Open Journal of Medicinal Chemistry
Vol.05 No.01(2015), Article ID:54436,7 pages
10.4236/ojmc.2015.51001
A Peptide Nucleic Acid to Reduce Type I Collagen Production by Fibroblast Cells
Yasutada Imamura1*, Suzuka Tsuboi1, Toru Sugiyama2, Atsushi Kittaka3, Yonchol Shin1
1Department of Applied Chemistry, Faculty of Engineering, Kogakuin University, Tokyo, Japan
2Department of Life Sciences, Graduate School of Arts and Sciences, The University of Tokyo, Tokyo, Japan
3Faculty of Pharmaceutical Sciences, Teikyo University, Tokyo, Japan
Email: *yimamura@cc.kogakuin.ac.jp
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 8 February 2015; accepted 3 March 2015; published 6 March 2015
ABSTRACT
In this study, we prepared a peptide nucleic acid (PNA) against the gene coding for the human alpha 1 chain of type I collagen. This PNA was incorporated into normal human fibroblast cells by electroporation, leading to a decrease in the mRNA level of the gene. Furthermore, mRNA for the alpha 2 chain of type I collagen was also reduced. The production of collagen protein exhibited a similar profile to the changes in mRNA. These results indicate that PNA targeting COL1A1 is effective as an antigene reagent, and opens the possibility of future clinical applications in fibroproliferative disorders.
Keywords:
Collagen, PNA, Antigene, Fibrosis, Vimentin

1. Introduction
The most abundant extracellular matrix protein in tissues such as the skin and bones is type I collagen [1] . The type I collagen molecule is assembled from two α1 chains and one α2 chain, which are encoded by the COL1A1 and COL1A2 genes respectively. Expression of these two genes is coordinated under most physiological and pathological conditions [2] . Excessive production of type I collagen is a hallmark of fibroproliferative disorders, such as systemic sclerosis and nephrogenic systemic fibrosis [3] [4] . Therefore, therapeutic strategies targeting these disorders commonly involve those to reduce collagen production [5] [6] .
The transcriptional regulation of these genes involves various factors, including cytokines such as TGF-β and PDGF and their signal transduction pathways, as well as various transcription factors and their interplays. For example, the transcription factor Sp1, the Smad3/4 complex and the co-activators p300/CBP mediate the response of the COL1A2 promoter to TGF-β stimulation [2] [5] [7] . The expression of collagen genes can also be regulated at the post-transcriptional level, with changes to mRNA stability contributing to turnover and steady state levels [3] [8] .
Peptide nucleic acids (PNAs) are analogs of DNA or RNA in which the pentose-phosphate backbone is replaced by an oligomer of N-(2-aminoethyl)glycine [9] . These molecules are relatively resistant to biodegradation and anneal by Watson-Crick base-pairing to complementary RNA or DNA sequences with a much higher affinity than the corresponding phosphodiester oligomers [10] . This renders them effective tools to target specific nucleic acid sequences.
The base-paired structure of DNA presents kinetic and thermodynamic obstacles to the hybridization of an antigene PNA to its complementary site within genomic DNA. However, formation of the open complex with RNA polymerase before the start of transcription enables binding of an antigene PNA to its target site, resulting in the formation of a hybrid PNA-DNA duplex that can inhibit transcription. PNA consisting of a sequence complementary to that of the DNA at a transcription start site could affect transcription of the downstream gene [11] .
Specific post-transcriptional inhibition of gene expression can be achieved with RNA interference (RNAi). Here, sequence-specific degradation of target mRNA is triggered by non-translated double-stranded RNA molecules dubbed small interfering RNA [12] [13] . In contrast to RNAi, PNA-based strategies for inhibiting gene expression involve the direct interaction of a PNA molecule with the DNA template and disruption of the RNA polymerization reaction. The melting of double-stranded DNA at the transcription start site is therefore required for effective PNA binding. As this strategy only affects active transcription, and thus de novo RNA synthesis, completed mRNA molecules or those already under construction are unaffected [11] .
In this study, we describe a PNA molecule that targets the transcription initiation site of the COL1A1 gene and evaluate the ability of this PNA to impair production of type I collagen in normal human fibroblast cells. The potential use of this PNA as an antigene reagent is discussed.
2. Materials and Methods
2.1. Cell Culture
The human diploid fetal lung fibroblast cell line TIG-1 was obtained from the Japanese Collection of Research Bioresources Cell Bank and grown in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich Japan, Tokyo, Japan) containing 10% fetal bovine serum (FBS; Equitech-Bio, Inc., Kerrville, TX, USA), 50 units/mL penicillin G, and 50 μg/mL streptomycin sulfate (Gibco BRL, Grand Island, NY, USA) on 100-mm culture dishes kept at 37˚C in a humidified atmosphere of 5% CO2 and 95% air. At confluence, cells were removed from the dishes by treatment with 0.025% trypsin-0.02% ethylenediamine tetra-acetic acid in calcium- and magnesium-free phosphate-buffered saline (PBS(-); Nissui Pharmaceutical, Tokyo, Japan) and were subcultivated at a 1:4 split ratio.
2.2. Incorporation of PNA into Cells
Cells were transfected using the Neon Transfection system (Life Technologies, Tokyo, Japan) according to the manufacturer’s protocol. For a well in a 6-well plate, 200 nM PNA per 1 × 106 cells were subjected to two pulses of 1350 V at a pulse width of 20 ms. Treated cells were transferred into 2 mL prewarmed medium containing 10% FBS and 2 mM ascorbate 2-phosphate. The medium was changed the next day and thereafter every 3 days. For the enzyme-linked immunosorbent assay (ELISA), FBS-free medium with 2 mM ascorbate 2-phos- phate was substituted after the first day.
2.3. Fluorescence Microscopy for Type I Collagen
Cells were fixed with 4% paraformaldehyde in 100 mM phosphate buffer (pH 7.4) for 10 min at room temperature and then permeabilized by treatment with 0.2% Triton-X100 in PBS(-) for 2 min at room temperature. After washing with PBS(-), cells were stained with 0.5 µg/ml anti-human type I collagen monoclonal antibody F-56 (Daiichi Fine Chemical Co. Ltd., Toyama, Japan) for 2 h at room temperature. After washing with 0.1% bovine serum albumin (BSA) in PBS(-), a secondary antibody conjugated with Alexa Fluor 488 (Life Technologies) was applied for 1 h at room temperature. After washing again with 0.1% BSA in PBS(-), cells were examined by fluorescence microscopy. Nuclei were stained with 4’,6-diamidino-2-phenylindole, dihydrochloride (Life Technologies).
2.4. ELISA for Measurement of Type I Collagen in Conditioned Medium
Conditioned medium from cell culture diluted 1:5 with PBS(-) was placed in wells of a microplate for 2 h at room temperature. After washing with PBS(-), each well was blocked with 1:4 diluted Block-Ace (DS Pharma Biomedical Co., Ltd., Osaka, Japan) overnight at 4˚C. After washing with PBS(-), 20 ng/ml anti-human type I collagen monoclonal antibody F-56 was added to each well for 1 h at room temperature. After washing with PBS(-) again, a secondary goat anti-mouse IgG HRP conjugated antibody (Thermo Fisher Scientific, Yokohama, Japan) was applied and incubation was continued for 30 min at room temperature. After washing, the horseradish peroxidase substrate (2,2’-azino-di[3-ethylbenzthiazoline sulfonate]; KPL) was added and incubation was continued for 20 min. The absorbance at 405 nm was then recorded.
2.5. Synthesis of PNA Oligomer
Reagents for PNA synthesis were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan) unless otherwise noted. PNA oligomers were manually synthesized from commercial PNA monomers (PolyOrg, Leominster, MA, USA) and Fmoc-Lys(Boc)-OH (Merck Millipore, Billerica, MA, USA) using standard Fmoc chemistry. Fmoc-PAL-PEG-PS resins (Life Technologies) were used as solid support. 1-[Bis(dimethylamino)me- thylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (Life Technologies), N, N-diisopro- pylethylamine, and 2,6-lutidine (Kanto Chemical, Tokyo, Japan) in N-Methylpyrrolidone were used for coupling each monomer. Completion of the coupling reaction was monitored by the Kaiser test. Piperidine (20%) in DMF was used for deprotection of the Fmoc-protecting group from the terminal amino group. The unreacted amino groups were capped with acetic anhydride. After coupling the last monomer, the resins were treated with Trifluoroacetic acid containing 13.9% m-cresol and 2.8% H2O to cleave the oligomers from the resins as well as to remove the protecting groups from the nucleobases and amino group (Boc) of lysines. The solvent was removed, and the residue was washed thrice with diethyl ether and dissolved in H2O. The final product was purified by reversed-phase HPLC and characterized by ESI-TOF mass spectrometry. Calcd for [M+3H]3+ 1453.6087, found 1453.6053.
2.6. Real-Time PCR Analysis of TIG-1 Cells for Measurement of Collagen Gene mRNA Expression
Extraction of RNA from TIG-1 cells with or without PNA introduction was carried out using the RNA isolation reagent RNAiso Plus (Takara Bio, Shiga, Japan) according to the manufacturer's instructions, with 0.4 mL of the reagent used for each 35-mm culture dish.
Synthesis of cDNA from total RNA samples was carried out using the ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan) with 0.5 µg total RNA/10 µL reaction volume. The primer for reverse transcription in the kit contains oligodT and random primers. The cDNA (1.0 µL RT sample) was amplified using THUNDERBIRD SYBR qPCR Mix (Toyobo) in a total volume of 20 µL. Standard real-time PCR conditions for the system (Mx3005P, Agilent Santa Clara, CA, USA) were used: an initial denaturation step for 10 min at 95˚C, followed by 40 cycles of denaturation at 95˚C for 30 s, annealing at 60˚C for 1 min, and elongation at 72˚C for 1 min. Melting curves were generated after amplification. The nucleotide sequences of the primers selected for the real-time PCR are shown as follows: forward and reverse primers for cDNAs of human type I collagen apha1 (COL1A1) and apha2 (COL1A2), COL1A1_Fw: GTCGAGGGCCAAGACGAAG, COL1A1_Rv: CAGATCAC- GTCATCGCACAAC, COL1A2_Fw: CGGACGACCTGGTGAGAGA, COL1A2_Rv: CATTGTGTCCCCT- AATGCCTT; those for cDNAs of human type IV collagen apha1 (COL4A1) and alpha2 (COL4A2), COL4A1_Fw: GGATGCTGTTGAAAGGTGAAAGA, COL4A1_Rv: GGTGGTCCGGTAAATCCTGG, and COL4A2_Fw: TTGGCGGGTGTGAAGAAGTTT, COL4A2_Rv: CCTTGTCTCCTTTACGTCCCTG ; those for cDNA of human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), GAPDH_Fw: CATGAGAAGTAT- GACAACAGCCT, GAPDH_Rv: AGTCCTTCCACGATACCAAAGT, respectively. Each sample was amplified in triplicate and the cycle threshold values were calculated by the MxPro QPCR Software and normalized against the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
2.7. Analysis of Genomic DNA
Genomic DNA from TIG-1 cells was extracted according to the SDS/Phenol method [14] . Approximately 0.5 µg DNA was used for PCR to amplify the region surrounding the transcription initiation site of the COL1A1 gene. PCR was carried out using TaKaRa Ex Taq DNA polymerase according to the manufacturer’s instructions (Takara Bio). The primers were forward 5’-CCAACCCTTCCACCTTTGGAAGT-3’, and reverse 5’-CCGG- AGGTCCACAAAGCTGAA-3’. PCR conditions were: an initial denaturation and polymerase activation step for 5 min at 95˚C, followed by 30 cycles of denaturation at 95˚C for 1 min, annealing at 55˚C for 1 min, and elongation at 72˚C for 1 min, with final elongation at 72˚C for 15 min. Amplified DNA fragments were subjected to DNA sequencing service from Bio Matrix Research, Inc. (Chiba, Japan).
3. Results
DNA sequencing of the transcription initiation site of the COL1A1 gene in TIG-1 cells confirmed that these cells possessed the same sequence in this region as detailed in the NCBI database (NCBI Reference Sequence: NG_007400.1; data not shown). PNA was designed to target 15 base sequences in the sense strand from −5 to +10 bases of the transcription start site and to be complementary to the template strand except for flanking with Lys at the 5’ end and Lys-NH2 at the 3’ end (Figure 1).
PNA (200 nM) targeting the COL1A1 gene was introduced into TIG-l cells by electroporation. As TIG-1 cells secrete both type I and type IV collagen, the mRNA expressions of COL1A1, COL1A2, COL4A1, and COL4A2 genes were measured using real-time PCR. Figure 2 demonstrates a time course of mRNA expression levels of collagen mRNA genes following introduction of PNA. The mRNA levels present in cells, which were electroporated without PNA and cultured for 1 day, were set as the values to which mRNA levels under other conditions were compared. Expression of COL1A1 was decreased at days 3 and 4, but had fully recovered by day 5. Similar behavior was observed for COL1A2, except partial recovery occurred at day 4. The reduction ratio of COL1A1 and COL1A2 was roughly 40%. There was no reduction in the expression levels of COL4A1 or COL4A2, which served as a control.
The relative amount of secreted type I collagen was measured by ELISA using an anti-type I collagen antibody. The antibody was conformation-dependent and specific to the triple helical molecule. Conditioned medium from a 3-day culture under conditions indicated in Figure 3 was subjected to the assay. The value which was obtained for type I collagen secretion levels from cells not exposed to PNA was set at 100%. A clear reduction in type I collagen secretion was observed over days 3 - 6 of culture and thereafter a recovery to 100% was observed.
Cells treated with or without PNA were immunostained using an anti-type I collagen antibody. As shown in Figure 4, PNA treatment had minimal impact on type I collagen levels on day 1 of culture after PNA introduction. However, by day 3, type I collagen staining became very faint.
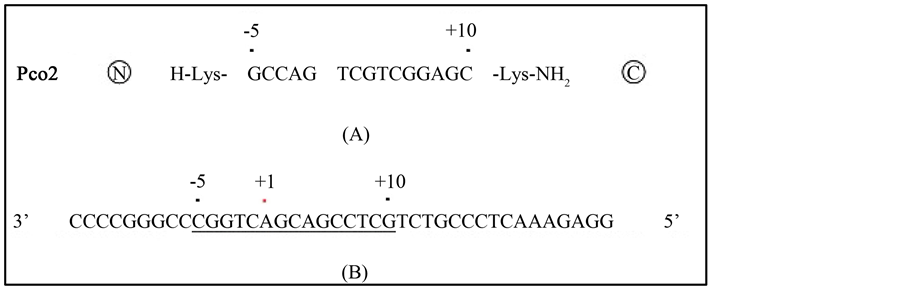
Figure 1. Sequences of the PNA and target DNA sequences surrounding the transcription initiation site of COL1A1. A schematic representation of the PNA named Pco2 is depicted in A. A Lysyl residue at the N-terminus and a Lysyl amide residue at the C-terminus flank the ends of the PNA. The antisense sequence target at the transcription initiation site of COL1A1 ranges from positions −14 to +26 bases and is shown in B. This PNA contains 15 bases and the sequences are complementary to those from −5 to +10 bases. The genome sequence and mRNA sequences are adopted from NCBI Reference Sequences NG_007400.1 and NM_000088.3, respectively
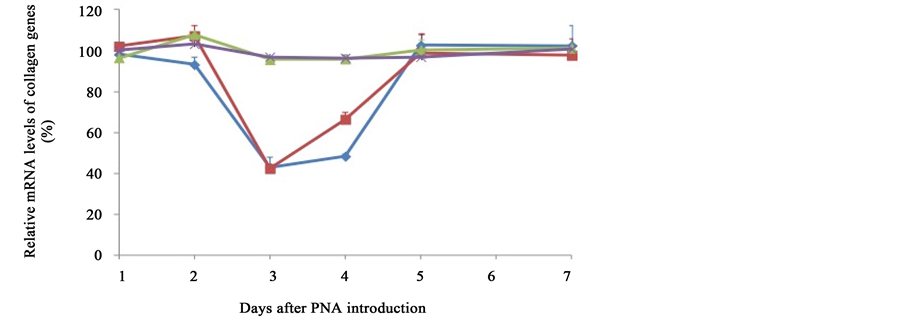
Figure 2. Time course of the expressions of several collagen mRNAs following introduction of the PNA. Expression levels of several collagen mRNA species (COL1A1, blue diamond; COL1A2, red square; COL4A1, green triangle; COL4A2, purple cross) were measured by real-time PCR using samples from cells cultured for the indicated period following PNA introduction. The amounts of expression for each gene relative to GAPDH are expressed as a percentage of that of control cells that were treated in the same way without PNA and had been cultured for an identical number of days.
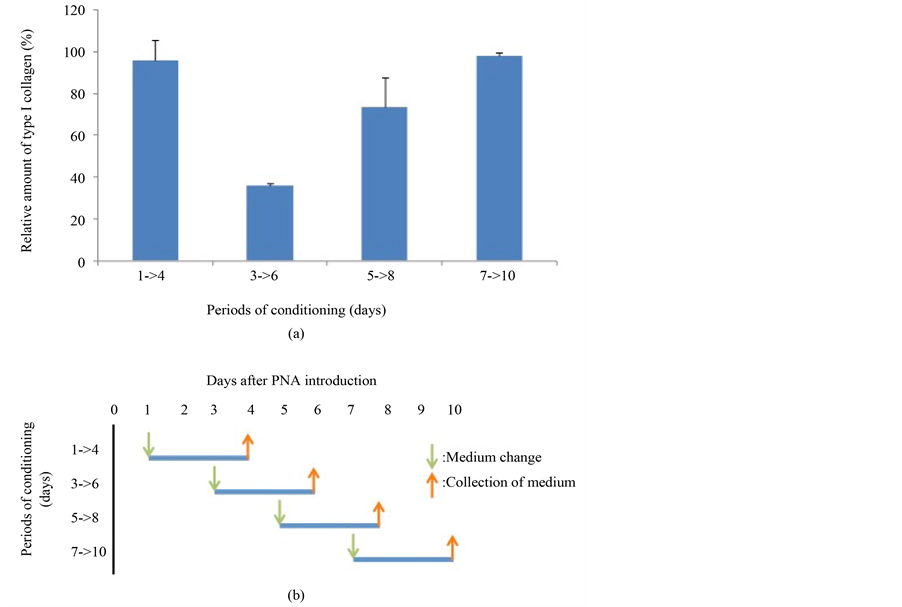
Figure 3. Measurement of type I collagen secretion following PNA introduction. Cells were treated with PNA or with PBS as controls and cultured. Days indicate the period following electroporation. The medium was changed to serum-free medium containing 2 mM ascorbate 2-phosphateon on the first day as indicated and was allowed to condition for 3 days. Following collection, media samples were subjected to ELISA using anti-type I collagen antibody. Levels of type I collagen are expressed as a percentage of those of control cells treated and cultured in the same way except that PBS replaced PNA.
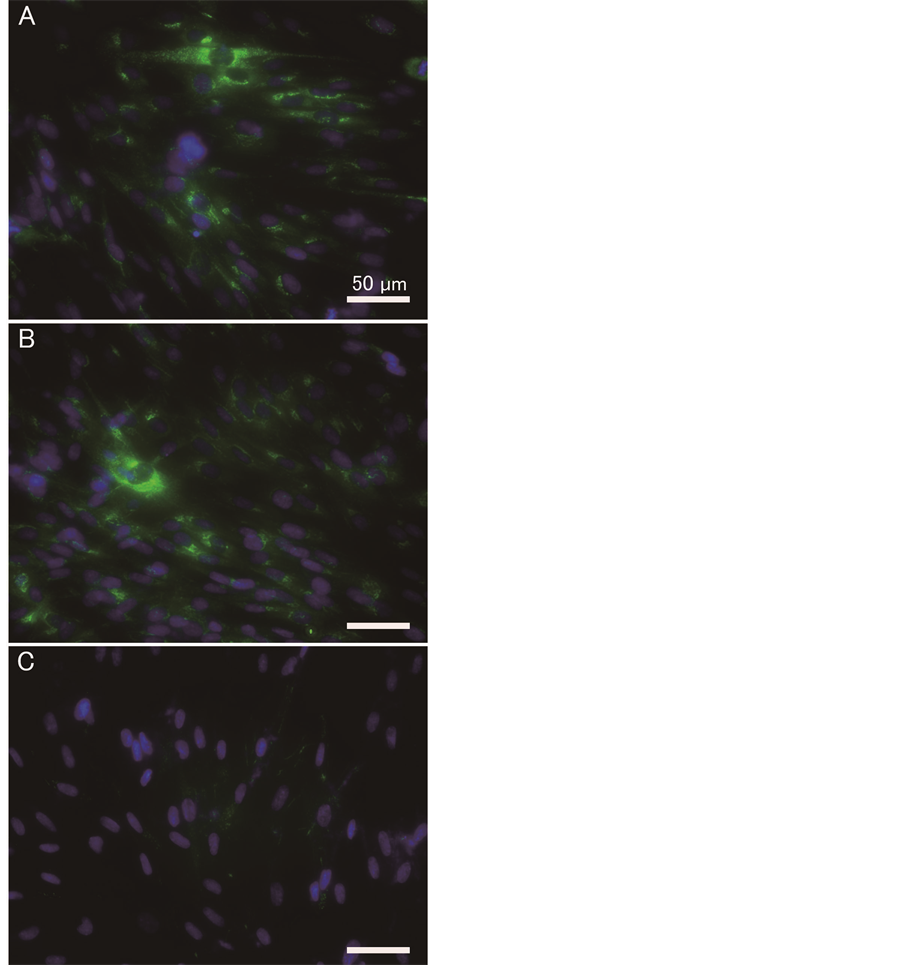
Figure 4. Immunostaining of cells for type I collagen antibody following PNA introduction. Cells were treated as described in Figure 3 and immunostained with anti-type I collagen antibody: (A) no PNA for 1 day culture; (B) 200 nM PNA for 1 day culture; (C) 200 nM PNA for 3 days culture. Staining of type I collagen and nuclei are depicted as green and blue respectively. Bars in the figure indicate 50 µm.
4. Discussion
The PNA constructed was designed to bind to the template strand of the transcription initiation site in the human COL1A1 gene where the open complex with RNA polymerase would be formed before the start of transcription. This binding would stop de novo transcription of the gene. In practice, we determined that the level of COL1A1 mRNA expression decreased transiently but was able to recover to the control level.
Incorporation of PNA into cells using the Neon Transfection system caused minimal damage to cells as determined by microscopic observation, and almost no effects on the mRNA levels of COL4A1 and COL4A2 were detected. The extent of the reduction in type I collagen mRNA levels should be determined by the incorporation ratio of the PNA into the cells. Since the transfection ratio of an expression plasmid encoding a fluorescent protein was roughly 50% in TIG-1 cells (data not shown), the reduction of the COL4A1 mRNA level to 40% could be well explained if it was assumed that PNA incorporation was similar to that of the plasmid. Within cells, the affinity of a PNA molecule consisting of 15 bases to bind to a complementary sequence would be sufficiently high [15] .
Following electroporation of PNA, a short time is needed for the reduction of mRNA to occur, and this lag period depends on the turnover time of the mRNA. The half life of COL1A1 mRNA is reported to be 4 - 24 h depending on the source [3] [5] , and these differences are likely attributable to the measurement method employed. Our data first detected a reduction in mRNA levels 2 days after PNA incorporation, which was longer than the half lives reported previously. Another report of a PNA-based inhibition strategy has also shown a similar time to take effect [16] .
Following PNA treatment, COL1A2 mRNA exhibited similar behaviors of reduction and recovery as COL1A1 mRNA. Recently, vimentin filaments have been shown to be associated with and stabilize COL1A1 and COL1A2 mRNA in a stem-loop in the 5’ untranslated regionand LARP6-dependent manner [8] . Since PNA designed to target the transcription start site of the gene may effectively suppress overall expression of the whole gene, there would be little chance of production of even the 5’ terminal region mRNA. Absence of COL1A1 mRNA could lead to failure of the complex formation and consequently destabilize COL1A2 mRNA. COL1A2 mRNA in excess of COL1A1 mRNA would therefore be degraded, and this is consistent with the observation that both mRNAs exhibited similar time-dependent behavior [8] .
The antigene strategy targeting COL1A1 results in reduction of the type I collagen protein. The type I collagen molecule is a heterotrimer and consists of two polypeptides of an alpha 1(I) chain and one of alpha 2(I). Stoichiometry of the trimer molecule has been thought to arise on the basis of interaction between three polypeptides inside endoplasmic reticulum just after translation. However, Challa and Stefanovic [8] have implicated an interaction between COL1A1 and COL1A2 mRNA in association with vimentin in the cytoskeleton. Although the number of different mRNAs that participate in the association is not yet clear, this phenomenon could partially contribute to the stoichiometry of the polypeptides.
In this paper, we have shown that PNA targeting COL1A1 is effective as an antigene reagent. Since PNA- based strategies preferentially affect transcriptionally active genes, activated cells in fibrotic disorders that produce excessive type I collagen are potential targets. The selective and potentially reversible nature of the gene suppression induced by PNA is an important characteristic for the future clinical application.
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Number 23650294 (Y.I.) and by a grant from the Strategic Research Foundation Grant-Aided Project for Private Universities from the Ministry of Education, Culture, Sport, Science, and Technology, Japan (MEXT), 2014-2018 (S1411005) (Y.I.).
References
- Myllyharju, J. and Kivirikko, K.I. (2001) Collagens and Collagen-Related Diseases. Annals of Medicine, 33, 7-21. http://dx.doi.org/10.3109/07853890109002055
- Ramirez, F., Tanaka, S. and Bou-Gharios, G. (2006) Transcriptional Regulation of the Human α2(I) Collagen Gene (COL1A2), an Informative Model System to Study Fibrotic Diseases. Matrix Biology, 25, 365-372. http://dx.doi.org/10.1016/j.matbio.2006.05.002
- Eckes, B., Mauch, C., Hüppe, G. and Krieg, T. (1996) Differential Regulation of Transcription and Transcript Stability of Pro-Alpha 1(I) Collagen and Fibronectin in Activated Fibroblasts Derived from Patients with Systemic Scleroderma. The Biochemical Journal, 315, 549-554.
- Trojanowska, M., LeRoy, E.C., Eckes, B. and Krieg, T. (1998) Pathogenesis of Fibrosis: Type 1 Collagen and the Skin. Journal of Molecular Medicine, 76, 266-274. http://dx.doi.org/10.1007/s001090050216
- Louneva, N., Saitta, B., Herrick, D.J. and Jimenez, S.A. (2003) Transcriptional Inhibition of Type I Collagen Gene Expression in Scleroderma Fibroblasts by the Antineoplastic Drug Ecteinascidin 743. The Journal of Biological Chemistry, 278, 40400-40407. http://dx.doi.org/10.1074/jbc.M301964200
- Rosenbloom, J., Mendoza, F.A. and Jimenez, S.A. (2013) Strategies for Anti-Fibrotic Therapies. Biochimica et Biophysica Acta (BBA)―Molecular Basis of Disease, 1832, 1088-1103. http://dx.doi.org/10.1016/j.bbadis.2012.12.007
- Massagué, J., Seoane, J. and Wotton, D. (2005) Smad Transcription Factors. Genes & Development, 19, 2783-2810. http://dx.doi.org/10.1101/gad.1350705
- Challa, A.A. and Stefanovic, B. (2011) A Novel Role of Vimentin Filaments: Binding and Stabilization of Collagen mRNAs. Molecular and Cellular Biology, 31, 3773-3789. http://dx.doi.org/10.1128/MCB.05263-11
- Hyrup, B. and Nielsen, P.E. (1996) Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications. Bioorganic & Medicinal Chemistry, 4, 5-23. http://dx.doi.org/10.1016/0968-0896(95)00171-9
- Egholm, M., Buchardt, O., Christensen, L., Behrens, C., Freier, S.M., Driver, D.A., et al. (1993) PNA Hybridizes to Complementary Oligonucleotides Obeying the Watson-Crick Hydrogen-Bonding Rules. Nature, 365, 566-568. http://dx.doi.org/10.1038/365566a0
- Janowski, B.A., Kaihatsu, K., Huffman, K.E., Schwartz, J.C., Ram, R., Hardy, D., et al. (2005) Inhibiting Transcription of Chromosomal DNA with Antigene Peptide Nucleic Acids. Nature Chemical Biology, 1, 210-215. http://dx.doi.org/10.1038/nchembio724
- Fire, A., Xu, S.Q., Montgomery, M.K., Kostas, S.A., Driver, S.E. and Mello, C.C. (1998) Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis elegans. Nature, 391, 806-811. http://dx.doi.org/10.1038/35888
- Hammond, S. M. (2005) Dicing and Slicing. FEBS Letters, 579, 5822-5829. http://dx.doi.org/10.1016/j.febslet.2005.08.079
- Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning. 2nd Edition, Cold Spring Harbor Laboratory Press, New York.
- Cutrona, G., Carpaneto, E.M., Ponzanelli, A., Ulivi, M., Millo, E., Scarfì, S., et al. (2003) Inhibition of the Translocated C-Myc in Burkitt’s Lymphoma by a PNA Complementary to the E mu Enhancer. Cancer Research, 63, 6144-6148.
- Macadangdang, B., Zhang, N., Lund, P.E., Marple, A.H., Okabe, M., Gottesman, M.M. et al. (2011) Inhibition of Multidrug Resistance by SV40 Pseudovirion Delivery of an Antigene Peptide Nucleic Acid (PNA) in Cultured Cells. PLoS ONE, 6, e17981. http://dx.doi.org/10.1371/journal.pone.0017981
NOTES

*Corresponding author.