Open Journal of Medicinal Chemistry
Vol.4 No.2(2014), Article
ID:45519,22
pages
DOI:10.4236/ojmc.2014.42003
Efficient Utilization of 6-Aminouracil to Synthesize Fused and Related Heterocyclic Compounds and Their Evaluation as Prostate Cytotoxic Agents with Cathepsin B Inhibition
Marwa Taha Mostafa Sarg1*, Shereen Said El-Shaer2
1Department of Organic Chemistry, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, Egypt
2Department of Biochemistry, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, Egypt
Email: *m.t.sarg@hotmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 6 April 2014; revised 21 April 2014; accepted 28 April 2014
ABSTRACT
6-aminouracil 1 was utilized to introduce different heterocyclic rings at C-6 position through various synthetic strategies. The synthesized compounds bear rings that are either directly attached to the uracil back bone as in compounds 6, 12a-c and 15, or attached through an amino bridge as compounds 3a-c, 5a, b, 7a, b, 9 and 10, or through an imino bridge as compound 18. Also, compounds 4, 8, 11a-c, 14, 16 and 17 bearing biologically active side chains were synthesized. In addition to, compounds 13, 19, 20, 21 and 22 bear fused rings to the uracil backbone. All synthesized compounds were evaluated for their anticancer activity against prostate PC3 cell line using in-vitro sulforhodamine-B (SRB) method, from which compounds 3a, c, 4, 5a, b, 6, 7a, b, 11a-c, 12a, b, 17 and 20 were the most active. These active compounds were further evaluated for their ability to inhibit cathepsin B enzyme by using enzyme-linked immunosorbent assay, which revealed that compounds 5a, b, 7a, 11a, 12a and 17 exhibited more than 50% inhibition of cathepsin B. Among which the phenyl thiourea derivative 17 was the most active exhibiting 82.3% inhibition, while the reference doxorubicin exerted 18.7% inhibition.
Keywords:Uracil, Cytotoxicity, PC3 Cell Line, Cathepsin B

1. Introduction
Cancer is thought to reflect a multistep process, resulting from accumulation of inherited and/or acquired defects in genes involved in the positive or negative regulation of cell proliferation and survival [1] . Among the widespread carcinoma is prostate cancer which is the most common tumor in men and the second cause of cancer-related death among them [2] [3] . However, pathways that play a role in prostate cancer tumorigenesis include chronic inflammation, immunosuppression and angiogenesis [4] .
Angiogenesis could be defined as a biological process involving the division and migration of endothelial cells, leading to the formation of microvasculature [5] [6] . This process facilitates tumor growth by providing oxygenation to the tumor through a series of steps including endothelial cell proliferation, motility of endothelial cells through the extracellular matrix toward the angiogenic stimuli and capillary differentiation [4] . Therefore, changes in lysosomal trafficking and content that support invasion and angiogenesis could account for disorders in the regulation of apoptotic and non-apoptotic cell death, particularly through the apparent cellular release of a class of proteases, cathepsins which are usually sequestered within the lysosomal lumen [7] [8] . Lysosomal cysteine cathepsins belong to a family of 11 human proteolytic enzymes. Some of them correlate with progression in a variety of cancers and therefore are considered as potential therapeutic targets [9] .
In cancer, the cellular localization of lysosomal cysteine cathepsins is often altered. Intracellular, cell surface and secreted cysteine cathepsins are involved in distinct tumorigenic processes in-vivo, such as angiogenesis, invasion through extracellular matrices and metastasis [10] -[12] . The precise role of each cathepsin in carcinogenesis remains unclear [13] . Cathepsin B has been shown to play a dominant role in executing the apoptotic program in several tumor cell lines [14] as it reveals that cathepsin B may play a role in malignancy as an executioner of apoptosis in cytotoxic signaling cascades and as a mediator of tumor invasion [8] . Moreover, it has been observed that increased cathepsin B protein levels, pericellular localization and secretion are thought to contribute to prostate cancer invasion and metastasis with cathepsin B acting both directly and indirectly on extracellular matrix (ECM) remodeling and degradation [15] .
Nucleoside analogues have very well recognized activity as anti-metabolites against several types of neoplastic cells [16] [17] , in which 5-fluorouracil and mercaptopurine are typical pyrimidine and purine analogues, respectively [18] . Recently, uracil derivatives substituted either at C-5 or C-6 positions as well as their nucleosides have proved to exhibit significant status in the field of chemotherapy [19] -[26] . However, significant attention has been paid to nucleoside analogues having a heterocyclic ring as a substituent [27] -[29] . Modifications are mainly introduced on the C-5 atom of the uracil ring among which the introduction of a heterocyclic system to the uracil moiety exhibited increased anticancer activity [29] [30] .
In this investigation, construction of several heterocyclic rings either directly attached or attached through an amino or imino bridges to the C-6 atom of the uracil ring were performed using different strategies utilizing 6-aminouracil as a building block. Among the registered active anticancer rings were pyrimidine [31] [32] , furan [33] [34] , pyrrole [35] , thiophene [36] , benzoxazine [37] , quinoxaline [38] , triazole [39] [40] , thiazolidine [41] as well as thienopyrimidine [42] [43] . This active building unit; 6-aminouracil was also used to synthesize some fused rings to the uracil moiety. The synthesized compounds were screened for their anticancer activity against prostate cancer PC3 cell line, from which the most active compounds were subjected to biochemical screening against cathepsin B which is considered as a potential target for prostate cancer therapy.
2. Discussion
2.1. Chemistry
The synthetic strategies adopted for the synthesis of the target compounds are depicted in Schemes 1-4. The starting material; 6-aminouracil was chosen as an active substrate [44] -[47] upon which different reaction conditions were applied utilizing its 6-amino function in the synthesis of various 6-heterocyclic uracil and fused uracil derivatives.
“(Scheme 1)” comprises the coupling of 6-aminouracil 1 with 6-substituted-4-chloro-2-thioxo-1, 2-dihydropyrimidine-5-carbonitriles 2a-c [48] under basic conditions [49] with subsequent elimination of a hydrochloride molecule to yield 6-substituted-4-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)-2-thioxo-1,2-dihydropyrimidine-5-carbonitriles 3a-c. 1H NMR spectra of compounds 3a & 3c exhibited four deuterium oxide exchangeable singlets at δ 6.14 - 6.20, δ 10.01 - 10.25, δ 10.49 - 11.73 and δ 10.61 - 11.92 ppm attributed to
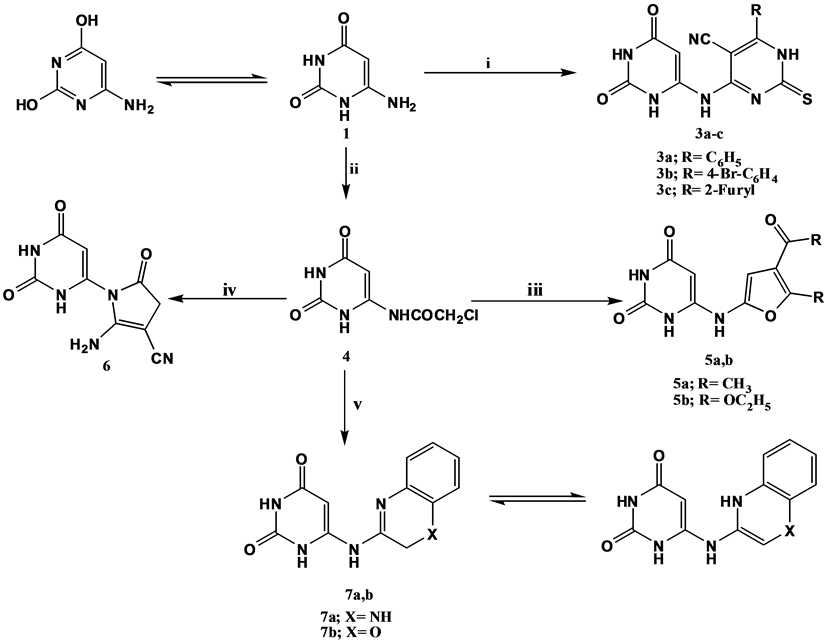
Scheme 1. Synthetic pathways for compounds 3a-c, 4, 5, 6 and 7a, b. Reagents & conditions: i: 4-Chlorodihydropyrimidine derivatives 2a-c/DMF/piperidine/Reflux; ii: ClCH2COCl/K2CO3/DMF/R.T.; iii: acetyl acetone or diethyl malonate/sod. ethoxide/Abs.ethanol/R.T.; iv: malononitrile/fusion; v: o-pheneylene diamine or o-aminophenol/Abs. ethanol/TEA/Reflux.
uracil-C6-NH, uracil-N1-H, uracil-N3-H and pyrimidine-N1-H; respectively. However, the 1H NMR spectrum of compound 3b revealed only two deuterium oxide exchangeable singlets each integrated for one proton at δ 6.17 and δ 10.04 ppm corresponding to uracil-C6-NH and uracil-N1-H; respectively. In addition to a deuterium oxide exchangeable singlet integrated for two protons at δ 10.56 ppm due to uracil-N3-H and pyrimidine-N1-H protons. Furthemore, the amino function of compound 1 was chloroacetylated by using equimolar amount of chloroacetyl chloride under anhydrous conditions at room temperature to yield the useful intermediate 6-chloroacetylaminouracil 4. The 1H NMR spectrum of compound 4 revealed a singlet integrated for two protons at δ 4.85 ppm attributed to CH2Cl protons. Compound 4 was utilized for the synthesis of several rings attached to the uracil nucleus, among which was the reaction of compound 4 in presence of sodium ethoxide with activated methylene bearing compounds namely; acetyl acetone and diethyl malonate [50] to yield the corresponding substituted 2-furan-2-ylaminouracil derivatives 5a & b; respectively. The preparation of such compounds is expected to proceed via nucleophilic substitution of the chloride atom by the active methylene bearing compounds under the reaction basic conditions followed by tautomerism then subsequent elimination of a water molecule leading to ring closure and formation of the furan ring. The 1H NMR spectrum of compound 5a revealed two singlets at δ 1.62 and δ 4.39 ppm corresponding to furyl-C5-CH3 protons and COCH3 protons; respectively. In addition to, a singlet at δ 4.28 ppm attributed to furyl-C3 proton, while the electron impact mass spectrum of compound 5b revealed the molecular ion peak at m/z 309 (2.40) and the base peak at m/z 55 (100).
Moreover, compound 4 was further utilized to synthesize the pyrrolidinone derivative 6 through its treatment with malononitrile in presence of a strong base [51] [52] . IR spectrum of compound 6 exhibited a strong absorption band at 2200 cm−1 due to cyano group, while its electron impact mass spectrum showed a peak at m/z 234 (3.84) corresponding to M+1 and a base peak at m/z 64 (100). Fulfilling the aforementioned strategy, the reactive intermediate; 6-chloroacetylaminouracil 4 was refluxed with o-phenylene diamine and o-aminophenol in presence of triethyl amine [50] to yield dihydroquinoxalin-2-ylamino derivative 7a and the benzo[b][1,4]oxazin- 3-ylamino derivative 7b; respectively. The reaction mechanism is believed to proceed through Schiff base formation between the chloroacetamido carbonyl function and the amino group of the selected reagent, followed by intramolecular cyclization via elimination of a hydrochloride molecule to afford the target compounds 7a and 7b. The 1H NMR spectra of both compounds revealed the presence of tautomerism as 1H NMR spectrum of 7a showed two singlets at δ 4.71 and δ 4.85 ppm attributed to quinoxalinyl-C3-CH2 and quinoxalinyl-C3-CH tautomers; respectively. In addition to a deuterium oxide exchangeable singlet at δ 8.14 ppm due to quinoxalinyl-N-H. Also, 1H NMR spectrum of compound 7b revealed a deuterium oxide exchangeable singlet at δ 4.41 ppm due to benzoxazine-N4-H tautomer. In addition to two singlets at δ 4.68 and δ 6.37 ppm corresponding to benzoxazine-C2-CH2 and benzoxazine-C2-CH tautomers; respectively.
6-Aminouracil 1 was further utilized for the synthesis of thiophene-2-carbonitrile derivative 9 “(Scheme 2)”. This was accomplished through the acetylation of compound 1 with acetic anhydride/glacial acetic mixture to yield 6-acetylaminouracil 8 [53] that was further subjected to Gewald reaction [54] [55] via its treatment with sulphur and malononitrile in dry dimethyl formamide which acted as a base and solvent at the same time. The synthesis of 3-aminothiophene-2-carbonitrile derivative 9 is assumed to proceed via Gewald reaction in which malononitrile condenses first with the acetamide ketone yielding a Knoevenagel-Cope condensation product which is then thiolated at the active methyl group with elemental sulphur followed by ring closure [55] (Figure 1). The electron impact mass spectrum of compound 9 showed the molecular ion peak at m/z 249 (10.42) and the base peak at m/z 58 (100). The reactive o-aminonitrile functions in compound 9 were subjected to cyclocondensation through treatment of compound 9 with formamide [55] to afford thienopyrimidine derivative 10. The IR spectrum of compound 10 was devoid of the absorption band due to cyano group of its precursor. However, its 1H NMR spectrum revealed a deuterium oxide exchangeable singlet at δ 5.88 ppm attributed to NH2
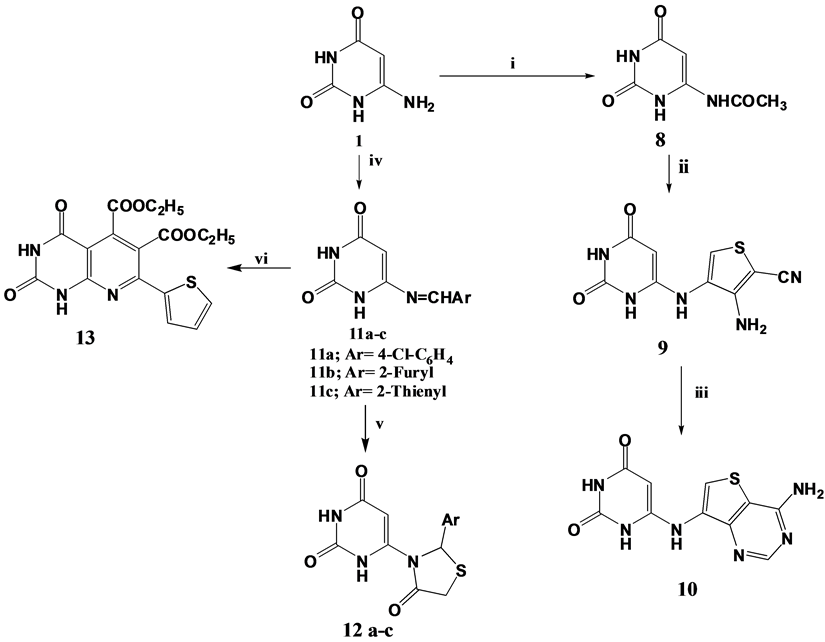
Scheme 2. Synthetic pathways for compounds 8, 9, 10, 11a-c, 12a-c, and 13. Reagents & conditions: i: (CH3CO)2O/gl.CH3COOH/steam bath; ii: malononitrile/sulphur/DMF/Reflux; iii: HCONH2/Reflux; iv: ArCHO/K2CO3/DMF/Reflux; v: HSCH2COOH/ZnCl2/Benzene/Reflux; vi: diethyl acetylene dicarboxylate/ Fusion.

Figure 1. Reaction mechanism for the synthetic route of compound 9.
protons. In addition to two singlets at δ 7.95 and δ 8.54 ppm corresponding to thienopyrmidine C6 & C2 protons; respectively. Moreover, 6-aminouracil 1 was reacted with aromatic aldehydes namely; 4-chlorobenzaldehyde, furan-2-carboxaldehyde and thiophene-2-carboxaldehyde in presence of anhydrous potassium carbonate to give the corresponding Schiff bases 11a-c [56] . It is worth mentioning that, in a previous report [57] the reaction of 6-aminouracil 1 with aromatic aldehydes was suggested to afford the corresponding condensation products 11 or 11’ (Figure 2). However, this report revealed that, the formation of the imino compound 11’ was not plausible as the imino group is much more nucleophilic than the CH in position 5 of 6-aminouracil. Furthermore, the prepared Schiff bases were used as key precursors for the synthesis of different compounds, in which the azomethine moiety underwent cycloaddition upon treatment with thioglycolic acid in presence of anhydrous zinc chloride [58] -[60] to yield 6-thiazolidinylpyrimidine derivatives 12a-c. The 1H NMR spectrum of compound 12c revealed the presence of two singlets at δ 3.65 and δ 4.37 ppm attributed to thiazolidine-C4-CH2 and thiazolidine-C2-CH protons; respectively. Aligned with the aim of the work, compound 11c was fused with diethyl acetylene dicarboxylate to yield the pyridopyrimidine derivative 13 that was prepared through cycloaddition of compound 11c on the triple bond of the reagent utilizing the methine protons of C5 uracil and the Schiff base. 1H NMR spectrum of compound 13 revealed the presence of two multiplets at δ 1.00 - 1.18 ppm and δ 3.60 - 3.90 ppm attributed to methyl and methylene protons of the two ethyl ester functions; respectively.
Diazotization of 6-aminouracil 1 was accomplished with sodium nitrite and hydrochloric acid at 0˚C - 5˚C which was followed by coupling with sodium azide to furnish 6-azidouracil 14 [61] “(Scheme 3)”. The azide function of compound 14 was coupled with malononitrile in basic medium followed by subsequent nucleophilic cycloaddition on one of the cyano functions [62] to afford the corresponding 5-amino-1,2,3-triazole-4-carbonitrile derivative 15. The IR spectrum of compound 15 lacked the absorption band attributed to azide group and displayed an absorption band at 2204 cm−1 corresponding to cyano group. Furthermore, its 1H NMR spectrum revealed the presence of three deuterium oxide exchangeable singlets at δ 7.28, δ 9.51 and δ 10.76 ppm attributed to triazole-NH2, uracil-N1-H and uracil-N3-H; respectively, while its electron impact mass spectrum exhibited its molecular ion peak at m/z 219 (5.14). Furthermore, the amino group in compound 1 was subjected to acylation with 2,4-dimethoxybenzoyl chloride in presence of piperidine as catalyst which afforded the 2,4-dimethoxybenzamide derivative 16. The 1H NMR spectrum of compound 16 revealed two singlets at δ 4.42 and δ 6.20 ppm attributed to methoxy protons and phenyl-C3 proton; respectively. In addition to a multiplet at δ 6.30 - 6.38 ppm due to phenyl-C5,6 protons.

Figure 2. Possible reaction products of compound 11.
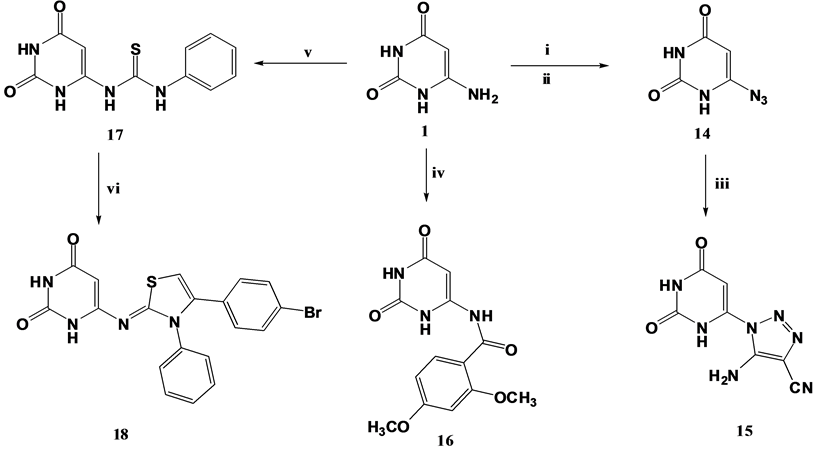
Scheme 3. Synthetic pathways for compounds 14, 15, 16, 17 and 18. Reagents & conditions: i: NaNO2/ HCl/0˚C; ii: NaN3/0˚C; iii: malononitrile/sod. ethoxide/Abs. ethanol/Reflux; iv: 2,4-di(OCH3)2C6H3COCl/ piperidine/DMF/Reflux; v: C6H5NCS/pyridine/Reflux; vi: 4-bromophenacyl bromide/Na acetate/Abs. ethanol/Reflux.
Moreover, the behavior of compound 1 towards isothiocyanate reagent was investigated to proceed typical to literature [63] [64] . Compound 1 was refluxed with phenyl isothiocyanate in pyridine to yield the acyclic thiourea derivative 17 which its 1H NMR spectrum revealed the presence of two deuterium oxide exchangeable singlets at δ 8.90 and δ11.63 ppm due to uracil-C6-NH and NH-phenyl protons; respectively. In addition to, other deuterium oxide exchangeable singlets due to uracil-N1 and N3 protons. In an attempt to fulfill the research goal, the thiourea derivative 17 was treated with 4-bromophenacyl bromide in absolute ethanol containing sodium acetate as a base to yield the thiazole derivative 18 that is believed to be formed through nucleophilic substitution reaction involving the reactive SH thiourea tautomer and 4-bromophenacyl bromide with elimination of a hydrobromide molecule followed by tautomerism and cyclocondensation (Figure 3) to yield compound 18 which its structure was established on its elemental and spectral data, as its 1H NMR spectrum revealed the presence of two doublets at δ 7.29 and δ 7.95 ppm due to C2,6 and C3,5 protons of 4-bromophenyl ring; respectively. In addition to, two singlets at δ 7.58 and δ 7.99 ppm attributed to thiazolidine C5 and uracil C5 protons; respectively.

Figure 3. Reaction mechanism for the synthetic route for compound 18.
Furthermore, compound 1 was further utilized to synthesize different substituted pyrimidopyrimidine derivatives 19, 20 and 21 “(Scheme 4)”. This was accomplished through refluxing compound 1 with ethanolamine and paraformaldehyde in absolute ethanol [65] to yield the tetrahydropyrimido[4,5-d]pyrimidinedione derivative 19. The IR spectrum of compound 19 showed strong absorption bands at 2949, 2916 and 2872 cm−1 due to aliphatic CH functions, while its 1H NMR spectrum exhibited two singlets at δ 3.96 and δ 4.85 ppm due to tetrahydropyrimidine C4 and C2 protons; respectively. In addition to, a deuterium oxide exchangeable singlet at δ 4.42 ppm due to OH proton. Moreover, formation of pyrimido[1,6-a]pyrimidine-2,6,8-trione derivative 20 was achieved through fusion of compound 1 with ethyl acetoacetate that was believed to be formed via Michael type addition of compound 1 on ethyl acetoacetate followed by ethanol elimination to give the acyclic intermediate 20’ which then underwent tautomerism and cyclization via elimination of a water molecule. The electron impact mass spectrum of compound 20 showed a peak at m/z 192 (0.34) corresponding to M-1, in addition to the base peak at m/z 60 (100). Also, 2-aminodihydropyrimido[1,6-a]pyrimidine-3-carbonitrile derivative 21 was synthesized through treatment of compound 1 with ethoxymethylene malononitrile [66] in absolute ethanol containing piperidine as catalyst. The reaction is suggested to proceed through Micheal type addition followed by elimination of an ethanol molecule to yield the acyclic intermediate which underwent intramolecular cyclization via nucleophilic addition on one cyano function and subsequent tautomerism leading to formation of compound 21 as demonstrated in (Figure 4). IR spectrum of compound 21 exhibited a strong absorption band at 2200 cm−1 due to cyano group. Besides, its 1H NMR spectrum revealed the presence of a deuterium oxide exchangeable singlet at δ 3.75 ppm due to NH2 protons. In addition to a singlet at δ 7.65 ppm due to pyrimidine C6 proton. Finally, compound 1 was refluxed with acetyl isothiocyanate in dry acetone [63] to yield the pyrimido[1,6-a][1,3,5]

Scheme 4. Synthetic pathways for compounds 19, 20, 21 and 22. Reagents & conditions: i: H2NCH2CH2OH/ HCHO/Abs. ethanol/Reflux; ii: CH3COCH2COOC2H5/fusion; iii: Ethoxymethylene malononitrile/piperidine/ Abs. ethanol/Reflux; iv: CH3COCl/NH4SCN/Acetone/Reflux.
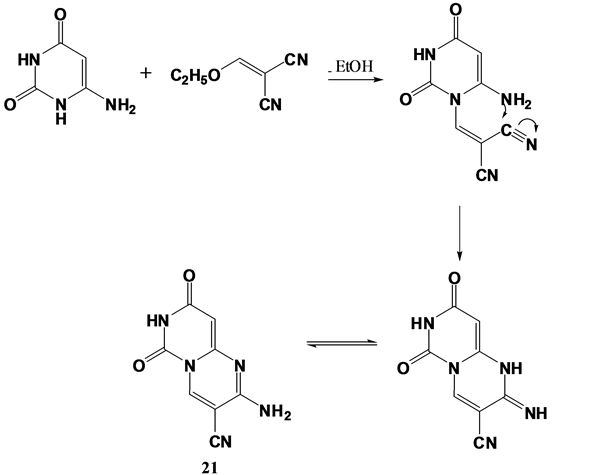
Figure 4. Reaction mechanism for the synthetic route for compound 21.
triazine derivative 22 that is assumed to be formed through formation of the thiourea intermediate 22’ that underwent tautomerism and subsequent cyclocondensation to yield compound 22. 1H NMR spectrum of compound 22 exhibited a singlet at δ 2.05 ppm due to methyl protons. In addition to, a deuterium oxide exchangeable singlet at δ 6.66 ppm attributed to triazine-N1 proton.
2.2. Biological Screening
2.2.1. Anticancer Screening
Cytotoxicity of all the synthesized compounds was evaluated against prostate cancer; PC3 cell line by using in-vitro sulforhodamine-B (SRB) method. The screening was performed by evaluation of the growth inhibition percent using five different doses from which their IC50 values were calculated. Also, we take doxorubicin as a reference drug in which its IC50 was 0.93 as presented in Table1

Table 1. Five doses growth inhibition percent and IC50 values of the tested compounds against PC3 cell line.
Aligned with the aim of this work; to synthesize and study the effect of various heterocyclic rings upon attachment to C6 position or fusion to the uracil backbone, the IC50 of 6-aminouracil 1 against PC3 cell line was evaluated which was found to be 362 μM. The attachment of a pyrimidine-2-thione ring through an amino bridge as in compounds 3a and 3c, greatly improved the anticancer activity as they exhibited IC50 43.95 and 79.20 μM; respectively. Also, chloroacetylation of 6-aminouracil as in compound 4 further improved the activity showing IC50 21.21 μM. However, attachment of different substituted furan rings through an amino bridge as in compounds 5a and 5b exhibited moderate anticancer activity with IC50 7.02 and 8.57 μM; respectively, compared to doxorubicin (IC50 0.93 μM), while direct attachment of a pyrrolidinone ring as in compound 6 showed IC50 38.73 μM.
Furthermore, attachment of a quinoxaline ring through an amino bridge as in compound 7a exhibited strong activity with IC50 2.31 μM, while replacing quinoxaline with 1,4-benzoxazine moiety as in 7b showed IC50 36.61 μM. Also, it is worth mentioning that attachment of a thiophene ring through an amino bridge as in compound 9 greatly diminished the activity showing IC50 547.40 μM which upon fusion to a pyrimidine ring to yield the thienopyrimidine derivative 10, the activity was slightly improved to exhibit IC50 146 μM. Moreover, the intermediate Schiff bases 11a-c showed moderate to low activity showing IC50 15.66 - 151 μM that upon cyclization to yield compounds 12a-c bearing a thiazolidinone ring directly attached to the uracil ring, showed enhancement in the activity of compounds 12a and 12b with IC50 8.84 and 29.54 μM; respectively, while compound 12c with a thiophene moiety attached to the thiazolidinone ring; exerted very weak activity with IC50 272.80 μM. However, cyclization of the Schiff base 11c into the pyridopyrimidine derivative 13 exhibited marked anticancer activity with IC50 1.89 μM which is nearly half potent to that of doxorubicin.
It is to be noted that, the presence of an azide function as in compound 14, a directly attached triazole ring as in compound 15 or a substituted benzamide side chain as in compound 16 attached to C6 position of uracil backbone, resulted in diminished anticancer activity. However, the presence of a phenyl thiourea side chain as in compound 17 exerted highly potent anticancer activity showing IC50 0.03 μM in comparison to doxorubicin IC50 0.93 μM, which upon cyclization to yield substituted thiazole derivative attached to the uracil ring through an imino bridge as in compound 18 resulted in slight decrease of anticancer activity with IC50 3.67 μM. Moreover, fusion of the uracil ring to perhydropyrimidine, 4-aminopyrimidine-5-carbonitrile and 2-thioxo[1,3,5]triazine rings as in compounds 19, 21 and 22; respectively, resulted in diminished anticancer activity, although that, fusion of pyrimidine moiety to the uracil backbone as in compound 20 exerted good anticancer activity with IC50 63.68 μM.
2.2.2. Cathepsin B Assay
The possible link between cathepsins and cancer and the role of cathepsin B in metastatic potential was postulated since many years [67] . Cathepsin B, either the mRNA or the protein, was often detected in higher amounts in malignant tumors than in benign ones or in normal tissues [68] . Moreover, a positive correlation between cathepsin B expression and metastasis of carcinoma cells to lymph nodes has been shown in prostate cancer [69] .
Current prostate cancer specific treatments are limited and development of new therapeutics is needed, specifically therapies against pathways that mediate the aggressiveness of the disease. A clinical approach based on targeting of proteases involved in pathomechanism of given diseases also stimulates the interest as anticancer strategy alternative, or supplementary, to surgical intervention and radiotherapy [70] . In various types of mouse cancer models deletion of cathepsin B led to suppression of the aggressiveness of the respective cancer phenotype. Moreover, delivery of broad spectrum cysteine cathepsin inhibitors in the tumor microenvironment disrupts the permissive ecosystem of the cancer and results in impaired growth or even in regression of the tumor [9] . A logical consequence of these results would be to further pursue selective inhibition of cathepsin B. In this investigation, the synthesized compounds exhibit a good inhibition of cathepsin B in vitro. Thus, we can hypothesize that this inhibition could be due to one or more of the mechanisms by which cathepsin B contribute to metastasis facilitating cell migration and invasiveness.
In general, loss of cell-cell and cell-matrix adhesion and degradation of extracellular matrix components are involved in invasion and migration [71] of cancerous cells. The tumor and metastasis-promoting effect of secreted cysteine cathepsins is thought to be caused by their ability to degrade extracellular matrix molecules, which in turn enables cancer cells to invade into the surrounding tissue and to metastasize [72] . In addition, proteases promote tumor growth by processing growth factors, cytokines, and chemokines or increasing their bioavailability by releasing them from the extracellular matrix [73] -[75] . Furthermore, they may play roles in the regulation of the action of certain growth factors, growth factor-binding proteins and growth factor receptors [13] , all are vital participants in human cancer growth. Therefore, proteases are functionally embedded in the complex proteolytic and cellular network [76] -[78] . Also, the localization of cysteine cathepsins on the tumor cell membrane might be mediated through an association of individual cysteine cathepsins with binding partners in membrane microdomains. The membrane microdomains include lipid rafts. The association on the tumor cell membrane of cathepsin B with caveolae, a subset of lipid rafts that contain the structural protein caveolin, is mediated by a direct interaction of procathepsin B with the light chain of the annexin II heterotetramer (AIIT) [10] .
Previous studies have implicated tumor cell cathepsin B in degradation of ECM proteins including fibronectin, laminin and collagen IV [79] [80] . In accordance to these studies, Cathepsin B has been shown to be implicated in the degradation of basement membrane proteins and invasion of the adjunct stroma in several experimental tumor models, and the over expression of cathepsin B shows a close association with disease progression in various kinds of human malignancy, including prostate cancer [81] . Active cathepsin B is also secreted from tumors, a mechanism likely to be facilitated by lysosomal exocytosis or extracellular processing by surface activators [82] .
Additionally, and in support of our study, inhibition of cathepsin B has been demonstrated to reduce collagen I degradation by prostate carcinoma cell lines [83] . Consistent with cathepsin B role in invasion, [84] demonstrated a critical function of tumor-derived cathepsin B in metastasis in the 4T1.2 model and revealed that preventing ECM degradation by cathepsin B inhibition is likely to reduce metastatic burden.
Therefore, the most active synthesized compounds against prostate PC3 cell line; 3a, 3c, 4, 5a, 5b, 6, 7a, 7b, 11a, 11b, 11c, 12a, 12b, 13, 17, 18 and 20 were further evaluated for their ability to inhibit cathepsin B by using enzyme-linked immunosorbent assay (ELISA) kit (Wuhan ElAab Science Co., Ltd, Wuhan, China), as shown in Table2
The presented results revealed that, all the tested compounds showed better cathepsin B inhibition than the
Table 2 . Growth inhibition percent of the selected compounds against cathepsin B enzyme.
reference drug doxorubicin (18.7%), except the 2-thioxopyrimidine derivative 3c. However, several compounds exhibited more than 50% inhibition of cathepsin B, among which are the furan derivatives 5a and 5b, quinoxaline derivative 7a, 4-chlorophenyl Schiff base derivative 11a and the thiazolidinone derivative 12a. In addition to, the phenyl thiourea derivative 17 which is the most active compound exerting 82.3% inhibition of cathepsin B.
In conclusion, our study reveals that these synthesized compounds have the ability to inhibit cathepsin B which is associated particularly with metastasis of prostate cancer and that inhibition could possibly lead to changes of tumor microenvironment and to decreased cell survival, therefore, to the impairment of metastatic seeding and onset. Further research on prostate cancer will however be crucial to generate therapeutic strategies based on the use of available or novel potential inhibitors of cathepsin B activity.
3. Experimental
3.1. Chemistry
All melting points were measured on Electro thermal LA 9000 SERIS, Digital Melting point Apparatus and are uncorrected. IR spectra (KBr) were recorded on FT-IR 200 spectrophotometer (ύ cm−1), pharmaceutical analytical unit, Faculty of Pharmacy, Al-Azhar University. 1H-NMR spectra were recorded in (DMSO-d6) at 300 MHz on a Varian Gemini NMR spectrometer (δ ppm) using TMS as an internal standard, Main defense chemical laboratory, Cairo. Mass spectra were recorded on GC Ms-QP 5050A mass spectrometer at 70 eV and microanalytical data were performed on Elementar Vario EI III CHN analyzer at the microanalytical unit, in Regional center for Mycology and Biotechnology, Al-Azhar University. Thin layer chromatography was performed on precoated (0.25mm) silica gel GF254 plates (E. Merck, Germany). Compounds were detected with 254 nm UV lamp.
3.1.1. General Method for Synthesis of Compounds 3a-c
A mixture of 6-aminouracil 1 (1.27 g, 10 mmol.) and 4-chlorodihydropyrimidine derivative 2a-c [48] (5 mmol) was refluxed in dry DMF (10 mL) containing piperidine (2 - 3 drops) for 6 hr. The reaction was triturated with water, the solid obtained was filtered, washed with water, dried then crystallized from DMF/H2O to yield compounds 3a-c.
1) 4-(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)-6-phenyl-2-thioxo-1,2-dihydro-pyrimidine-5-carbonitrile; 3a:
Light brown powder, yield 55%, m.p. < 300˚C; IR (KBr) cm−1: 3336, 3174 (NH); 3050 (CH-aromatic); 2210 (CN); 1729 (C=O); 1639 (C=N); 1521, 1386, 1165, 1017 (I, II, III, IV bands N-C=S). 1H NMR (DMSO-d6) δ ppm: 6.20 (s, 1 H, uracil-C6-NH, D2O exchangeable); 6.60 - 6.90 (m, 5 H, C6H5); 7.95 (s, 1 H, uracil-C5-H); 10.25 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.49 (s, 1 H, uracil-N3-H, D2O exchangeable); 10.61 (s, 1 H, pyrimidine-N1-H, D2O exchangeable). Anal. Calcd for C15H10N6O2S (338.34): C, 53.25; H, 2.98; N, 24.84. Found: C, 53.31; H, 3.04; N, 24.92.
2) 4-(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)-6-(4-bromophenyl)-2-thioxo-1,2-dihydropyrimidine- 5-carbonitrile; 3b:
Dark brown powder, yield, 63%, m.p. < 300˚C; IR (KBr) cm−1: 3419 (NH); 2922 (CH-aromatic); 2216 (CN); 1710 (C=O); 1627 (C=N); 1545, 1243, 1163, 1064 (I, II, III, IV bands N-C=S). 1H NMR (DMSO-d6) δ ppm: 6.17 (s, 1 H, uracil-C6-NH, D2O exchangeable); 7.12 - 7.40 (m, 2 H, 4-Br-C6H4-C2,6-H); 7.71 - 7.79 (m, 2 H, 4-Br-C6H4-C3,5-H); 8.57 (s, 1 H, uracil-C5-H); 10.04 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.56 (s, 2 H, uracil-N3-H & pyrimidine-N1-H, D2O exchangeable). Anal. Calcd for C15H9BrN6O2S (417.24): C, 43.18; H, 2.17; N, 20.14. Found: C, 43.17; H, 2.21; N, 20.18.
3) 4-(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)-6-(2-furyl)-2-thioxo-1,2-dihydro-pyrimidine-5-carbonitrile; 3c:
Light orange crystals, yield, 55%, m.p. < 300˚C; IR (KBr) cm−1: 3451, 3152 (NH); 3048 (CH-aromatic); 2219 (CN); 1735, 1670 (two C=O); 1632 (C=N); 1578, 1321, 1133, 1035 (I, II, III, IV bands N-C=S); 1222, 1078 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 6.14 (s, 1 H, uracil-C6-NH, D2O exchangeable); 6.88 (dd, 1 H, J = 3.3, 1.5 Hz, furyl-C4-H); 7.43 (d, 1 H, J = 3.3 Hz, furyl-C5-H); 7.76 (d, 1 H, J = 3.3 Hz, furyl-C3-H); 8.19 (s, 1 H, uracil-C5-H); 10.01 (s, 1 H, uracil-N1-H, D2O exchangeable); 11.73 (s, 1 H, uracil-N3-H, D2O exchangeable); 11.92 (s, 1 H, pyrimidine-N1-H, D2O exchangeable). Anal. Calcd for C13H8N6O3S (328.31): C, 47.56; H, 2.46; N, 25.60. Found: C, 47.69; H, 2.52; N, 25.74.
3.1.2. Synthesis of 2-Chloro-N-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)acetamide; 4
A mixture of 6-aminouracil 1 (1.27 g, 10 mmol.), chloroacetyl chloride (1.13 g, 0.8 mL, 10 mmol.) and anhydrous potassium carbonate (1.38 g, 10 mmol.) was stirred at room temperature for 24 hr. The reaction mixture was triturated with water, dried and crystallized from DMF/H2O.
Canary yellow crystals, yield, 75%, m.p. < 300˚C; IR (KBr) cm−1: 3393, 3259 (NH); 3093 (CH-aromatic); 2865 (CH-aliphatic); 1734, 1651 (two C=O); 1528 (C=C); 778 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 4.85 (s, 2 H, CH2Cl); 7.42 (s, 1 H, uracil-C6-NH, D2O exchangeable); 7.95 (s, 1 H, uracil-C5-H); 9.74 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.76 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C6H6ClN3O3 (203.58): C, 35.40; H, 2.97; N, 20.64. Found: C, 35.43; H, 3.02; N, 20.85.
3.1.3. General Method for Synthesis of Compounds 5a, b
To an ethanolic solution of sodium ethoxide [prepared from sodium metal (0.05 g, 2.2 mmol.) and 15 mL absolute ethanol] was added (5 mmol.) of the appropriate activated methylene bearing compounds namely; acetyl acetone and dimethyl malonate. After stirring for 15 min. at room temperature, compound 4 (0.2 g, 1 mmol.) was added and stirring was continued for 24 hr. The solvent was evaporated under reduced pressure and the remainder was poured onto ice cold water (25 mL) and neutralized with diluted HCl. The solid product was collected by filtration, washed with water, dried and crystallized from DMF/H2O.
1) 6-(4-Acetyl-5-methylfuran-2-ylamino)pyrimidine-2,4(1H,3H)dione; 5a Light green crystals, yield, 86%, m.p. < 300 ˚C; IR (KBr) cm−1: 3357, 3191 (NH); 2940 (CH-aliphatic); 1710 (C=O); 1561 (C=C); 1286, 1094 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 1.62 (s, 3 H, furyl-C5-CH3); 4.28 (s, 1 H, furyl-C3-H); 4.39 (s, 3 H, COCH3); 6.30 (s, 1 H, uracil-C6-NH, D2O exchangeable); 8.40 (s, 1 H, uracil-C5-H); 9.05 (s, 1 H, uracil-N1-H, D2O exchangeable); 9.49 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C11H11N3O4 (249.22): C, 53.01; H, 4.45; N, 16.86. Found: C, 52.98; H, 4.50; N, 16.97.
2) Ethyl 5-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)-2-ethoxyfuran-3-carboxylate; 5b Pale yellow crystals, yield, 38%, m.p. < 300˚C; IR (KBr) cm−1: 3358 (NH); 3000 (CH-aromatic); 2936 (CH-aliphatic); 1730 (C=O); 1563 (C=C); 1294, 1099 (C-O-C). MS (m/z, %): 309 (M+, 2.40); 55 (100). Anal. Calcd for C13H15N3O6 (309.27): C, 50.49; H, 4.89; N, 13.59. Found: C, 50.52; H, 4.91; N, 13.68.
3.1.4. Synthesis of 2-Amino-1-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-5-oxo-4,5-dihydro-1Hpyrrole-3-carbonitrile; 6
An equimolar mixture of the 6-chloroacetylaminouracil 4 (2.04 g, 10 mmol.) and malononitrile (0.66 g, 10 mmol.) was fused for 20 min. The reaction mixture was triturated with ethanol and the product was collected, washed with ethanol, dried then crystallized from DMF/H2O.
Light brown powder, yield, 74%, m.p. < 300˚C; IR (KBr) cm−1: 3380, 3320, 3180 (NH2, NH); 3002 (CH-aromatic); 2200 (CN); 1720 (C=O); 1530 (C=C). MS (m/z, %): 234 (M+1, 3.84); 64 (100). Anal. Calcd for C9H7N5O3 (233.18): C, 46.36; H, 3.03; N, 30.03. Found: C, 46.41; H, 3.05; N, 30.14.
3.1.5. General Method for Synthesis of Compounds 7a, b
To a suspension of compound 4 (2.04 g, 10 mmol.) in absolute ethanol (30 mL), an equimolar amount of the appropriate aromatic amine; namely o-phenylene diamine and o-aminophenol, two drops of triethyl amine were added and the suspension was heated under reflux for 12 hr., then left to cool. The solid product was collected by filtration, washed with ethanol, dried and crystallized from DMF/H2O.
1) 6-(3,4-Dihydroquinoxalin-2-ylamino)pyrimidine-2,4(1H,3H)-dione; 7a Light green crystals, yield, 44%, m.p. < 300˚C; IR (KBr) cm−1: 3364, 3178 (NH); 2922 (CH-aromatic); 1707 (C=O); 1618 (C=N); 1506 (C=C). 1H NMR (DMSO-d6) δ ppm: 4.71 (s, 1 H, quinoxalinyl-C3-CH2 tautomer); 4.85 (s, 1 H, quinoxalinyl-C3-C=CH tautomer); 7.35 (s, 1 H, uracil-C6-NH, D2O exchangeable); 7.62 - 7.64 (m, 2 H, quinoxalinyl-C5,8-H); 7.67 (s, 1 H, uracil-C5-H); 7.75 (t, 1 H, J=7.73 Hz, quinoxalinyl-C6-H); 7.97 (t, 1 H, J = 7.73 Hz, quinoxalinyl-C7-H); 8.14 (s, 1 H, quinoxalinyl-N-H, D2O exchangeable); 10.65 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.80 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C12H11N5O2 (257.25): C, 56.03; H, 4.31; N, 27.22. Found: C, 56.05; H, 4.37; N, 27.29.
2) 6-(2H-Benzo[b][1,4]oxazin-3-ylamino)pyrimidine-2,4(1H,3H)-dione; 7b Dark orange crystals, yield, 63%, m.p. < 300˚C; IR (KBr) cm−1: 3408, 3177 (NH); 2921 (CH-aromatic); 1711 (C=O); 1626 (C=N); 1460 (C=C); 1287, 1051 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 4.41 (s, 1/2 H, benzoxazine-N4-H tautomer, D2O exchangeable); 4.68 (s, 1 H, benzoxazine-C2-CH2); 6.22 (s, 1 H, uracil-C6-NH, D2O exchangeable); 6.37 (s, 1/2 H, benzoxazine-C2-C=CH-O tautomer); 7.30 - 7.75 (m, 4 H, benzoxazine-C5,6,7,8-H); 8.25 (s, 1 H, uracil-C5-H); 10.08 (s, 2 H, uracil-N1-H & N3-H, D2O exchangeable). Anal. Calcd for C12H10N4O3 (258.23): C, 55.81; H, 3.90; N, 21.70. Found: C, 55.87; H, 3.89; N, 21.81.
3.1.6. Synthesis of 3-Amino-4-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-ylamino)thiophene-2- carbonitrile; 9
An equimolar mixture of 6-acetylaminouracil 8 [53] , malononitrile and sulphur (2 mmol.) was refluxed in dry DMF (15 mL) for 6 hr. The reaction mixture was concentrated, allowed to cool then triturated with cold water to yield the target compound which was filtered, washed with water, dried and crystallized from DMF/H2O.
Chocolate brown crystals, yield, 46%, m.p. < 300˚C; IR (KBr) cm−1: 3359, 3186 (NH); 3035 (CH-aromatic); 2210 (CN); 1716 (C=O); 1513 (C=C); 1267, 1052 (C-S-C). MS (m/z, %): 249 (M+., 10.42); 58 (100). Anal. Calcd for C9H7N5O2S (249.25): C, 43.37; H, 2.83; N, 28.10. Found: C, 43.40; H, 2.87; N, 28.17.
3.1.7. Synthesis of 6-(4-aminothieno[3,2-d]pyrimidin-7-ylamino)pyrimidine-2,4(1H, 3H)-dione; 10
Compound 9 (0.2 g, 5 mmol) was refluxed in excess formamide (10 mL) for 5 hr. The reaction mixture was concentrated under reduced pressure then poured onto crushed ice. The obtained product was filtered, washed with water, dried and crystallized from DMF/H2O.
Dark brown crystals, yield, 45%, m.p. < 300˚C; IR (KBr) cm−1: 3359, 3182 (NH2, NH); 2921 (CH-aromatic); 1688 (C=O); 1632 (C=N); 1510 (C=C); 1290, 1044 (C-S-C). 1H NMR (DMSO-d6) δ ppm: 5.88 (s, 2 H, NH2, D2O exchangeable); 7.37 (s, 1 H, uracil-C6-NH, D2O exchangeable); 7.83 (s, 1 H, uracil-C5-H); 7.95 (s, 1 H, thienopyrimidine-C6-H); 8.54 (s, 1 H, thienopyrimidine-C2-H); 10.14 (s, 2 H, uracil-N1-H & N3-H, D2O exchangeable). Anal. Calcd for C10H8N6O2S (276.27): C, 43.47; H, 2.92; N, 30.42; S, 11.61. Found: C, 43.53; H, 2.97; N, 30.57; S, 11.69.
3.1.8. General Method for Synthesis of Compounds 11a-c
A mixture of 6-aminouracil 1 (1.27 g, 10 mmol.) and the appropriate aromatic aldehyde (20 mmol.) namely; 4-chlorobenzaldehyde, furan-2-carboxaldehyde, thiophen-2-carboxaldehyde, in presence of anhydrous potassium carbonate (1.38 g, 10 mmol.) was refluxed in dry DMF (25 mL) for 5 hr. The reaction mixture was allowed to cool, then poured onto crushed ice to yield a solid product that was collected by filtration. The obtained product was washed with ice-cold water, dried then crystallized from DMF/H2O.
1) 6-(4-Chlorobenzylideneamino)pyrimidine-2,4(1H,3H)-dione; 11a Yellow crystals, yield, 75%, m.p. < 300˚C [56] ; IR (KBr) cm−1: 3438, 3150 (NH); 3088 (CH-aromatic); 2921, 2855 (CH-aliphatic); 1715 (C=O); 1625 (C=N); 1490 (C=C); 831 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 7.10 (s, 1 H, arylidine-CH); 7.19 (d, 2 H, J = 8.4 Hz, 4-Cl-C6H4-C2,6-H); 7.26 (d, 2 H, J = 8.4 Hz, 4-Cl-C6H4-C3,5-H); 7.95 (s, 1 H, uracil-C5-H); 9.13 (s, 1 H, uracil-N1-H, D2O exchangeable); 9.89 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C11H8ClN3O2 (249.65): C, 52.92; H, 3.23; N, 16.83. Found: C, 52.94; H, 3.23; N, 16.91.
2) 6-(Furan-2-ylmethyleneamino)pyrimidine-2,4(1H,3H)-dione; 11b Dark brown powder, yield, 35%, m.p. < 300˚C [56] ; IR (KBr) cm−1: 3413, 3143 (NH); 3050 (CH-aromatic); 2924, 2856 (CH-aliphatic); 1718 (C=O); 1625 (C=N); 1495 (C=C); 1294, 1064 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 4.85 (s, 1 H, furyl-C4-H); 5.89 (s, 1 H, furyl-C3-H); 6.21 (s, 1 H, furyl-C5-H); 7.30 (s, 1 H, arylidine-CH); 7.95 (s, 1 H, uracil-C5-H); 9.86 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.22 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C9H7N3O3 (205.17): C, 52.69; H, 3.44; N, 20.48. Found: C, 52.67; H, 3.49; N, 20.54.
3) 6-(Thiophen-2-ylmethyleneamino)pyrimidine-2,4(1H,3H)-dione; 11c Dark yellow powder, yield, 64%, m.p. < 300˚C [56] ; IR (KBr) cm−1: 3450, 3150 (NH); 3091 (CH-aromatic); 2923, 2857 (CH-aliphatic); 1717 (C=O); 1636 (C=N); 1543, 1495 (C=C); 1294, 1099 (C-S-C). 1H NMR (DMSO-d6) δ ppm: 7.08 (s, 1 H, arylidine-CH); 7.17 - 7.20 (m, 1 H, J = 5.4 Hz, thienyl-C4-H); 7.50 (d, 1 H, J = 5.4 Hz, thienyl-C5-H); 7.72 (d, 1 H, J = 5.4 Hz, thienyl-C3-H); 7.96 (s, 1 H, uracil-C5-H); 9.03 (s, 1 H, uracilN1-H, D2O exchangeable); 9.96 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C9H7N3O2S (221.24): C, 48.86; H, 3.19; N, 18.99. Found: C, 49.02; H, 3.22; N, 19.12.
3.1.9. General Method for Synthesis of Compounds 12a-c
The appropriate Schiff base 11a-c (2 mmol.) was refluxed with thioglycolic acid (0.37 g, 0.28 mL, 4 mmol.) containing anhydrous zinc chloride (0.27 g, 2 mmol.) in dry benzene (20 mL) for 30 hr. The reaction mixture was cooled and the solid product was collected, washed with benzene and crystallized from DMF/H2O.
1) 6-[2-(4-Chlorophenyl)-4-oxothiazolidin-3-yl]pyrimidine-2,4(1H,3H)-dione; 12a Dark brown powder, yield, 60%, m.p. 230˚C [58] ; IR (KBr) cm−1: 3464 (OH tautomer); 3205 (NH); 3091 (CH-aromatic); 2923 (CH-aliphatic); 1700 (C=O); 1565 (C=C); 1380, 1021 (C-S-C). 1H NMR (DMSO-d6) δ ppm: 3.65 (s, 2 H, thiazolidine-C4-CH2); 4.93 (s, 1 H, thiazolidine-C2-CH); 5.11 (s, 1 H, uracil-C5-H); 7.09 (d, 2 H, J = 8.6 Hz, 4-Cl-C6H4-C2,6-H); 7.33 (d, 2 H, J = 8.6 Hz, 4-Cl-C6H4-C3,5-H); 11.10 (s, 1 H, uracil-N1-H, D2O exchangeable); 11.80 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C13H10ClN3O3S (323.75): C, 48.23; H, 3.11; N, 12.98; S, 9.90. Found: C, 48.29; H, 3.09; N, 13.13; S, 9.97.
2) 6-[2-(Furan-2-yl)-4-oxothiazolidin-3-yl]pyrimidine-2,4(1H,3H)-dione; 12b Dark brown powder, yield, 34%, m.p. < 300˚C [58] ; IR (KBr) cm−1: 3200 (NH); 3090 (CH-aromatic); 2919, 2861 (CH-aliphatic); 1701 (C=O); 1534 (C=C); 1395, 1050 (C-S-C); 1262, 1050 (C-O-C). MS. (m/z, %): 279 (M+, 0.18); 278 (M+-1, 0.27); 71 (100). Anal. Calcd for C11H9N3O4S (279.27): C, 47.31; H, 3.25; N, 15.05; S, 11.48. Found: C, 47.43; H, 3.29; N, 15.19; S, 11.62.
3) 6-[4-Oxo-2-(thiophen-2-yl)thiazolidin-3-yl]pyrimidine-2,4(1H,3H)-dione; 12c Light brown powder, yield, 36%, m.p. < 300˚C; IR (KBr) cm−1: 3163 (NH); 3039 (CH-aromatic); 2843 (CH-aliphatic); 1697 (C=O); 1453 (C=C); 1386, 1071 (C-S-C). 1H NMR (DMSO-d6) δ ppm: 3.65 (s, 2 H, thiazolidine-C4-CH2); 4.37 (s, 1 H, thiazolidine-C2-CH); 4.66 (d, 1 H, J = 6 Hz, thienyl-C3-H); 4.92 - 5.11 (m, 1 H, thienyl-C4-H); 5.56 (d, 1 H, J = 6 Hz, thienyl-C5-H); 8.53 (s, 1 H, uracil-C5-H); 11.53 (s, 1 H, uracil-N1-H, D2O exchangeable); 11.92 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C11H9N3O3S2 (295.34): C, 44.73; H, 3.07; N, 14.23; S, 21.71. Found: C, 44.85; H, 3.12; N, 14.51; S, 21.84.
3.1.10. Synthesis of Diethyl 2,4-Dioxo-7-(thiophen-2-yl)-1,2,3,4-tetrahydropyrido[2,3-d] pyrimidine-5,6-dicarboxylate; 13
The Schiff base derivative 11c (0.3 g, 1 mmol.) was fused with excess diethyl acetylene dicarboxylate (1 mL) for 1 hr. The reaction mixture was allowed to cool then triturated with ethanol. The obtained product was filtered, washed with ethanol, left to dry then crystallized from DMF/H2O.
Light brown powder, yield, 43%, m.p. charrs at 280˚C; IR (KBr) cm−1: 3210, 3120 (NH); 3030 (CH-aromatic); 2925, 2812 (CH-aliphatic); 1718 (C=O); 1616 (C=N); 1495 (C=C); 1375, 1067 (C-S-C); 1293, 1261, 1067, 1033 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 1.00 - 1.18 (m, 6 H, two CH3); 3.60 - 3.90 (m, 4 H, two CH2); 6.70 - 6.90 (m, 1 H, thienyl-C4-H); 7.09 (d, 1 H, J = 5.4 Hz, thienyl-C3-H); 7.90 - 8.02 (m, 1 H, thienyl-C5-H); 10.03 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.20 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C17H15N3O6S (389.38): C, 52.44; H, 3.88; N, 10.79. Found: C, 52.58; H, 3.92; N, 11.02.
3.1.11. Synthesis of 6-Azidopyrimidine-2,4(1H, 3H)-dione; 14
To an ice cold mixture of 6-aminouracil 1 (0.5 g, 4 mmol.), water (10 mL) and concentrated hydrochloric acid (3 mL), sodium nitrite solution (0.27 g, 4 mmol. in 1 mL H2O) was added drop wise; while stirring at 0˚C. Stirring was continued for 15 min., then an ice cold aqueous solution of sodium azide (0.26 g, 4 mmol.) was added drop wise. The reaction mixture was stirred for another 30 min. then filtered to yield purple colored powder of compound 13.
Purple powder, yield, 72%, m.p. charrs at 240˚C without melting [61] ; IR (KBr) cm−1: 3430 (OH tauotomer); 3292, 3182 (two NH); 3015 (CH-aromatic); 2150 (N3); 1672 (C=O); 1512, 1428 (N=N); 1543 (C=C). MS. (m/z, %): 153 (M+, 1.81); 71 (100). Anal. Calcd for C4H3N5O2 (153.10): C, 31.38; H, 1.98; N, 45.74. Found: C, 31.42; H, 2.02; N, 45.81.
3.1.12. Synthesis of 5-Amino-1-(2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-1H-1,2,3-triazole- 4-carbonitrile; 15
Compound 14 ( 0.3 g, 1.96 mmol.) was refluxed with malononitrile (0.26 g, 3.92 mmol.) in an ethanolic solution of sodium ethoxide (0.1 g, 1.96 mmol.) for 12 hr. The reaction mixture was filtered while hot and the filtrate was concentrated and allowed to cool to yield compound 15 which was collected, washed with ethanol and crystallized from ethanol.
Dark orange crystals, yield, 81%, m.p. < 300˚C. IR (KBr) cm−1: 3450, 3334 (NH2, NH); 2204 (CN); 1710 (C=O); 1646 (C=N). 1H NMR (DMSO-d6) δ ppm: 7.28 (s, 2 H, NH2, D2O exchangeable); 8.50 (s, 1 H, uracil-C5-H); 9.51 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.76 (s, 1 H, uracil-N3-H, D2O exchangeable). MS. (m/z, %): 219 (M+, 5.14); 92 (100). Anal. Calcd for C7H5N7O2 (219.16): C, 38.36; H, 2.30; N, 44.74. Found: C, 38.45; H, 2.36; N, 44.89.
3.1.13. Synthesis of N-(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-2,4-dimethoxybenzamide; 16
An equimolar mixture of 6-aminouracil 1 (0.2 g, 1.57 mmol.) and 2,4-dimethoxybenzoyl chloride (0.32 g, 1.57 mmol.) was refluxed in dry DMF (10 mL) containing 2 - 3 drops of piperidine for 6 hr. The reaction mixture was concentrated then allowed to cool. The separated product was collected, washed with ethanol then crystallized from DMF/H2O.
Light brown crystals, yield, 32%, m.p. < 300˚C. IR (KBr) cm−1: 3325, 3175 (NH); 3018 (CH-aromatic); 2924, 2850 (CH-aliphatic); 1729 (C=O); 1632 (C=N); 1455 (C=C); 1290, 1088 (C-O-C). 1H NMR (DMSO-d6) δ ppm: 4.42 (s, 6 H, two CH3); 6.20 (s, 1 H, C6H3-C3-H); 6.30 - 6.38 (m, 2 H, dimethoxy-C6H3-C5,6-H); 6.72 (s, 1 H, uracil-C6-NH, D2O exchangeable); 7.95 (s, 1 H, uracil-C5-H); 10.15 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.43 (s, 1 H, uracil-N3-H, D2O exchangeable). MS. (m/z, %): 219 (M+, 4.16); 65 (100). Anal. Calcd for C13H13N3O5 (291.26): C, 53.61; H, 4.50; N, 14.43. Found: C, 54.13; H, 3.91; N, 14.72.
3.1.14. Synthesis of 1-(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-3-phenylthiourea; 17
An equimolar mixture of 6-aminouracil 1 (0.5 g, 4 mmol.) and phenyl isothiocyanate (0.53 g, 0.4 mL, 4 mmol.) was refluxed in dry pyridine (10 mL) for 60 hr. The reaction mixture was allowed to cool, poured onto crushed ice then neutralized by using 10% hydrochloric acid to yield the desired product. The product was filtered, washed with water, dried and crystallized from DMF/H2O.
Dark yellow crystals, yield, 55%, m.p. 105˚C. IR (KBr) cm−1: 3400, 3195 (NH); 3035 (CH-aromatic); 1680 (C=O); 1538, 1328, 1244, 1050 (I, II, III, IV bands N-C=S). 1H NMR (DMSO-d6) δ ppm: 7.10 - 7.21 (m, 1 H, C6H5-C4-H); 7.23 - 7.47 (m, 2 H, C6H5-C2,6-H); 7.53 (d, 2 H, J = 8.1 Hz, C6H5-C3,5-H); 8.54 (s, 1 H, uracil-C5-H); 8.90 (s, 1 H, uracil-C6-NH, D2O exchangeable); 10.01 (s, 1 H, uracil-N1-H, D2O exchangeable); 11.16 (s, 1 H, uracil-N3-H, D2O exchangeable); 11.63 (s, 1 H, C6H5-NH, D2O exchangeable). Anal. Calcd for C11H10N4O2S (260.27): C, 50.37; H, 3.84; N, 21.36. Found: C, 50.49; H, 3.82; N, 21.52.
3.1.15. Synthesis of 6-[4-(4-Bromophenyl)-3-phenylthiazol-2(3H)-ylideneamino]pyrimidine- 2,4(1H, 3H)-dione; 18
An equimolar mixture of thiourea derivative 17 (0.26 g, 1 mmol.) and 4-bromophenacyl bromide (0.28 g, 1 mmol.) was refluxed in absolute ethanol (30 mL) containing sodium acetate (1 g, 12 mmol.) for 12 hr. The reaction mixture was allowed to cool then filtered. The filtrate was concentrated then diluted with ice cold water. The obtained product was filtered, washed with water, dried and crystallized from DMF/H2O.
Dark yellow crystals, yield, 40 %, m.p. charrs at 290˚C. IR (KBr) cm−1: 3186 (NH); 3058, 3031 (CH-aromatic); 1705 (C=O); 1579 (C=N); 1484 (C=C); 1389, 1068 (C-S-C). 1H NMR (DMSO-d6) δ ppm: 6.96 - 7.06 (m, 1 H, C6H5-C4-H); 7.29 (d, 2 H, J = 8.4 Hz, 4-Br-C6H4-C2,6-H); 7.42 - 7.49 (m, 2 H, C6H5-C3,5-H); 7.58 (s, 1 H, thiazolidine-C5-H); 7.68 - 7.86 (m, 2 H, C6H5-C2,6-H); 7.95 (d, 2 H, J = 8.4 Hz, 4-Br-C6H4-C3,5-H); 7.99 (s, 1 H, uracil-C5-H); 11.10 (s, 1 H, uracil-N1-H, D2O exchangeable); 11.84 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C19H13BrN4O2S (441.30): C, 51.71; H, 2.97; N, 12.70. Found: C, 51.84; H, 3.01; N, 12.84.
3.1.16. Synthesis of 6-(2-Hydroxyethyl)-5,6,7,8-tetrahyropyrimido[4,5-d]pyrimidine-2, 4(1H, 3H)- dione; 19
A mixture of ethanolamine (0.31 g, 0.31 mL, 5 mmol) and paraformaldehyde (0.84 g, 7 mmol.) was added to a suspension of 6-aminouracil 1 ( 0.39 g, 3 mmol.) in absolute ethanol (20 mL). The reaction mixture was heated under reflux for 12 hr. then allowed to attain room temperature. The obtained precipitate was filtered, washed with ethanol, dried and crystallized from DMF/H2O.
White powder, yield, 25%, m.p. 280˚C. IR (KBr) cm−1: 3350 (OH); 3208, 3126 (NH); 2949, 2916, 2872 (CH-aliphatic); 1704 (C=O); 1585 (C=C). 1H NMR (DMSO-d6) δ ppm: 3.40 - 3.62 (m, 4 H, N-CH2-CH2-OH); 3.96 (s, 2 H, tetrahydropyrimidine-C4-H); 4.42 (s, 1 H, OH, D2O exchangeable); 4.85 (s, 2 H, tetrahydropyrimidine-C2-H); 6.38 (s, 1 H, tetrahydropyrimidine-N1-H, D2O exchangeable); 9.81 (s, 1 H, uracil-N1-H, D2O exchangeable); 10.05 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C8H12N4O3 (212.21): C, 45.28; H, 5.70; N, 26.40. Found: C, 45.79; H, 4.85; N, 26.83.
3.1.17. Synthesis of 4-Methyl-1H-pyrimido[1,6-a]pyrimidine-2,6,8(7H)-trione; 20
An equimolar mixture of 6-aminouracil 1 (0.50 g, 4 mmol.) and ethyl acetoacetate (0.51 g, 0.5 mL, 4 mmol.) was fused for 6 hr. The reaction mixture was triturated with ethanol, filtered and dried. The obtained product was crystallized from DMF/H2O.
Pale yellow crystals, yield, 52%, m.p. < 300˚C. IR (KBr) cm−1: 3439 (broad OH tautomer); 3280, 3160 (NH); 2950 (CH-aliphatic); 1700 (C=O); 1640 (C=N); 1561 (C=C). MS. (m/z, %): 192 (M-1, 0.34); 60 (100). Anal. Calcd for C8H7N3O3 (193.16): C, 49.74; H, 3.65; N, 21.75. Found: C, 49.88; H, 3.71; N, 21.92.
3.1.18. Synthesis of 2-Amino-6,8-dioxo-7,8-dihydro-6H-pyrimido[1,6-a]pyrimidine-3-carbonitrile; 21
An equimolar mixture of compound 1 (0.50 g, 4 mmol.) and 2-(ethoxymethylene)malononitrile [66] (0.48 g, 4 mmol.) was refluxed in absolute ethanol (20 mL) containing a catalytic amount of piperidine (2 - 3 drops) for 12 hr. The reaction mixture was left to cool and the obtained product was filtered, washed with ethanol and crystallized from DMF/H2O.
Yellow crystals, yield, 45%, m.p. 98˚C. IR (KBr) cm−1: 3374, 3197 (NH2, NH); 3083, 3022 (CH-aromatic); 2220 (CN); 1725 (C=O); 1630 (C=N); 1460 (C=C). 1H NMR (DMSO-d6) δ ppm: 3.75 (s, 2 H, NH2, D2O exchangeable); 7.65 (s, 1 H, pyrimidine-C6-H); 7.95 (uracil-C5-H); 10.79 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C8H5N5O2 (203.16): C, 47.30; H, 2.48; N, 34.47. Found: C, 47.46; H, 2.45; N, 34.61.
3.1.19. Synthesis of 4-Methyl-2-thioxo-1H-pyrimido[1,6-a][1,3,5]triazine-6,8(2H,7H)-dione; 22
To a solution of acetyl isothiocyanate (1.01 g, 10 mmol.) [prepared by refluxing acetyl chloride ( 0.79 g, 0.72 mL, 10 mmol.) with ammonium thiocyanate (0.76 g, 10 mmol.) in dry acetone] in dry acetone (50 mL), compound 1 (1.27 g, 10 mmol.) was added. The reaction was refluxed for 12 hr., allowed to cool then filtered. The filtrate was concentrated and the obtained product was collected then crystallized from acetone.
Orange crystals, yield, 30%, m.p. charrs at 215˚C. IR (KBr) cm−1: 3395, 3312 (NH); 3070 (CH-aromatic); 2917, 2850 (CH-aliphatic); 1699 (C=O); 1640 (C=N); 1543 (C=C); 1515, 1367, 1168, 1052 (I, II, III, IV bands N-C=S). 1H NMR (DMSO-d6) δ ppm: 2.05 (s, 3 H, CH3); 6.66 (s, 1 H, triazine-N1-H, D2O exchangeable); 8.36 (s, 1 H, uracil-C5-H); 11.06 (s, 1 H, uracil-N3-H, D2O exchangeable). Anal. Calcd for C7H6N4O2S (210.21): C, 40.00; H, 2.88; N, 26.65. Found: C, 40.13; H, 2.94; N, 26.84.
3.2. Biological Screening
3.2.1. Anticancer Screening
1) Cell culture PC3 human prostate cancer cells were grown in RPMI-1640 medium, supplemented with 10% heat inactivated FBS, 100 units/mL of penicillin and 100 mg/mL of streptomycin and maintained at 37˚ in a humidified atmosphere containing 5% CO2. The cells were maintained as “monolayer culture” by serial subculturing.
2) SRB cytotoxicity assay Cytotoxicity was determined using SRB method as previously described by Skehan et al. [85] . Exponentially growing cells were collected using 0.25% Trypsin-EDTA and seeded in 96-well plates at 1000 - 2000 cells/well in RPMI-1640 supplemented medium. After 24 h, cells were incubated for 72 h with various concentrations of the tested compounds. Following 72 h treatment, the cells will be fixed with 10% trichloroacetic acid for 1 h at 4˚C. Wells were stained for 10 min at room temperature with 0.4% SRB dissolved in 1% acetic acid. The plates were air dried for 24 h and the dye was solubilized with Tris-HCl for 5 min on a shaker at 1600 rpm. The optical density (OD) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (ChroMate-4300, FL, USA). The IC50 values were calculated according to the equation for Boltzman sigmoidal concentration-response curve using the nonlinear regression fitting models (Graph Pad, Prism Version 5).
3.2.2. Cathepsin B Assay
1) Cell culture PC3 human prostate cancer cells were grown in RPMI-1640 medium supplemented with 10% heat inactivated FBS, 100 units/mL of penicillin and 100 mg/mL of streptomycin and maintained at 37˚ in a humidified atmosphere containing 5% CO2. Exponentially growing cells were seeded in 6-well plates at (4 × 105) cells/well in RPMI-1640 supplemented medium. After 24 h, cells were incubated for 24 h with the tested compounds at their median inhibitory concentrations (IC50). Then, cells were washed with 1x PBS, scraped, homogenized in ice-cold PBS using Dounce homogenizer and stored overnight at −20˚C. After two freeze-thaw cycles, the cells were centrifuged at 5000 × g for 5 minutes. Supernatants were collected and assayed immediately.
2) ELISA assay Cathepsin-B (CTS-B) was quantified in the collected samples using enzyme-linked immunosorbent assay kit (Wuhan ElAab Science Co., Ltd, Wuhan, China) according to the manufacturer instructions. Briefly, the assay was based on the sandwich technique in which specific antibodies to CTS-B were pre-coated onto 96-well plate. The specific detection antibodies were biotinylated. The test samples and biotinylated detection antibodies were added sequentially followed by washing. Avidin-Peroxidase Complex was added and unbound conjugates were washed. A substrate solution was added to the wells to determine the bound enzyme activity. The color development was stopped by sulphuric acid, and the absorbance was read at 450 nm using an ELISA microplate reader (ChroMate-4300, FL, USA). The intensity of the color is directly proportional to the concentration of CTS-B in the sample.
4. Conclusion
Upon the different prepared synthesized compounds bearing different heterocyclic rings either directly attached or attached through an amino or imino bridge to C-6 position of uracil backbone, the most active anticancer compounds against prostate PC3 cell line were those bearing rings as 2-thioxopyrimidine-5-carbonitrile 3a and 3c, quinoxaline 7a, 1,4-benzoxazine 7b, thiazolidinone 12a, 12b and thiazolidine ring 18. In addition to uracil bearing side chains at C-6 position as the chloroacetylamino derivative 4 and the phenylthiourea derivative 17. However, among fusion of different heterocycles to uracil ring, only the pyridopyrimidine derivative 13 and the pyrimidopyrimidine trione derivative 20 were considerably active. All these compounds exhibited better inhibition of cathepsin B than the reference drug doxorubicin did, except compound 3c, while compounds 5a, 5b, 7a, 11a, 12a and 17 exerted more than 50% inhibition of cathepsin B that is associated with metastasis of prostate cancer.
References
- El-Deeb, I.M. and Lee, S.H. (2010) Design and Synthesis of New Anticancer Pyrimidines with Multiple-Kinase Inhibitory Effect. Bioorganic & Medicinal Chemistry, 18, 3860-3874.
- Jemal, C., Siegel, R., Xu, J. and Ward, E. (2010) Cancer Statistics. Ca—A Cancer Journal for Clinicians, 60, 277-300. http://dx.doi.org/10.3322/caac.20073
- Merino, M., Pinto, A., González, R. and Espinosa, E. (2011) Antiangiogenic Agents and Endothelin Antagonists in Advanced Castration Resistant Prostate Cancer. European Journal of Cancer, 47, 1846-1851.http://dx.doi.org/10.1016/j.ejca.2011.04.020
- VanCleave, T.T., Moore, J.H., Benford, M.L., Brock, G.N., Kalbfleisch, T., Baumgartner, R.N., Lillard Jr., J.W., Kittles, R.A. and Kidd, L.C.R. (2009) Interaction among Variant Vascular Endothelial Growth Factor (VEGF) and Its Receptor in Relation to Prostate Cancer Risk. The Prostate, 70, 341-352.http://dx.doi.org/10.1002/pros.21067
- Folkman, J. (1972) Angiogenesis in Psoriasis: Therapeutic Implications. Journal of Investigative Dermatology, 59, 40-43.http://dx.doi.org/10.1111/1523-1747.ep12625746
- Giavazzi, R. and Taraboletti, G. (1999) Angiogenesis and Angiogenesis Inhibitors in Cancer. Forum (Genova), 9, 261-272.
- Kroemer, G. and Jaattela, M. (2005) Lysosomes and Autography in Cell Death Control. Nature Reviews Cancer, 5, 886-897.http://dx.doi.org/10.1038/nrc1738
- Oliveira, C.R., Barbosa, C.M.V., Nascimento, F.D., Lanetzki, C.S., Meneghin, M.B., Pereira, F.E.G., Paredes-Gamero, E.J., Ferreira, A.T., Rodrigues, T., Queiroz, M.L.S., Caires, A.C.F., Tersariol, I.L.S. and Bincoletto, C. (2009) Pre-Clinical Antitumour Evaluation of Biphosphinic Palladacycle Complex in Human Leukaemia Cells. Chemico-Biological Interactions, 177, 181-189. http://dx.doi.org/10.1016/j.cbi.2008.10.034
- Reinheckel, T., Peters, C., Krüger, A., Turk, B. and Vasiljeva, O. (2012) Differential Impact of Cysteine Cathepsins on Genetic Mouse Models of De Novo Carcinogenesis: Cathepsin B as Emerging Therapeutic Target. Frontiers in Pharmacology, 3, 133. http://dx.doi.org/10.3389/fphar.2012.00133
- Mohamed, M.M. and Sloane, B.F. (2006) Cysteine Cathepsins: Multifunctional Enzymes in Cancer. Nature Reviews Cancer, 6, 764-775. http://dx.doi.org/10.1038/nrc1949
- Gocheva, V. and Joyce, J.A. (2007) Cysteine Cathepsins and the Cutting Edge of Cancer Invasion. Cell Cycle, 6, 60-64.http://dx.doi.org/10.4161/cc.6.1.3669
- Vasiljeva, O., Reinheckel, T., Peters, C., Turk, D., Turk, V. and Turk, B. (2007) Emerging Roles of Cysteine Cathepsins in Disease and Their Potential as Drug Targets. Current Pharmaceutical Design, 13, 387-403.http://dx.doi.org/10.2174/138161207780162962
- Doxakis, A., Maria, A., Savvas, P. and Zafiroula, I.K. (2013) Assessment of the Roles of Cathepsins B, H and L in the Progression of Colorectal Cancer. Journal of Cancer Therapy, 4, 1-7. http://dx.doi.org/10.4236/jct.2013.46A2001
- Foghsgaard, L., Wissing, D., Mauch, D., Lademann, U., Bastholm, L., Boes, M., Elling, F., Leist, M. and Jaattela, M. (2001) Cathepsin B Acts as a Dominant Execution Protease in Tumour Cell Apoptosis Induced by Tumour Necrosis Factor. The Journal of Cell Biology, 153, 999-1010. http://dx.doi.org/10.1083/jcb.153.5.999
- Beckham, T.H., Lu, P., Cheng, J.C., Zhao, D., Turner, L.S., Zhang, X., Hoffman, S., Armeson, K.E., Liu, A., Marrison, T., Hannun, Y.A. and Liu, X. (2012) Acid Ceramidase-Mediated Production of Sphingosine 1-Phosphate Promotes Prostate Cancer Invasion through Upregulation of Cathepsin B. International Journal of Cancer, 131, 2034-2043.http://dx.doi.org/10.1002/ijc.27480
- El Gemeie, G.H. (2003) Thioguanine, Mercaptopurine: Their Analogs and Nucleosides as Antimetabolites. Current Pharmaceutical Design, 9, 2627-2642.http://dx.doi.org/10.2174/1381612033453677
- Parker, W.B., Secrist, J. and Waud, W.R. (2004) Purine Nucleoside Antimetabolites in Development for the Treatment of Cancer. Current Opinion in Investigational Drugs, 5, 592-596.
- Nussbaumer, S., Bonnabry, P., Veuthey, J.L. and Fleury-Souverain, S. (2011) Analysis of Anticancer Drugs: A Review. Talanta, 85, 2265-2289.http://dx.doi.org/10.1016/j.talanta.2011.08.034
- Heidelberger, C. (1984) Pyrimidine and Pyrimidine Antimetabolites in Cancer Medicine. Lea and Febiger, Philadelphia, 801-824.
- Kundu, N.G., Das, P., Balzarini, J. and De Clercq, E. (1997) Palladium-Catalyzed Synthesis of [E]-6-(2-Acylvinyl) Uracils and [E]-6-(2-Acylvinyl)-1-[(2-hydroxyethoxy)methyl]uracils—Their Antiviral and Cytotoxic Activities. Bioorganic & Medicinal Chemistry, 5, 2011-2018.http://dx.doi.org/10.1016/S0968-0896(97)00114-4
- Sigmond, J. and Peters, G.J. (2005) Pyrimidine and Purine Analogues, Effects on Cell Cycle Regulation and the Role of Cell Cycle Inhibitors to Enhance Their Cytotoxicity. Nucleosides, Nucleotides and Nucleic Acids, 24, 1997-2022. http://dx.doi.org/10.1080/15257770500269556
- Prekupec, S., Makuc, D., Plavec, J., Suman, L., Kralj, M., Pavelic, K., Balzarini, J., De Clercq, E., Mintas, M. and Raic-Malic, S. (2007) Novel C-6 Fluorinated Acyclic Side Chain Pyrimidine Derivatives: Synthesis, 1H and 13C NMR Conformational Studies, and Antiviral and Cytostatic Evaluations. Journal of Medicinal Chemistry, 50, 3037-3045.http://dx.doi.org/10.1021/jm0614329
- Gazivoda, T., Sokcevic, M., Kralj, M., Suman, L., Pavelic, K., De Clercq, E., Andrei, G., Snoeck, R., Balzarini, J., Mintas, M. and Raic-Malic, S. (2007) Synthesis and Antiviral and Cytostatic Evaluations of the New C-5 Substituted Pyrimidine and Furo[2,3-d]pyrimidine 4’,5’-Didehydro-L-ascorbic Acid Derivatives. Journal of Medicinal Chemistry, 50, 4105-4112.http://dx.doi.org/10.1021/jm070324z
- Gazivoda, T., Raic-Malic, S., Marjanovic, M., Kralj, M., Pavelic, K., Balzarini, J., De Clercq, E. and Mintas, M. (2007) The Novel C-5 Aryl, Alkenyl, and Alkynyl Substituted Uracil Derivatives of L-Ascorbic Acid: Synthesis, Cytostatic, and Antiviral Activity Evaluations. Bioorganic & Medicinal Chemistry, 15, 749-758.http://dx.doi.org/10.1016/j.bmc.2006.10.046
- Spácilová, L., Dzubák, P., Hajdúch, M., Krupkováa, S., Hradil, P. and Hlavác, J. (2007) Synthesis and Cytotoxic Activity of Various 5-[Alkoxy-(4-nitro-phenyl)-methyl]uracils in Their Racemic Form. Bioorganic & Medicinal Chemistry Letters, 17, 6647-6650. http://dx.doi.org/10.1016/j.bmcl.2007.09.022
- Kraljevic, T.G., Kristafor, S., Suman, L., Kralj, M., Ametamey, S.M., Cetina, M. and Raic-Malic, S. (2010) Synthesis, X-Ray Crystal Structure Study and Antitumoral Evaluations of 5,6-Disubstituted Pyrimidine Derivatives. Bioorganic & Medicinal Chemistry, 18, 2704-2712. http://dx.doi.org/10.1016/j.bmc.2010.02.023
- Lee, Y.S. and Kim, B.H. (2002) Heterocyclic Nucleoside Analogues: Design and Synthesis of Antiviral, Modified Nucleosides Containing Isoxazole Heterocycles. Bioorganic & Medicinal Chemistry Letters, 12, 1395-1397.http://dx.doi.org/10.1016/S0960-894X(02)00182-8
- Lee, Y.S., Park, S.M. and Kim, B.H. (2009) Synthesis of 5-Isoxazol-5-yl-2’-deoxyuridines Exhibiting Antiviral Activity against HSV and Several RNA Viruses. Bioorganic & Medicinal Chemistry Letters, 19, 1126-1128.http://dx.doi.org/10.1016/j.bmcl.2008.12.103
- Jakubiec, D. and Walczak, K.Z. (2011) A Convenient Synthesis of 5-(1,2,4-Oxadiazol-5-yl)pyrimidine-2,4(1H, 3H)- diones. Tetrahedron Letters, 52, 6890-6891. http://dx.doi.org/10.1016/j.tetlet.2011.10.033
- Lee, Y.S., Park, S.M., Kim, H.M., Park, S.K., Lee, K., Lee, C.W. and Kim, B.H. (2009) C5-Modified Nucleosides Exhibiting Anticancer Activity. Bioorganic & Medicinal Chemistry Letters, 19, 4688-4691.http://dx.doi.org/10.1016/j.bmcl.2009.06.072
- Soliman, R., Habib, N.S., Ismail, K., Moustafa, A., Sarg, M.T. and Fanaki, N.H. (2003) Synthesis of Novel Pyrimidine-2,4-diones and 2-Thioxopyrimidine-4-ones as Potential Anticancer and Antimicrobial Agents. Bollettino Chimico Farmaceutico, 142, 167-174.
- Habib, N.S., Soliman, R., Ismail, K., Hassan, A.M. and Sarg, M.T. (2003) Synthesis of Novel Pyrimidines, 1,2,4-Triazolo[4,3-a]pyrimidin-7-ones and Pyrimidino[2,1-c][1,2,4]triazin-8-ones for Their Antimicrobial and Anticancer Activities. Bollettino Chimico Farmaceutico, 142, 396-405.
- Cui, Z., Li, Y., Huang, J., Cui, J., Wang, R. and Yang, X. (2010) New Class of Potent Antitumour Acylhydrazone Derivatives Containing Furan. European Journal of Medicinal Chemistry, 45, 5576-5584.http://dx.doi.org/10.1016/j.ejmech.2010.09.007
- Abdel-Hamid, H.F., Soliman, A., Helaly, F.M. and Ragab, S. (2011) Cytotoxic Potency and Induced Biochemical Parameters in Mice Serum of New Furan Derivatives against Liver Cancer Cell Line. Acta Poloniae Pharmaceutica, 68, 499-505.
- Idhayadhulla, A., Kumar, R.S., Nasser, A.J.A. and Manilal, A. (2013) Synthesis of Some New Pyrrole and Pyridine Derivatives and Their Antimicrobial, Anticancer Activities. International Journal of Biological Chemistry, 7, 15-26. http://dx.doi.org/10.3923/ijbc.2013.15.26
- Alsaid, M.S., El-Gazzar, M.G. and Ghorab, M.M. (2013) Anticancer Activity of Novel Thiophenes Containing a Biological Active Diphenyl Sulfone, Diazepine, Piperidine, Oxazepine, Acrylaldehyde and Sulfonamide Moieties. Drug Research, 63, 263-269.
- Garg, V., Kumar, A., Chaudhary, A., Agrawal, S., Tomar, P. and Sreenivasan, K.K. (2013) Synthesis, Biological Evaluation and Molecular Docking Studies of 1,3-Benzoxazine Derivative as Potential Anticancer Agents. Medicinal Chemistry Research, 22, 5256-5266.
- Lee, S.H., Kim, N., Kim, S.J., Song, J., Gong, Y.D. and Kim, S.Y. (2013) Anti-Cancer Effect of Quinoxaline Derivatives GK13 as a Transglutaminase 2 Inhibitor. Journal of Cancer Research and Clinical Oncology, 139, 1279-1294.http://dx.doi.org/10.1007/s00432-013-1433-1
- Bathula, S.N.P. and Vadla, R. (2011) Bioactivity of 1,4-Disubstituted-1,2,3-triazoles as Cytotoxic Agents against the Various Human Cell Lines. Asian Journal of Pharmaceutical and Clinical Research, 4, 66-67.
- Kurumurthy, C., Rao, P.S., Swamy, B.V., Kumar, G.S., Rao, P.S., Narsaiah, B., Velatooru, L.R., Pamanji, R. and Rao, J.V. (2011) Synthesis of Novel Alkyltriazole Tagged Pyrido[2,3-d]pyrimidine Derivatives and Their Anticancer Activity. European Journal of Medicinal Chemistry, 46, 3462-3468.http://dx.doi.org/10.1016/j.ejmech.2011.05.011
- El-Gaby, M.S.A., Ismail, Z.H., Abdel-Gawad, S.M., Aly, H.M. and Ghorab, M.M. (2009) Synthesis of Thiazolidine and Thiophene Derivatives for Evaluation as Anticancer Agents. Phosphorus, Sulfur and Silicon and the Related Elements, 184, 2645-2654. http://dx.doi.org/10.1080/10426500802561096
- Abu-Hashem, A.A., El-Shehry, M.F. and Badria, F.A. (2010) Design and Synthesis of Novel Thiophenecarbohydrazide, Thienopyrazole and Thienopyrimidine Derivatives as Antioxidant and Antitumour Agents. Acta Pharmaceutica, 60, 311-323.
- Zhu, W.F., Zhai, X., Li, S., Cao, Y.Y., Gong, P. and Liu, Y.J. (2012) Synthesis and Cytotoxic Activity of Novel 2,6- Disubstituted-4-morpholinothieno[3,2-d]pyrimidines as Potent Anti-Tumour Agents. Chinese Chemical Letters, 23, 703-706. http://dx.doi.org/10.1016/j.cclet.2012.04.012
- Bernier, J.L., Henichart, J.P., Warin, V., Trentesaux, C. and Jardillier, J.C. (1985) 5-Cinnamoyl-6-aminouracil Derivatives as Novel Anticancer Agents: Synthesis, Biological Evaluation and Structure-Activity Relationships. Journal of Medicinal Chemistry, 28, 497-502. http://dx.doi.org/10.1021/jm00382a020
- Dabiri, M., Arvin-Nezhad, H., Khavasi, H.R. and Bazgir, A. (2007) A Novel and Efficient Synthesis of Pyrimido[4, 5-d]pyrimidine-2,4,7-trione and Pyrido[2,3-d:6,5-d]dipyrimidine-2,4,6,8-tetrone Derivatives. Tetrahydron, 63, 1770-1774. http://dx.doi.org/10.1016/j.tet.2006.12.043
- Essa, F.B., Bazbouz, A. and Al-Lababidi, S. (2010) Preparation, Chemical and Biological Study of Acyclovir Analogue Starting from 6-Aminouracil. Oriental Journal of Chemistry, 26, 1281-1292.
- Samai, S., Nandi, G.C., Chowdhury, S. and Singh, M.S. (2011) L-Proline Catalyzed Synthesis of Densely Functionalized Pyrido[2,3-d]pyrimidines via Three-Component One-Pot Domino Knoevenagel Aza-Diels-Alder Reaction. Tetrahydron, 67, 5935-5941. http://dx.doi.org/10.1016/j.tet.2011.06.051
- Fargualy, A.M., Habib, N.S., Ismail, K.A., Hassan, A.M.M. and Sarg, M.T.M. (2013) Synthesis, Biological Evaluation and Molecular Docking Studies of Some Pyrimidine Derivatives. European Journal of Medicinal Chemistry, 66, 276-295. http://dx.doi.org/10.1016/j.ejmech.2013.05.028
- Fathalla, O.A., Zeid, I.F., Haiba, M.E., Soliman, A.M., Abd-Elmoez, S.I. and El-Serwy, W.S. (2009) Synthesis, Antibacterial and Anticancer Evaluation of Some Pyrimidine Derivatives. World Journal of Chemistry, 4, 127-132.
- Bondock, S., Khalifa, W. and Fadda, A.A. (2011) Synthesis and Antimicrobial Activity of Some New 4-Hetarylpyrazole and Furo[2,3-c]pyrazole Derivatives. European Journal of Medicinal Chemistry, 46, 2555-2561.http://dx.doi.org/10.1016/j.ejmech.2011.03.045
- Alqasoumi, S.I., Ghorab, M.M., Ismail, Z.H., Abdel-Gawad, S.M., El-Gaby, M.S.A. and Aly, H.M. (2009) Novel Antitumor Acetamide, Pyrrole, Pyrrolopyrimidine, Thiocyanate, Hydrazone, Pyrazole, Isothiocyanate and Thiophene Derivatives Containing a Biologically Active Pyrazole Moiety. Arzneimittel Forschung, 59, 666-671.
- Aly, H.M. (2011) Novel Pyrrolidinone and Pyrazolo[1,5-a][1,3,5]triazine Derivatives Bearing a Biologically Active Sulfamoyl Moiety as a New Class of Antitumor Agents. Monatshefte für Chemie—Chemical Monthly, 142, 935-941. http://dx.doi.org/10.1007/s00706-011-0517-3
- Shim, J.L., Niess, R. and Broom, A.D. (1972) Acylation of Some 6-Aminouracil Derivatives. The Journal of Organic Chemistry, 37, 578-581. http://dx.doi.org/10.1021/jo00969a013
- Gewald, K.J. (1966) Heterocyclen aus CH-aciden nitrilen. IV. Addition von Schwefelkohlenstoff an Alkyliden Malonitrile. Journal für Praktische Chemie, 31, 205-213.
- Aly, H.M., Saleh, N.M. and Elhady, H.A. (2011) Design and Synthesis of Some New Thiophene, Thienopyrimidine and Thienothiadiazine Derivatives of Antipyrine as Potential Antimicrobial Agents. European Journal of Medicinal Chemistry, 46, 4566-4572. http://dx.doi.org/10.1016/j.ejmech.2011.07.035
- Shingare, M.S. and Ingle, D.B. (1976) Synthesis of Pyrimidine Schiff Bases as Anticancer Agents. Journal of Indian Chemical Society, LIII, 1036-1037.
- Mohamed, N.R., El-Saidi, M.M.T., Ali, Y.M. and Elnagdi, M.H. (2007) Utility of 6-Amino-2-thiouracil as a Precursor for the Synthesis of Bioactive Pyrimidine Derivatives. Bioorganic & Medicinal Chemistry, 15, 6227-6235.http://dx.doi.org/10.1016/j.bmc.2007.06.023
- Shingare, M.S. (1983) Synthesis of New Thiazolidinones from Pyrimidine Schiff Bases and Their Biological Activity. Indian Journal of Chemistry B, 22, 714-715.
- Archana, Srivastava, V.K. and Kumar, A. (2002) Synthesis of Newer Thiadiazolyl and Thiazolidinoyl Quinazolin-4 (3H)-ones as Potential Anticonvulsant Agents. European Journal of Medicinal Chemistry, 37, 873-882.http://dx.doi.org/10.1016/S0223-5234(02)01389-2
- Patel, N.B. and Patel, V.N. (2010) Synthesis, Antibacterial and Antifungal Activities of Novel Thiazolidinyl Sulfonamides of Quinazolin-4(3H)ones. Pharmaceutical Chemistry Journal, 44, 438-445.http://dx.doi.org/10.1007/s11094-010-0486-6
- Abdou, W.M., Fahmy, A.F.M. and Kamel, A.A. (2002) A Facile Synthesis of Pyrrolo[3,2-d]pyrimidines from 6-Azidouracils and Ylide Phosphoranes. Heteroatom Chemistry, 13, 357-365. http://dx.doi.org/10.1002/hc.10048
- Pokhodylo, N.T., Matiychuk, V.S. and Obushak, M.D. (2009) Synthesis of Ethyl 4,5-Disubstituted 2-Azido-3-thiophenecarboxylates and Use in the Synthesis of Thieno[3,2-e][1,2,3]triazolo[1,5-a]pyrimidin-5(4H)-ones. Tetrahydron, 65, 2678-2683.http://dx.doi.org/10.1016/j.tet.2009.01.086
- Hafiz, I.S.A., Ramiz, M.M.M., Mahmoud, F.F. and Darwish, E.S. (2008) β-Enaminonitriles in Heterocyclic Synthesis: Synthesis of New Tetrahydropyridinetrione, Pyridopyrimidines, Pyridotriazines and Dihydropyridines. Journal of Chemical Sciences, 120, 339-345.http://dx.doi.org/10.1007/s12039-008-0057-5
- Rostom, S.A.F., El-Ashmawy, I.M., El Razik, H.A., Badr, M.H. and Ashour, H.M.A. (2009) Design and Synthesis of Some Thiazolyl and Thiadiazolyl Derivatives of Antipyrine as Potential Non-Acidic Anti-Inflammatory, Analgesic and Antimicrobial Agents. Bioorganic & Medicinal Chemistry, 17, 882-895.http://dx.doi.org/10.1016/j.bmc.2008.11.035
- Marugan, J.J., Zheng, W., Motabar, O., Southall, N., Goldin, E., Sidransky, E., Aungst, R.A., Liu, K., Sadhukhan, S.K. and Austin, C.P. (2010) Evaluation of 2-Thioxo-2,3,5,6,7,8-hexahydropyrimido[4,5-d]pyrimidine-4(1H)-one Analogues as GAA Activators. European Journal of Medicinal Chemistry, 45, 1880-1897. http://dx.doi.org/10.1016/j.ejmech.2010.01.027
- Ding, R., He, Y., Xu, J., Liu, H., Wang, X., Feng, M., Qi, C., Zhang, J. and Peng, C. (2012) Preparation and Bioevaluation of 99mTc Nitrido Radiopharmaceuticals with Pyrazolo[1,5-a]pyrimidine as Tumor Imaging Agents. Medicinal Chemistry Research, 21, 523-530. http://dx.doi.org/10.1007/s00044-011-9558-8
- Moin, K., Demchik, L., Mai, J., Duessing, J., Peters, C. and Sloane, B.F. (2000) Observing Proteases in Living Cells. Advances in Experimental Medicine and Biology, 477, 391-401. http://dx.doi.org/10.1007/0-306-46826-3_40
- Frohlich, E., Schlagenhauff, B., Mohrle, M., Weber, E., Klessen, C. and Rassner, G. (2001) Activity, Expression, and Transcription Rate of the Cathepsins B, D, H, and L in Cutaneous Malignant Melanoma. Cancer, 91, 972-982.
- Sinha, A.A., Quast, B.J., Wilson, M.J., Fernandes, E.T., Reddy, P.K., Ewing, S.L. and Gleason, D.F. (2002) Prediction of Pelvic Lymph Node Metastasis by the Ratio of Cathepsin B to Stefin A in Patients with Prostate Carcinoma. Cancer, 94, 3141-3149. http://dx.doi.org/10.1002/cncr.10604
- Bialas, A. and Kafarski, P. (2009) Proteases as Anti-Cancer Targets—Molecular and Biological Basis for Development of Inhibitor-Like Drugs against Cancer. Anti-Cancer Agents Medicinal Chemistry, 9, 728-762.http://dx.doi.org/10.2174/187152009789056877
- Lankelma, J., Voorend, D., Barwari, T., Koetsveld, J., Van der Spek, A., De Porto, A., Van Rooijen, G. and Van Noorden, C. (2010) Cathepsin L, Target in Cancer Treatment? Life Sciences, 86, 225-233.http://dx.doi.org/10.1016/j.lfs.2009.11.016
- Rothberg, J.M., Sameni, M., Moin, K. and Sloane, B.F. (2012) Live-Cell Imaging of Tumor Proteolysis: Impact of Cellular and Non-Cellular Microenvironment. Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics, 1824, 123-132.http://dx.doi.org/10.1016/j.bbapap.2011.07.025
- Van Damme, J., Struyf, S. and Opdenakker, G. (2004) Chemokine-Protease Interactions in Cancer. Seminars in Cancer Biology, 14, 201-208. http://dx.doi.org/10.1016/j.semcancer.2003.10.007
- Green, K.A. and Lund, L.R. (2005) ECM Degrading Proteases and Tissue Remodeling in the Mammary Gland. BioEssays, 27, 894-903.http://dx.doi.org/10.1002/bies.20281
- Joyce, J.A. and Pollard, J.W. (2009) Microenvironmental Regulation of Metastasis. Nature Reviews Cancer, 9, 239-252.http://dx.doi.org/10.1038/nrc2618
- López-Otin, C. and Hunter, T. (2010) The Regulatory Crosstalk between Kinases and Proteases in Cancer. Nature Reviews Cancer, 10, 278-292.
- Mason, S.D. and Joyce, J.A. (2011) Proteolytic Networks in Cancer. Trends in Cell Biology, 21, 228-237.http://dx.doi.org/10.1016/j.tcb.2010.12.002
- Turk, B., Turk, D.S. and Turk, V. (2012) Protease Signalling: The Cutting Edge. The EMBO Journal, 31, 1630-1643.
- Buck, M.R., Karustis, D.G., Day, N.A., Honn, K.V. and Sloane, B.F. (1992) Degradation of Extracellular-Matrix Proteins by Human Cathepsin B from Normal and Tumor Tissues. Biochemical Journal, 282, 273-278.
- Premzl, A., Zavasnik-Bergant, V., Turk, V. and Kos, J. (2003) Intracellular and Extracellular Cathepsin B Facilitate Invasion of MCF-10A neoT Cells through Reconstituted Extracellular Matrix in Vitro. Experimental Cell Research, 283, 206-214.http://dx.doi.org/10.1016/S0014-4827(02)00055-1
- Miyake, H., Hara, I. and Eto, H. (2004) Serum Level of Cathepsin B and Its Density in Men with Prostate Cancer as Novel Markers of Disease Progression. Anticancer Research, 24, 2573-2577.
- Podgorski, I. and Sloane, B.F. (2003) Cathepsin B and Its Role(s) in Cancer Progression. Biochemical Society Symposia, 70, 263-276.
- Podgorski, I., Linebaugh, B.E., Sameni, M., Jedeszko, C., Bhagat, S., Cher, M.L. and Solane, B.F. (2005) Bone Micro-environment Modulates Expression and Activity of Cathepsin B in Prostate Cancer. Neoplasia, 7, 207-223.http://dx.doi.org/10.1593/neo.04349
- Withana, N.P., Blum, G., Sameni, M., Slaney, C., Anbalagan, A., Olive, M.B., Bidwell, B.N., Edgington, L., Wang, L., Moin, K., Sloane, B.F., Anderson, R.L., Bogyo, M.S. and Parker, B.S. (2012) Cathepsin B Inhibition Limits Bone Metastasis in Breast Cancer. Cancer Research, 72, 1199-1209.http://dx.doi.org/10.1158/0008-5472.CAN-11-2759
- Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., Warren, J.T., Bokesch, H., Kenney, S. and Boyd, M.R. (1990) New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. Journal of the National Cancer Institute, 82, 1107-1112.http://dx.doi.org/10.1093/jnci/82.13.1107
NOTES

*Corresponding author.