Open Journal of Composite Materials
Vol.07 No.04(2017), Article ID:77982,6 pages
10.4236/ojcm.2017.74011
Computational and Experimental Analyses of Detachment Force at the Interface between Carbon Fibers and Epoxy Resin
Kazuki Mori1*, Nobuhiko Matsumoto2, Sukeharu Nomoto1, Kenji Tsuruta3
1ITOCHU Techno-Solutions Corp., Kasumigaseki, Chiyoda-ku, Tokyo, Japan
2Mitsubishi Gas Chemical Company, Inc., Higashiyawata, Hiratsuka-shi, Kanagawa, Japan
3Graduate School of Natural Science and Technology, Okayama University, Tsushima-naka, Kita-ku, Okayama, Japan

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: June 1, 2017; Accepted: July 24, 2017; Published: July 27, 2017
ABSTRACT
Herein, we used theoretical and experimental methods to investigate the shear fracture strengths of carbon fiber/epoxy resin interfaces. The shear strengths of carbon fiber and epoxy resin were measured using the microdroplet test, whereas interaction and binding energies were estimated using Ab initio and molecular dynamics methods. However, binding energies did not impact the shear strength volumes determined by microdroplet tests, i.e., bonds between functional groups of the carbon filer and the epoxy resin were difficult to break. On the other hand, the interaction energies calculated for epoxy monomers were in good agreement with experimental data. Moreover, we determined the relationship between the simulated interaction energy and the shear fracture strength volume obtained using the microdroplet test.
Keywords:
Carbon Fiber?Reinforced Composite, Ab initio Method, Molecular Dynamics Simulation, Microdroplet Test

1. Introduction
Lightweight high-strength materials used in the aerospace industry have recently found automotive applications, as exemplified by the growing popularity of polymer composites [1] [2] containing dispersed carbon nanotubes (CNTs). These composites, exhibiting high strength-to-weight ratios, were investigated by quantum chemical calculations and molecular dynamics (MD) simulations to elucidate the underlying reasons of their superior properties. In particular, the calculation model used to investigate interactions between polymer chains and nanotubes/graphene, both were set at the quantum chemical and force field level [3] - [9] . Salahoor et al. elucidated the mechanism of adhesion between graphene and epoxy matrix layers and determined the corresponding interfacial fracture energy [10] . Minoia et al. investigated the mechanism of interfacial interaction between polyethylene and CNTs using MD and periodic density functional theory calculations, focusing on CH-π interactions [11] . Conversely, the study of Chen et al. involved pulling out single nanotubes from the polymer matrix, comparing MD simulation results with experimental data. Estimating the mechanism of adhesion between graphene and epoxy matrix layers is important for analyses of the detachment force. However, it might be a good idea to estimate the adhesion between graphene and epoxy matrix layers based on the physical properties of the epoxy resin because it is inferred that the carbon composites contain the surface epoxy resin that interacts with carbon graphene and the bulk epoxy resin. Since the above simulations did not investigate the physical properties of the epoxy resin or corresponding monomers, this work employed numerical simulations to evaluate the physical properties of epoxy monomers and the energies of their binding to carbon fibers, thus determining the optimal structures of these mechanical composites. Simulations were performed using Ab initio and MD methods, and the obtained results were compared with those of microdroplet tests.
2. Experimental and Numerical Methods
2.1. Materials
Bisphenol A diglycidyl ether (BADGE), 1,6-hexanediol diglycidyl ether (HDGE), and 3,4-epoxycyclohexylmethyl-3',4'-epoxycyclohexanecarboxylate (EHMHC) were used as epoxy resin monomers, with cyclohexane-1,2,4-tricarboxylic acid- 1,2-anhydride (H-TMAn), 1,3-bis(aminomethyl)cyclohexane (BAC), and isophoronediamine (IPDA) utilized as curing agents (Figure 1).
2.2. Microdroplet Test
In the microdroplet test, performed using an HM410 instrument (Tohei Sangyo Co.), the melted resin was attached to a single carbon fiber extracted from the bulk. All samples were prepared at a constant temperature of 23˚C and a relative humidity of 55%.
2.3. Calculations
Compositional optimization was performed using the B3LYP hybrid density functional with a 6-31G(d) basis set, and the epoxy binding energy was estimated for the optimized structure. Charge densities for each optimized structure were calculated at MP2 (second-order Møller-Plesset perturbation, full)/6- 31G(d) levels and also using the ChelpG method [12] . In our Ab initio calculations, graphene was substituted by benzene to reduce the number of atoms in the model and thus facilitate computation. Similarly to previous studies [13]

Figure 1. Chemical structures of (a) BADGE; (b) HDGE; (c) EHMHC; (d) H-TMAn; (e) BAC; (f) IPDA.
[14] , a COOH functional group was attached to benzene and allowed to react with epoxy monomers, as shown in Figure 2, with its binding energy estimated by the Ab initio method. The above calculations were performed using Gaussian09 software [15] . k1 was showed the C?O bond of a COOH functional group. k2 was showed the C?O bond which was generated after carbon atom of epoxy ring attached a COOH functional group.
Interactions between epoxy compounds were modeled using GROMACS 5.1.2 [16] molecular modeling software implementing a version of AMBER03 [17] . All force field MD calculations were performed in an NPT ensemble at 300 K for 30 ns under the condition of a constant number of atoms and pressure using the Nose-Hoover thermostat [18] and the Parrinello-Rahman barostat [19] . Prior to NPT simulation, we conducted structural relaxation in an NVT ensemble at 800 K for 10 ns, utilizing a unit cell with 256 molecules. The LINCE algorithm [20] was applied to all constrained bonds in epoxy monomers. The simulation time step equaled 1 fs, and calculations utilized the epoxy monomer charge density estimated by the Ab initio method.
3. Results and Discussion
3.1. Microdroplet Test
The experimentally determined shear stress (Table 1) varied between 59 and 65 MPa, with the maximum corresponding to the BADGE/IPDA resin.
3.2. Epoxy Monomer Binding Energies
The C?O bond binding energies of k1 and k2 were estimated as shown in Figure 2, with the corresponding numerical values listed in Table 2.
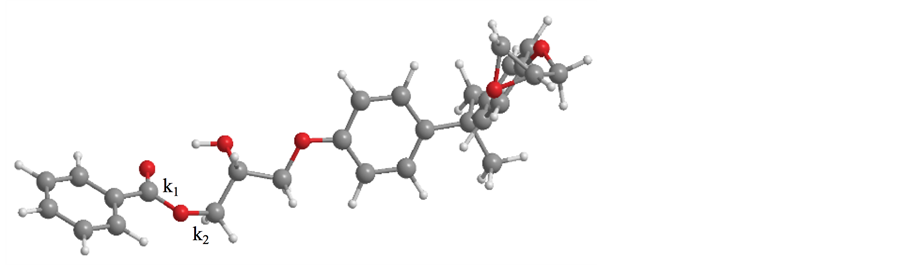
Figure 2. Binding constants k1 and k2.

Table 1. Microdroplet test results.
Table 2. Calculated epoxy monomer binding energies.
For BADGE-benzene, k1 and k2 were determined as 561 and 431 kcal∙mol?1∙ Å?2, respectively, with the corresponding values of HDGE-benzene equaling 372 and 282 kcal mol?1∙Å?2. For EHMHC-benzene, k1 was calculated as 331 kcal mol?1∙Å?2, whereas k2 could not be obtained due to the non-convergence of performed calculations. Consequently, we compared the calculated binding energies and the shear strength values determined by the microdroplet test, revealing that these two parameters were not correlated. Thus, bonds between the functional groups of the carbon filer and the epoxy resin were hard to break. In addition, the calculation of binding energies was very time-intensive due to the multi-atom nature of the simulated compounds, implying that the adopted calculation method was not suitable for estimating the carbon fiber/epoxy interfacial fracture toughness.
Subsequently, we estimated interactions for each epoxy compound monomer by MD simulations. Its interaction energy meant the intermolecular interaction energy between resins in a bulk phase. The intermolecular interaction energy increased with the hardness and/or heat resisting of material [21] . The energy of bulk epoxy monomers, i.e., the interaction energy, was obtained by averaging the energies of the last 10 ns of simulation time, as indicated by Equation (1).
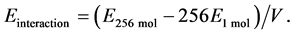 (1)
(1)
Since the above interactions become stronger with increasing number of atoms per molecule, the total energies of bulk epoxy monomers were divided by the number of molecules in the calculation cell to allow meaningful comparison, affording interaction energies of 39.54, 0.13, 51.04, 18.14, and 40.64 kJ∙mol?1∙nm?3 for BADGE/BAC, BADGE/H-TMAn, BADGE/IPDA, EHMHC/H- TMAn, and HDGE/H-TMAn combinations, respectively. The above interaction energies increased correspond to the shear strength value of microdroplet test, implying that the detachment force for the carbon fiber/epoxy resin interface is influenced not only by interfacial energy but also by the interaction energy of epoxy monomers.
4. Conclusion
We successfully utilized Ab initio and MD simulations to determine the intermolecular interaction energy for each epoxy monomer in bulk epoxy compound, observing a correlation between the simulated interaction energy and the shear strength determined by the microdroplet test and revealing that resin fragility possibly contributes to interfacial fracture toughness. Although we did not estimate carbon fiber/epoxy monomer interfacial energies, these energies, together with epoxy monomer interaction energies, allow the interfacial fracture toughness to be analyzed, which will be addressed in our future work.
Cite this paper
Mori, K., Matsumoto, N., Nomoto, S. and Tsuruta, K. (2017) Computational and Experimental Analyses of Detachment Force at the Interface between Carbon Fibers and Epoxy Resin. Open Journal of Composite Materials, 7, 179-184. https://doi.org/10.4236/ojcm.2017.74011
References
- 1. Moniruzzaman, M. and Winey, K.I. (2006) Polymer Nanocomposites Containing Carbon Nanotubes. Macromolecules, 39, 5194-5205.
https://doi.org/10.1021/ma060733p - 2. Cheng, Q., Wang, B., Zhang, C. and Liang, Z. (2010) Functionalized Carbon Nanotube Sheet/Bismaleimide Nanocomposites: Mechanical and Electrical Performance beyond Carbon-Fiber Composites. Small, 6, 763-767.
https://doi.org/10.1002/smll.200901957 - 3. Kar, T., Bettinger, H.F., Scheiner, S. and Roy, A.K. (2008) Noncovalent π-π Stacking and CH-π Interactions of Aromatics on the Surface of Single-Wall Carbon Nanotubes: An MP2 Study. Journal of Physical Chemistry C, 112, 20070-20075.
https://doi.org/10.1021/jp807809u - 4. Beigbeder, A., Linares, M., Devalckenaere, M., Degee, P., Claes, M., Beljonne, D., Lazzaroni, R. and Dubois, P. (2008) CH-π Interactions as the Driving Force for Silicone-Based Nanocomposites with Exceptional Properties. Advanced Materials, 20, 1003-1007.
https://doi.org/10.1002/adma.200701497 - 5. Wei, C. (2006) Radius and Chirality Dependent Conformation of Polymer Molecule at Nanotube Interface. Nano Letters, 6, 1627-1631.
https://doi.org/10.1021/nl0605770 - 6. Meyer, F., Minoia, A., Raquez, J.M., Spasova, M., Lazzaroni, R. and Dubois, P. (2010) Poly(Amino-methacrylate) as Versatile Agent for Carbon Nanotube Dispersion: An Experimental, Theoretical and Application Study. Journal of Materials Chemistry, 20, 6873-6880.
https://doi.org/10.1039/c0jm00386g - 7. Yang, H., Chen, Y., Liu, Y., Cai, W.S. and Li, Z.S. (2007) Molecular Dynamics Simulation of Polyethylene on Single Wall Carbon Nanotube. The Journal of Chemical Physics, 127, 094902.
https://doi.org/10.1063/1.2768060 - 8. Yang, M., Koutsos, V. and Zaiser, M. (2005) Interactions between Polymers and Carbon Nanotubes: A Molecular Dynamics Study. Journal of Physical Chemistry B, 109, 10009-10014.
https://doi.org/10.1021/jp0442403 - 9. Liu, Y. and Kumar, S. (2014) Polymer/Carbon Nanotube Nano Composite Fibers— A Review. ACS Applied Materials & Interfaces, 6, 6069-6087.
https://doi.org/10.1021/am405136s - 10. Salahshoor, H. and Rahbar, N. (2012) Nano-Scale Fracture Toughness and Behavior of Graphene/Epoxy Interface. Journal of Applied Physics, 112, 023510.
https://doi.org/10.1063/1.4737776 - 11. Nishio, M., Hirota, M. and Umezawa, Y. (1998) The CH/π Interaction: Evidence, Nature and Consequences. Wiley-VCH, New York.
- 12. Chirlian, L.E. and Francl, M.M. (1987) Atomic Charges Derived from Electrostatic Potentials: A Detailed Study. Journal of Computational Chemistry, 8, 894-905.
https://doi.org/10.1002/jcc.540080616 - 13. Kim, H.J., Koizhaiganova, R., Vasudevan, T., Sanjeeviraja, C. and Lee, M.-S. (2009) Single Step Synthesis of Poly (3-Octylthiophene)/Multi-Walled Carbon Nanotube Composites and Their Characterizations. Polymers for Advanced Technologies, 9, 736-741.
https://doi.org/10.1002/pat.1325 - 14. Kakarla, R.R., Kwang, P.L., Iyengar, G.A., Seok, K.M., Ali, M.S. and Chang, N.Y. (2006) Synthesis of Metal (Fe or Pd)/Alloy (Fe–Pd)-Nanoparticles-Embedded Multiwall Carbon Nanotube/Sulfonated Polyaniline Composites by γ Irradiation. Journal of Polymer Science Part A: Polymer Chemistry, 44, 3355-3364.
https://doi.org/10.1002/pola.21451 - 15. Frisch, M.J., et al. (2009) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford.
- 16. Berendsen, H.J.C., van der Spoel, D. and van Drunen, R. (1995) GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Computer Physics Communications, 91, 43-56.
https://doi.org/10.1016/0010-4655(95)00042-E - 17. Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Jr. Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W. and Kollman, P.A. (1995) A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society, 117, 5179-5197.
https://doi.org/10.1021/ja00124a002 - 18. Evans, D. and Holian, B. (1985) The Nose-Hoover Thermostat. The Journal of Chemical Physics, 83, 4069-4074.
https://doi.org/10.1063/1.449071 - 19. Parrinello, M. and Rahman, A. (1981) Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. Journal of Applied Physics, 52, 7182-7190.
https://doi.org/10.1063/1.328693 - 20. Hess, B., Bekker, H., Berendsen, H. and Fraaije, J. (1997) LINCS: A Linear Constraint Solver for Molecular Simulations. Journal Computational Chemistry, 18, 1463-1472.
https://doi.org/10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H - 21. Mori, K. and Sakakibara, K. (2011) Theoretical Study of Ionic Liquids on the Difference of Melting Points between Tertiary and Quaternary Ammonium Triflates. Chemistry Letters, 40, 690-692.
https://doi.org/10.1246/cl.2011.690