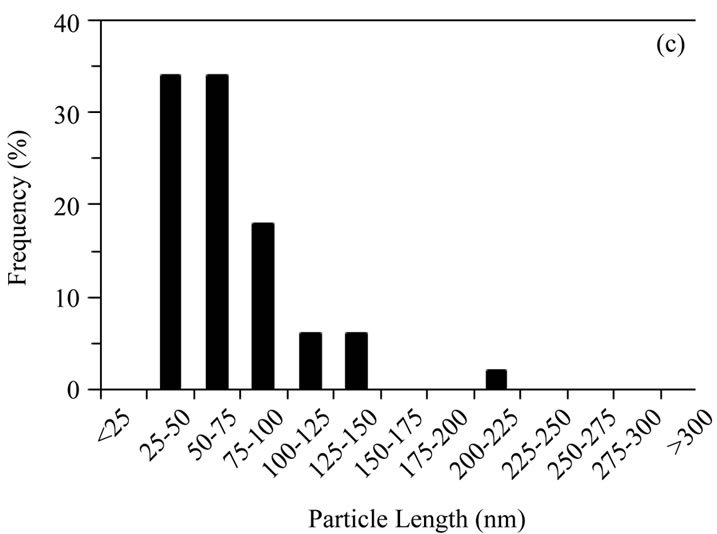
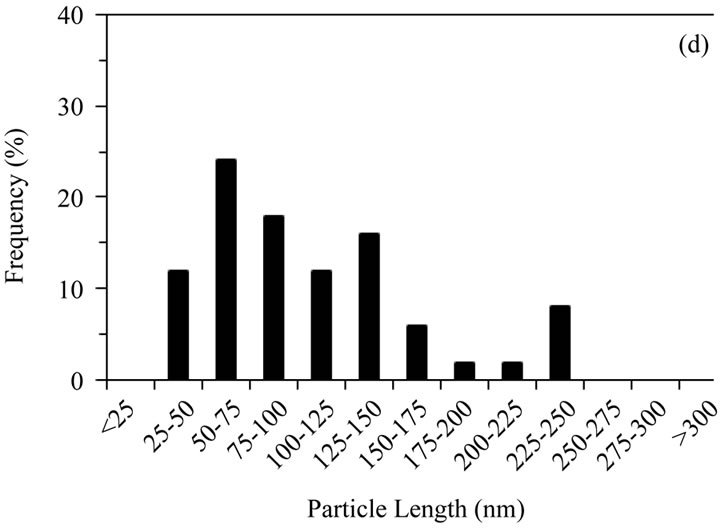
Figure 2. Distribution of particle lengths (50 particles) measured from TEM images of (a) Sigma-Aldrich HA; (b) pH 11 hydrothermal HA; (c) 500˚C sol-gel HA and (d) 700˚C sol-gel HA. The smaller particle size and size distribution of the sol-gel powders, compared to the hydrothermal powder, is retained even after calcination at 700˚C.
 (a)
(a)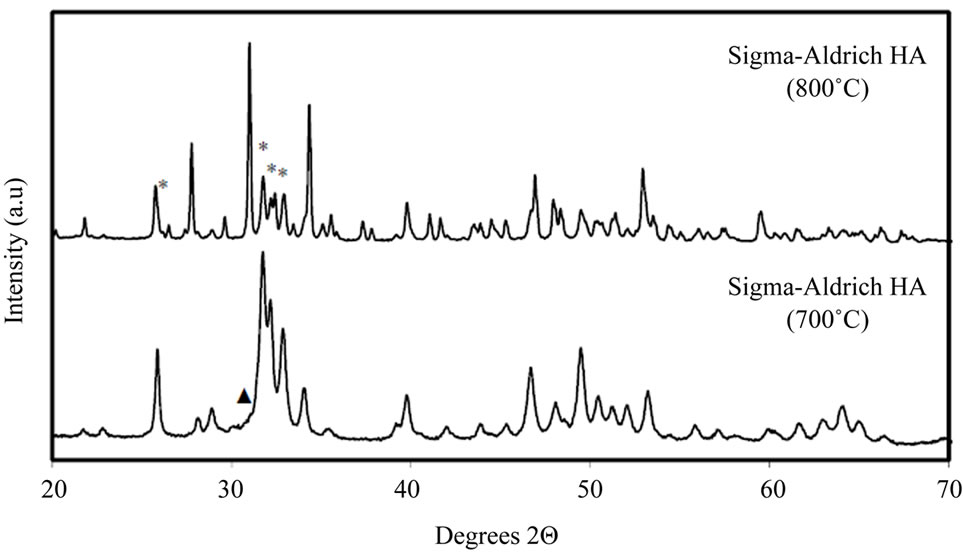 (b)
(b)
Figure 3. (a) Powder XRD patterns for all HA samples. All peaks labelled “*” in the Sigma-Aldrich pattern and all unlabelled peaks in subsequent patterns correspond to a stoichiometric HA phase [27]. The Sigma-Aldrich and hydrothermal HA powders show only HA reflections. Peaks labelled “▲” indicate β-TCP (β-Tricalcium phosphate, Ca3(PO4)2) [29]; peaks labelled “■” indicate DCPA (dicalcium phosphate anhydrate, CaHPO4) [30], peaks labelled “◊” indicate calcium oxide (CaO) [31] and peaks labelled “#” indicate calcium carbonate (CaCO3) [28]. (b) Powder XRD patterns for Sigma-Aldrich HA decomposed for 1 hour at 700˚C and 800˚C. Unlabelled peaks at 700˚C and peaks labelled “*” at 800˚C correspond to a stoichiometric HA phase [27]. Unlabelled peaks at 800˚C and peak labelled “▲” at 700˚C indicate β-TCP (β-Tricalcium phosphate, Ca3(PO4)2) [29]. At 800˚C most of the HA has decomposed to β-TCP.
calcium carbonate phase observed in the 500˚C sol-gel sample, which could be due to evaporative losses of the phosphite precursor reagent, would be expected to decompose to calcium oxide at 700˚C. This could explain the absence of CaCO3, and presence of CaO, in the 700˚C sol-gel powder [35].
The presence of carbonate or hydrated secondary phases was also investigated using FTIR, Figure 4 [36,37]. All four samples exhibited absorption bands at ~3570 cm–1 due to OH stretch; this was most intense in the hydrothermal powder and weakest in the commercial powder. The 500˚C sol-gel sample showed medium absorption bands at 1420 and 1456 cm–1, attributed to carbonate groups. Very faint bands in this region are also detected in the 700˚C sol-gel and hydrothermal powders. The remaining bands at lower wavenumber < 1100 cm–1 are due to lattice absorption and have previously been assigned to HA by [38].
Whilst it is difficult to assign individual bands unambiguously in this low frequency region, the stronger intensity of the ~870 cm–1 band in the sol-gel 500˚C sample may signify the presence of structural carbonate [38]. As already suggested, evaporation of unreacted triethyl phosphite from the sol or gel could constitute to the presence of CaCO3 in the 500˚C sol-gel powder, however carbonate could also form in a partially decomposed solgel system due to interaction between the inorganic product (HA) and the organic decomposition vapours (CO2-rich). In addition, the formation of an interfacial CaCO3 phase could occur as a result of interaction of the
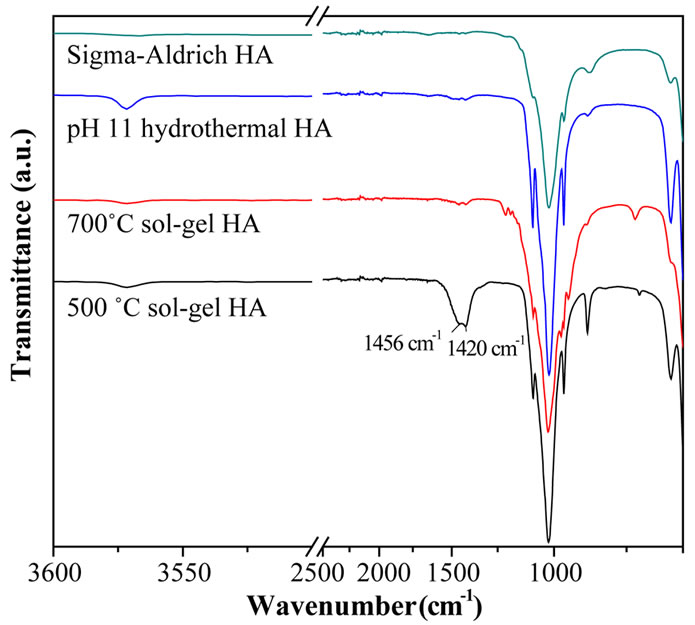
Figure 4. FTIR spectra of Sigma-Aldrich HA, pH 11 hydrothermal HA, 700˚C sol-gel HA and 500˚C sol-gel HA. The main lattice vibrations of HA are at <1200 cm–1 along with OH stretches at 3570 cm–1. Second phase carbonate stretches are clearly visible in the 500˚C sol-gel powder (at 1456 and 1420 cm–1).
nano-HA with atmospheric CO2 during storage in air and this might explain the low levels detected in the 700˚C sol-gel and hydrothermal powders. The TEM-EDX method allows compositional analysis to be carried out on individual particles, providing valuable information on fine-scale compositional homogeneity within a sample.
The TEM-EDX method allows compositional analysis to be carried out on individual particles, providing valuable information on fine-scale compositional homogeneity within a sample. However it is important to ensure that the sample is not subjected to an electron beam so intense that damage occurs and erroneous levels of Ca and P are detected. Therefore, a series of prior experiments were undertaken to establish the optimal electron fluence for obtaining reliable data. The electron fluence can be controlled using a condenser aperture, spot size and exposure time. To determine a threshold-fluence, whereby accurate and representative compositional data can be obtained by EDX without significant damage to the samples, two experiments were carried out using the commercial HA powder (Sigma-Aldrich, reagent grade) expanding on the method in [19]. First, a series of spot EDX measurements of different, small, particle clusters were made across the fluence range ~105 - 109 e nm–2 (by changing fluence and fluence rate) and the spectra quantified to obtain Ca/P ratios (Figure 5(a) and [27]).
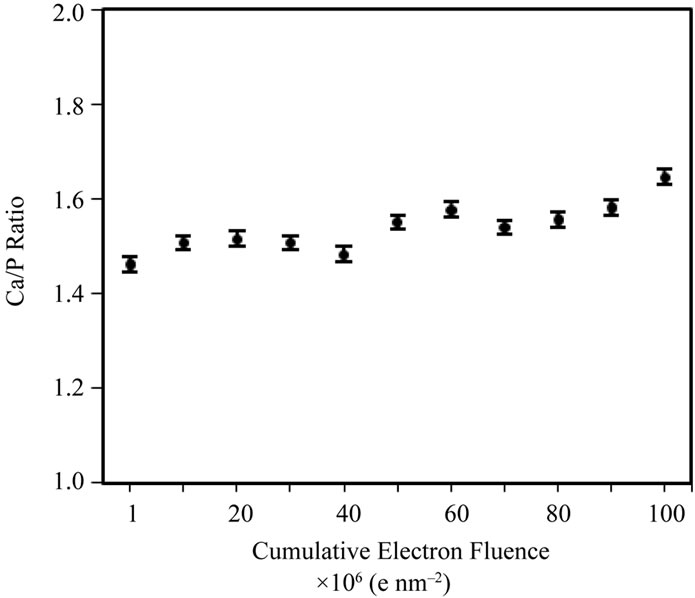
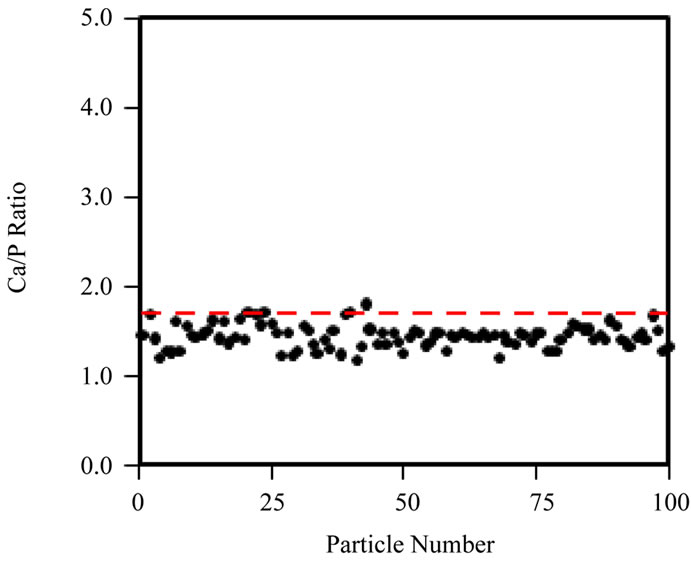
Secondly, spot EDX measurements were taken across a single cluster of particles, at a fixed irradiation area, to really isolate the effects of increasing fluence at a fixed fluence rate (105 e nm–2·s–1) on the Ca/P ratio (Figure 5(b)). Figure 5(a) shows that the Ca/P ratio remains stable up to ~107 e nm–2; beyond this point the Ca/P ratio rises and specimen alteration is observed (images not shown here). Prolonged irradiation of a single area of particles (Figure 5(b)) demonstrates a progressive rise in the Ca/P ratio (1.45 to 1.65) up to a final fluence of 108 e nm–2. The TEM-EDX analysis of all HA samples here forth uses a maximum threshold fluence of 107 e nm–2 to gather Ca/P ratios for 100 particles of each HA powder. The Ca/P ratios calculated from EDX peak ratios for 100 particles, when working below the threshold fluence for irradiation damage (107 e nm–2) are shown in Figure 6(a)-(d) and Table 1. The Sigma Aldrich powder displayed the least variability in particle Ca/P ratio, but the average Ca/P ratio was only 1.45 (standard deviation, S.D. = 0.16), significantly below the stoichiometric target of 1.67, but in agreement with previous reports [19]. The hydrothermal powder showed similar consistency in inter-particle Ca/P ratios, but the average Ca/P ratio, 1.61 (S.D. = 0.17), was in closer agreement with the stoichiometric target. Both sol-gel powders displayed a greater variability between particles, average Ca/P ratios were 1.72 for 500˚C and 1.65 for 700˚C, with standard deviations of 0.38 and 0.35 respectively. Figures 6(c) and (d) indicate that this was mainly due to a few particles with extreme Ca/P ratios, 3 - 4.5; this may be due to an overlap of more calcium rich phases; CaCO3 in the 500˚C sample and CaO in the 700˚C sample. This high level of variability within a small percentage of particles contrasts to the other “in-house” powder, as none of the 100 particles in the hydrothermal sample had a Ca/P ratio > 2.
Ca/P ratios measured in bulk using XRF by a commercial laboratory are compared to those from TEMEDX in Table 1. The XRF data confirms the low Ca/P ratio for the Sigma Aldrich powder, 1.45 by EDX compared to 1.5 by XRF. The XRF average values of Ca/P ratio for the ‘in-house’ powders were in agreement with the TEM-EDX data, as the XRF values were each within the range of one standard deviation of the average TEM-EDX value (standard deviations for the XRF powders were not reported by the commercial XRF facility). Overall, the results show that elemental analysis at the particle level is a valuable tool in assessing hydroxyapatite nanopowders.

Table 1. Comparison of Ca/P ratios obtained by analysis of TEM-EDX and XRF spectra. The two techniques give different Ca/P estimates but there is not a systematic differrence between the techniques across the four samples however both do indicate the hydrothermal HA to be nearstoichiometric i.e. Ca/P lies between 1.6 and 1.7.
 (a)
(a)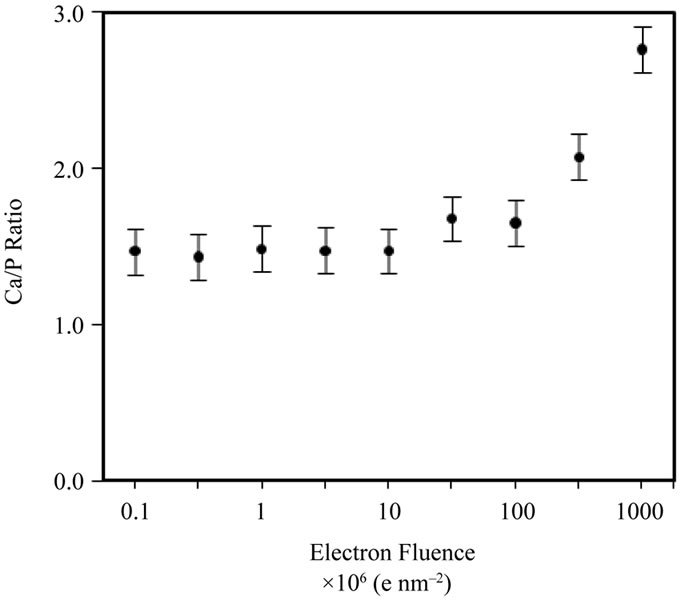 (b)
(b)
Figure 5. (a) TEM-EDX Ca/P ratio for different Sigma-Aldrich HA particle clusters as a function of electron fluence; (b) TEM-EDX Ca/P ratio data, over a fixed particle area (at a fixed fluence rate of 105 e nm–2·s–1) of the Sigma-Aldrich HA, as a function of cumulative fluence. There are no systematic changes to the Ca/P ratio below a fluence of 50 × 106 e nm–2.
 (a)
(a)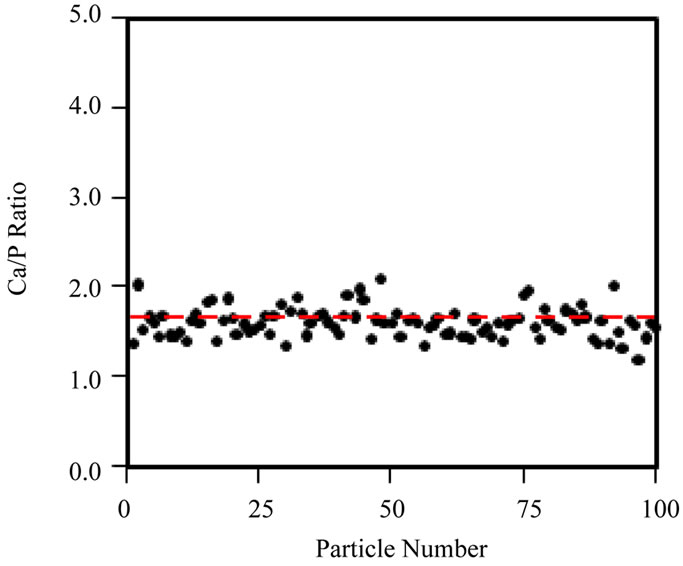 (b)
(b)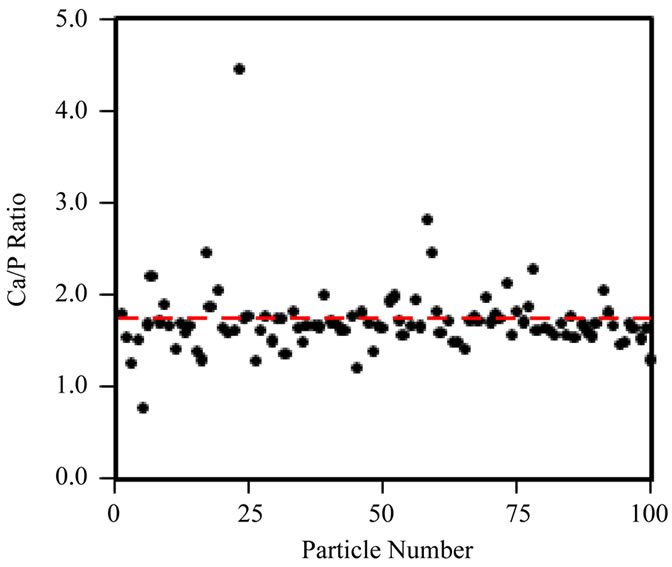 (c)
(c)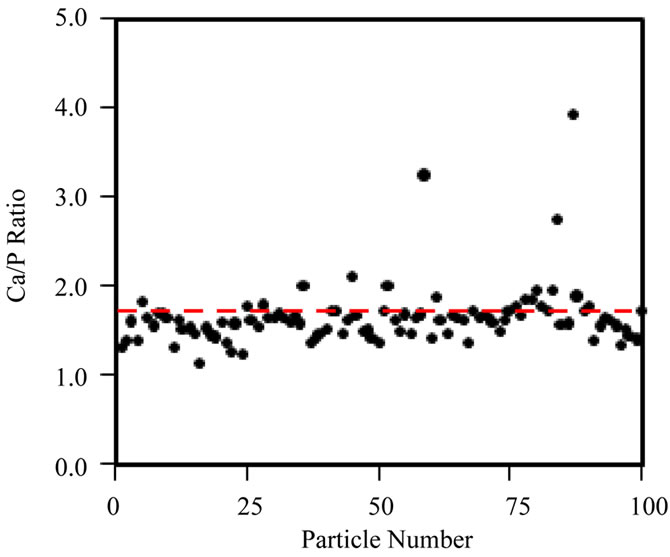 (d)
(d)
Figure 6. Ca/P ratio distribution of 100 individual particles, obtained with TEM-EDX acquired below the threshold fluence of 107 e nm–2, for (a) Sigma-Aldrich HA; (b) pH 11 hydrothermal HA; (c) 500˚C sol-gel HA and (d) 700˚C sol-gel HA. Red-dashed line indicates a Ca/P ratio = 1.67. Ca/P averages are displayed in Table 1, however neither the Sigma-Aldrich or hydrothermal HA powders contain particles with a Ca/P ratio  2.
2.
The technique of measuring individual particle Ca/P ratios, at controlled electron fluence, showed that the hydrothermally synthesised powder produced a consistent, single phase powder with “bulk” Ca/P ratio in close agreement with the value for stoichiometric hydroxyapatite, 1.67. By contrast the sol-gel produced powders are shown to be least uniform in Ca/P ratio, each containing a small percentage of Ca-rich particles. This may be linked to volatility of the phosphite precursor reagent during processing. A Ca-deficient composition was identified in the commercial powder, with Ca/P = ~1.5.
The need for careful consideration of the electron fluence in TEM-EDX analysis of HA has been demonstrated. Levels of compositional variability revealed by particle to particle quantification of the Ca/P ratio, at controlled electron fluence (≤107 e nm–2), are essential to collecting reliable analytical data and avoid radiolytic damage. This measurement is a valuable addition to the more typical characterisation methods employed for HA analysis.
This type of TEM-EDX analysis will be particularly useful for assessing the homogeneity of synthetic HAs that have been chemically modified to more closely match the composition of natural HA to gain further improvements in biocompatibility and osteointergration.
The work also demonstrates the importance of applying a combination of analytical techniques (XRD, FTIR, TEM-EDX and XRF) to adequately characterise nano hydroxyapatite powders. Individual techniques identified secondary phases in the sol-gel powders that would have an important bearing on the performance of this powder in applications such as tissue scaffolds.
4. Conclusions
Two solution-based techniques for the preparation of nano-particulate HA have been presented: hydrothermal and sol-gel synthesis routes. The physical and chemical properties of the powders have been compared to a commercial HA powder using a range of techniques. Hydrothermal synthesis produced single phase HA of needle-like nano-particles, the growth and morphological variation of which is restricted with the increased addition of ammonium hydroxide. The sol-gel synthesis method produced HA nano-particles which were a mixture of equiaxed and rod-like particles. A low-fluence TEM-EDX technique has been demonstrated, which is able to highlight particle to particle compositional variability. Hydrothermally produced HA had a near-stoichiometric Ca/P content; TEM-EDX revealed an average Ca/P ratio of 1.61 (S.D. = 0.17), with no detection of Ca-rich particles. The sol-gel method produced particles for which TEM-EDX, at the particle level, indicated average Ca/P ratios in close agreement with the stoichiometric target, 1.67. However a small, but significant, fraction of particles with much higher Ca/P ratios were detected. The sol-gel powders displayed average Ca/P ratios of 1.72 for 500˚C (S.D. = 0.38) and 1.65 for 700˚C (S.D. = 0.35). The commercial powder displayed a similar level of compositional consistency to the hydrothermal “in-house” sample, but the powder was deficient in calcium with a Ca/P ratio of only 1.45 (S.D. = 0.16).
5. Acknowledgements
For use of FTIR, gratitude must be expressed to Dr. Adrian Cunliffe and Mr. Stuart Micklethwaite of the Energy and Resources Research Institute, University of Leeds, and for his work with XRF, Mr. Nick Marsh of the Department of Geology, University of Leicester.
MB is in receipt of a DTA scholarship courtesy of the EPSRC and APB holds an EPSRC Advanced Research Fellowship (EP/E059678/1).
REFERENCES
- R. N. Correia, M. C. F. Magalhães, P. A. A. P. Marques and A. M. R. Senos, “Wet Synthesis and characterization of Modified Hydroxyapatite Powders,” Journal of Materials Science: Materials in Medicine, Vol. 7, No. 8, 1996, pp. 501-505. doi:10.1007%2FBF00705432
- S. M. Best, A. E. Porter, E. S. Thian and J. Huang, “Bioceramics: Past, Present and for the Future,” Journal of the European Ceramic Society, Vol. 28, No. 7, 2008, pp. 1319-1327 doi:10.1016/j.jeurceramsoc.2007.12.001
- D. M. Liu, T. Troczynski and W. J. Tseng, “Water-Based Sol-Gel Synthesis of Hydroxyapatite: Process Development,” Biomaterials, Vol. 22, No. 13, 2001, pp. 1721- 1730. doi:10.1016/S0142-9612(00)00332-X
- C. Ergun, “Effect of Ti Ion Substitution on the Structure of Hydroxylapatite,” Journal of the European Ceramic Society, Vol. 28, No. 11, 2008, pp. 2137-2149
- R. Z. LeGeros, “Properties of Osteoconductive Biomaterials: Calcium Phosphates,” Clinical Orthopaedics & Related Research, Vol. 395, No. 2002, 2002, pp. 81-98. doi:10.1097/00003086-200202000-00009
- R. Y. Zhang and P. X. Ma, “Poly(Alpha-Hydroxyl Acids) Hydroxyapatite Porous Composites for Bone-Tissue Engineering. I. Preparation and Morphology,” Journal of Biomedical Materials Research, Vol. 44, No. 4, 1999, pp. 446-455. doi:10.1002/(SICI)1097-4636(19990315)44:4<446::AID-JBM11>3.0.CO;2-F
- R. C. Thomson, M. J. Yaszemski, J. M. Powers and A. G. Mikos, “Hydroxyapatite Fiber Reinforced Poly(AlphaHydroxy Ester) Foams for Bone Regeneration,” Biomaterials, Vol. 19, No. 21, 1998, pp. 1935-1943. doi:10.1016/S0142-9612(98)00097-0
- J. S. Earl, D. J. Wood and S. J. Milne, “Nanoparticles for Dentine Tubule Infiltration: An in Vitro Study,” Journal of Nanoscience and Nanotechnology, Vol. 9, No. 11, 2009, pp. 6668-6674. doi:10.1166/jnn.2009.1330
- J. S. Earl, D. J. Wood and S. J. Milne, “Dentine Infiltration—A Cure for Sensitive Teeth,” American Ceramic Society Bulletin, Vol. 85, No. 7, 2006, pp. 22-25.
- W. Suchanek and M. Yoshimura, “Processing and Properties of Hydroxyapatite-Based Biomaterials for Use as Hard Tissue Replacement Implants,” Journal of Materials Research, Vol. 13, No. 1, 1998, pp. 94-117. doi:10.1557/JMR.1998.0015
- W. Habraken, J. G. C. Wolke and J. A. Jansen, “Ceramic Composites as Matrices and Scaffolds for Drug Delivery in Tissue Engineering,” Advanced Drug Delivery Reviews, Vol. 59, No. 4-5, 2007, pp. 234-248. doi:10.1016/j.addr.2007.03.011
- J. C. Elliott and R. A. Young, “Conversion of Single Crystals of Chlorapatite into Single Crystals of Hydroxyapatite,” Nature, Vol. 214, No. 5091, 1967, pp. 904- 906. doi:10.1038/214904b0
- B. Cengiz, Y. Gokce, N. Yildiz, Z. Aktas and A. Calimli, “Synthesis and Characterization of Hydroxyapatite Nanoparticles,” Colloids and Surfaces a-Physicochemical and Engineering Aspects, Vol. 322, No. 1-3, 2008, pp. 29-33. doi:10.1016/j.colsurfa.2008.02.011
- J. S. Earl, D. J. Wood and S. J. Milne, “Hydrothermal Synthesis of Hydroxyapatite,” Journal of Physics Conference Series, Vol. 26, 2006, p. 268. doi:10.1088/1742-6596/26/1/064
- I. S. Neira, F. Guitian, T. Taniguchi, T. Watanabe and M. Yoshimura, “Hydrothermal Synthesis of Hydroxyapatite Whiskers with Sharp Faceted Hexagonal Morphology,” Journal of Materials Science, Vol. 43, No. 7, 2008, pp. 2171-2178. doi:10.1007/s10853-007-2032-9
- R. E. Riman, W. L. Suchanek, K. Byrappa, C. W. Chen, P. Shuk and C. S. Oakes, “Solution Synthesis of Hydroxyapatite Designer Particulates,” Solid State Ionics, Vol. 151, No. 1-4, 2002, pp. 393-402. doi:10.1016/S0167-2738(02)00545-3
- S. J. Kalita and H. A. Bhatt, “Nanocrystalline Hydroxyapatite Doped with Magnesium and Zinc: Synthesis and Characterization,” Materials Science & Engineering CBiomimetic and Supramolecular Systems, Vol. 27, No. 4, 2007, pp. 837-848. doi:10.1016/j.msec.2006.09.036
- E. F. Brès, J. L. Hutchison, B. Senger, J. C. Voegel and R. M. Frank, “Hrem Study of Irradiation Damage in Human Dental Enamel Crystals,” Ultramicroscopy, Vol. 35, No. 3-4, 1991, pp. 305-322. doi:10.1016/0304-3991(91)90083-I
- P. Eddisford, A. Brown and R. Brydson, “Identifying and Quantifying the Mechanism of Electron Beam Induced Damage and Recovery in Hydroxyapatite,” Journal of Physics: Conference Series, Vol. 126, 2008, Article ID: 01208. doi:10.1088/1742-6596/126/1/012008
- J. S. Earl, “Hydroxyapatite Nanoparticles and Dentine Tubule Infiltration,” Ph.D. Thesis, Institute for Materials Research, University of Leeds, Leeds, 2007
- J. Christoffersen, M. R. Christoffersen, W. Kibalczyc and F.A. Andersen, “A Contribution to the Understanding of the Formation of Calcium Phosphates,” Journal of Crystal Growth, Vol. 94, No. 3, 1989, pp. 767-777. doi:10.1016/0022-0248(89)90102-4
- J. Rakovan, P. H. Ribbe and J. R. Rosso, “Growth and Surface Properties of Apatite,” Reviews in Mineralogy and Geochemistry, Mineralogical Society of America, Vol. 48, No. 1, 2002, pp. 51-86. doi:10.2138/rmg.2002.48.3
- J. L. Meyer and G. H. Nancollas, “The Effect of pH and Temperature on the Crystal Growth of Hydroxyapatite,” Archives of Oral Biology, Vol. 17, No. 11, 1972, pp. 1623-1627. doi:10.1016/0003-9969(72)90049-0
- C. S. Chai, K. A. Gross and B. Ben-Nissan, “Critical Ageing of Hydroxyapatite Sol-Gel Solutions,” Biomaterials, Vol. 19, No. 24, 1998, pp. 2291-2296
- J. Christoffersen, J. Dohrup and M. R. Christoffersen, “The Importance of Formation of Hydroxyl Ions by Dissociation of Trapped Water Molecules for Growth of Calcium Hydroxyapatite Crystals,” Journal of Crystal Growth, Vol. 186, No. 1-2, 1998, pp. 275-282. doi:10.1016/S0022-0248(97)00472-7
- M. H. Prado Da Silva, J. H. C. Lima, G. A. Soares, C. N. Elias, M. C. de Andrade, S. M. Best and I. R. Gibson, “Transformation of Monetite to Hydroxyapatite in Bioactive Coatings on Titanium,” Surface and Coatings Technology, Vol. 137, No. 2-3, 2001, pp. 270-276. doi:10.1016/S0257-8972(00)01125-7
- K. Sudarsanan and R. A. Young, “Significant Precision in Crystal Structural Details: Holly Springs Hydroxyapatite,” Acta Cryst, Vol. 25, No. 8, 1969, pp. 1534-1543. doi:10.1107/S0567740869004298
- H. E. Swanson and R. K. Fuyat, “Calcite,” Standard X-Ray Diffraction Patterns, Vol. 539, 1953, pp. 52-53.
- B. Dickens and W. E. Brown, “Crystallographic Studies of Role of Mg as a Stabilizing Impurity in BetaCa3(PO4)2 .1. Crystal-Structure of Pure Beta-Ca3(PO4)2,” Journal of Solid State Chemistry, Vol. 10, No. 3, 1974, pp. 232-248. doi:10.1016/0022-4596(74)90030-9
- M. Catti, G. Ferraris and A. Filhol, “Hydrogen-Bonding in Crystalline State—CaHPO4 (Monetite), P1bar or P1 - Novel Neutron-Diffraction Study,” Acta Crystallographica Section B-Structural Science, Vol. 33, No. 4, 1977, pp. 1223-1229. doi:10.1107/S0567740877005706
- I. Oftedal, “The Lattice Constants of CaO, CaSe, CaS, CaTe,” Zeitschrift für Physikalische Chemie (Leipzig), Vol. 128, 1927, pp. 154-158.
- M. H. Fathi and A. Hanifi, “Evaluation and Characterization of Nanostructure Hydroxyapatite Powder Prepared by Simple Sol-Gel Method,” Materials Letters, Vol. 61, No. 18, 2007, pp. 3978-3983. doi:10.1016/j.matlet.2007.01.028
- S. Raynaud, E. Champion, D. Bernache-Assollant and P. Thomas, “Calcium Phosphate Apatites with Variable Ca/P Atomic Ratio I. Synthesis, Characterisation and Thermal Stability of Powders,” Biomaterials, Vol. 23, No. 4, 2002, pp. 1065-1072. doi:10.1016/S0142-9612(01)00218-6
- A. J. Ruys, K. A. Zeigler, O. C. Standard, A. Brandwood, B. K. Milthorpe and C. C. Sorrell, “Hydroxyapatite sintering Phenomena: Densification and Dehydration Behaviour,” Ceramics: Adding the Value. International Ceramic Conference, AUSTCERAM-92, Melbourne, 16-21 August 1992, p. 605
- C. Ettarh and A. K. Galwey, “A Kinetic and Mechanistic Study of the Thermal Decomposition of Calcium Nitrate,” Thermochimica Acta, Vol. 288, No. 1-2, 1996, pp. 203- 219. doi:10.1016/S0040-6031(96)03052-3
- A. Chowdhury, J. Bould, M. G. S. Londesborough and S. J. Milne, “Fundamental Issues in the Synthesis of Ferroelectric Na0.5K0.5NbO3 Thin Films by Sol Gel Processing,” Chemistry of Materials, Vol. 22, No. 13, 2010, pp. 3862-3874. doi:10.1021/cm903697
- A. Chowdhury, P. R. Thompson and S. J. Milne, “TGA-FTIR Study of a Lead Zirconate Titanate Gel Made from a Triol-Based Sol-Gel System,” Thermochimica Acta, Vol. 475, No. 1-2, 2008, pp. 59-64. doi:10.1016/j.tca.2008.06.009
- J. A. M. van der Houwen, G. Cressey, B. A. Cressey and E. Valsami-Jones, “The Effect of Organic Ligands on the Crystallinity of Calcium Phosphate,” Journal of Crystal Growth, Vol. 249, No. 3-4, 2003, pp. 572-583. doi:10.1016/S0022-0248(02)02227-3