Open Journal of Medicinal Chemistry
Vol.3 No.1(2013), Article ID:29463,6 pages DOI:10.4236/ojmc.2013.31001
New Gene Therapy Strategies for the Deletion of Exon 44 of Dystrophin Gene Based on Gene Editing by TALENs
1Department of Molecular Diagnostics and Biopharmaceutics, College of Pharmaceutical Science, Soochow University, Suzhou, China
2State Key Laboratory of Reproductive Medicine, Nanjing Maternity and Child Health Care Hospital Affiliated to Nanjing Medical University, Nanjing, China
3University of Waterloo, Waterloo, Canada
4Gene Core, The Genomics Institute of the Novartis Research Foundation, San Diego, USA
5DNA Sequencing Core Laboratory, City of Hope National Medical Center, Duarte, USA
6Centre Hospitalier de l’Université Laval Research Center, Laval University, Quebec, Canada
Email: #kaili34@gmail.com
Received November 15, 2012; revised December 19, 2012; accepted December 29, 2012
Keywords: TALENs; DMD; Gene Therapy; Exon Skipping
ABSTRACT
Duchenne Muscular Dystrophy (DMD) is a severe childhood form of muscular dystrophy. Both the severe form and its milder form of Becker Muscular Dystrophy (BMD) are caused by the mutation of dystrophin gene. Different from some other genetic diseases such as hemophilia that can be treated by replacement therapy, there is no effective therapy for muscular dystrophy in conventional medication. Gene editing technology from the recently developed engineered nucleases such as TALENs has been successfully employed in genome modification of a variety of species, and will be applied in gene therapy of selected human diseases. The genetic basis of DMD and BMD indicates that DMD is a good target for gene therapy through returning the reading frame of dystrophin gene. Gene therapy strategies described here may apply to many other genetic diseases. Wider application of TALENs in gene therapy have the potential to dramatically prolong the lifespan of individuals with genetic diseases.
1. Potential of Engineered Nuclease TALENs in Gene Therapy
Gene therapy is a genetic intervention used for the prevention or treatment of human diseases by targeting and modifying selected genes. Lack of efficient gene editing enzymes and gene delivery strategies are the two major technological obstacles that prevent from a wide clinical application of gene therapy. Recently, the technological development of several engineered nucleases may partially help to overcome the aforementioned difficulties and pave a new way to gene therapy. Among the engineered nucleases, Zinc-finger nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs) have been successfully employed for both in vitro and ex vivo gene modifications. Based on these milestone events and their great potential, gene editing technology with ZFNs and TALENs was recognized as the Technology of the Year by the journal “Nature- Methods” in 2011 [1].
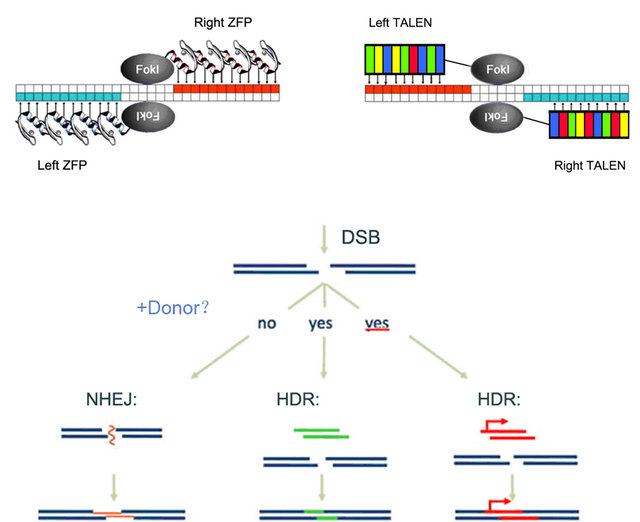
TALENs are made of a number of DNA binding modules and the nuclease domain from FokI restriction enzyme. Each protein module typically contains 34 amino acids, and the 12th and 13th residues are the key site of the target identification, known as the repeated variable di-residues (RVDs) [2-5] Currently, there are 7 recognized units decoded and 4 commonly used identification units: NI identifies A, HD identifies C, NG identifies T, and NN identifies G or A. When TALENs have repeated DNA binding elements in its DNA binding domain, they can then efficiently and precisely cleave specific DNA sequences and induce a double-strand break (DSB), followed by the homology-directed repair (HDR) or nonhomologous end joining (NHEJ) pathways [6,7]. These gene editing abilities of TALENs are illustrated in Figure 1.
ZFNs and TALENs recognize DNA targets through their DNA binding domains. While each zinc finger protein module identifies three nucleotides, a TALE module only recognizes one nucleotide through its 13th amino acid. The cleavage domains of homodimers or heterodimers form an endonuclease that digests the target sequence and creates a double strand break. Following
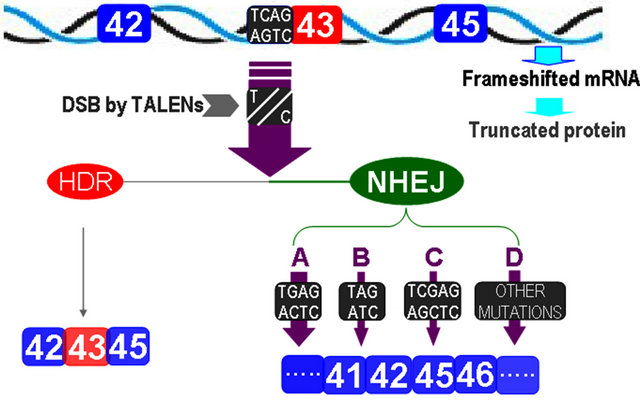
Figure 1. Structures and applications of ZFNs and TALENs.
the creation of double strand break, gene editing efficiencies increase by two log scales as compared to those formed through spontaneous gene recombination, which greatly facilitates gene disruption, gene correction, or gene insertion.
Gene disruption is the simplest means of gene editing. By causing a frameshift mutation, the disruption or abolishment of the function of selected genes can be easily obtained. Engineered nucleases-driven gene disruption has already been used for model organisms and a variety of mammalian cells. TALENs can also efficiently disrupt the zebrafish genes as well as the endogenous human genes NTF3 and CCR5 at efficiencies of up to 25% [8,9].
Gene addition is the second strategy useful for gene editing by TALENs. This approach consists of introducing DNA sequences into a predetermined genomic location. To make this strategy work, two essential properties should be considered [10]. Firstly, the inserted DNA sequences must functionally integrate within the modified cells. Secondly, the normal function and gene regulation in cells harboring the predetermined genomic location should not be blocked, or in other words, no off-target effect is required following the gene modification or gene addition. As an application example, TALEN expression constructs and the corresponding donor plasmids were introduced into human embryonic stem cell (ESC); the TALEN-mediated targeting of PPP1R12C [11] resulted in a robust transgene expression in pluripotent as well as in differentiated cells.
Gene correction is one of the most accepted strategies in classical gene therapy. Many therapeutic progresses using engineered nucleases have been reported in the past years. In a mouse model of haemophilia B [12], ZFNs co-delivered with an appropriately designed genetargeting vector stimulated gene correction in vivo. As a consequence, the gene therapy treatment was sufficient to correct the prolonged clotting times in this mouse model of haemophilia B, and remained a persistent effect after the induced liver regeneration. In addition, gene corrections using ZFNs and TALENs have also been achieved in tobacco, fruitflies, arabidopsis thaliana and mouse ES cells for a variety of gene abnormalities [13- 15].
Since its development in 2010, TALENs have become the hot enzymes used in genome modifications of a variety of species. The aforementioned three strategies used in genome modification, gene disruption, gene addition, and gene correction, can be applied in gene therapy of human diseases such as hemophilia, thalassemia, DMD, neuronal deafness, Stargardt’s syndrome, essential hypertension, TP53 gene mutation as well as lung, breast, cervix, prostate, colon, rectum cancers and leukemia although relatively long clinical trials are to be carried out first. The target disease list will get longer with a better understanding of the pathophysiology of human diseases. TALENs are a new type of “drugable” macromolecules. As compared to traditional drugs, the effectiveness and toxicity of TALENs are easy to determine by screening their gene editing spectra and off-target effects. With the availability of next generation sequencing technology, both gene editing spectra and off-target effects can be efficiently assayed using cultured human cells. Thus, engineered nucleases particularly the “drugable” TALENs are expected to be invaluable for future gene therapy. However, the gene editing technology is only one approach in gene therapy as the lack of gene delivery is still an obstacle to be overcome Fortunately, both lentiviral and adenoviral vectors are efficient enough to deliver ZFN and TALENs in vivo and ex vivo, particularly when used in reversed gene therapy that only requires a partial knockout of a selected gene in order to reach an observable improvement for clinical symptom.
2. Therapeutic Obstacles and Clinical Features of DMD
Duchenne Muscular Dystrophy (DMD) is an X-linked recessive inherited disease. As a severe childhood form of muscular dystrophy, it has a high birth prevalence in nearly 1:4000 [16]. Except for patients from inherited mutations, the high birth prevalence of DMD is also partially attributed to spontaneous mutations and an unending trail of new cases emphasizes the compelling need to find an effective treatment.
Many types of mutations have been identified in DMD, including point mutation, deletion, insertion, and duplication. However, the most common type of diseasecausing mutation of the DMD gene is deletion of one or more exons identified in approximately 60% to 65% of patients [17]. Among these deletions, the leading mutation is in exon 44 that is found in about 30% of all DMD deletions [18] and is attributed to the hot breakpoint in intron 43 within a THE-1 element [19]. Regardless of the complicated types of mutations, the genotype-phenotype relationship in muscular dystrophy patients showed that frameshift mutations are usually associated with DMD while mutations leading to BMD usually do not change the reading-frame of the transcripts. BMD patients typically die in the fourth and fifth decades.
The genotype-phenotype relationship indicated a therapeutic possibility of switching DMD to BMD. Relevant strategies tested until now include readthrough and exon skipping. The former is approached by using tRNA suppressor [20] and aminoglycoside antibiotics [21]. In 2003, Bertoni et al. [22] tested the ability of chimeric RNA/ DNA oligonucleotides (chimeraplasts) to alter key bases in specific splice sequences in the dystrophin gene to induce exon skipping. A similar strategy using morpholino phosphorodiamidate oligonucleotides obtained convincing results in mdx mouse [23]. These strategies were well tested in cultured cells, in mdx mouse model and in DMD dog models [24].
An observation encouraging the readthrough and exon skipping strategies in the treatment of DMD is the identification of dystrophin-positive fibers possibly raised from somatic reversion/suppression. Another mechanism to explain the positively staining fibers is a second site in-frame deletion [25]. On a therapeutic point of view, DMD patients bearing dystrophin-positive fibers are well suited to either readthrough or exon skipping therapies without or virtually without immune rejection. These two strategies are different from therapy using mini dystrophin gene as the latter may lead to immune rejection that eventually attenuates the efficacy of the treatment or even worsens the disease procedure of DMD.
Recently, Mendell et al. published an updated review about gene therapy for muscular gene therapy [26]. As stated by Mendell et al., current treatment options for DMD are restricted and only glucocorticoids have consistently demonstrated efficacy in any of the muscular dystrophies [26]. Clearly, efficient treatments to DMD are still urgently needed. Based on the recent data (2012-10-12) recorded in the website of “www.clinicaltrials.gov”, there are 402 ongoing registered clinical trials when searched using the keyword of DMD. When narrowed down to DMD gene therapy, there are 27 clinical trails and only 7 of them are currently recruiting patients. For the specific gene therapy strategy of DMD exon skipping the database recorded, only 6 clinical trails recorded with 4 being active. The slow progress in developing practically available gene therapy for DMD may soon gain momentum with the development of TALENs. It is not too optimistic to expect the availability of selected clinically effective gene therapies for DMD and for many other genetic diseases in the next decade since more fundamental research and clinical trials are in progress or have been completed.
3. Multiple Gene Therapy Strategies Available in Switching DMD to BMD
As discussed in the previous section, the genotype-phenotype relationship in dystrophin gene mutation-related diseases has demonstrated a therapeutic algorithm by switching frameshifted mutation into inframe mutation, and thereby changing the clinical outcome of DMD to BMD.
Two genetic consequences occur following the deletion of exon 44 in dystrophin gene: loss of the related nucleotides in the transcript and shift the reading frame of the transcript. Accordingly, gene therapy strategies targeting the treatment of exon 44 deletion need to deal with both frameshift and loss of nucleotides in the transcript whenever possible. Here we suggest some rules in designing gene therapy strategies using the deletion of exon 44 as an example. We believe that these rules are not only well applicable to dystrophin mutations, but they are also widely applicable to those using gene editing in the treatment of human diseases.
Practical rules applicable in gene therapy using gene editing technology are listed below:
1) Gene normalization, either through gene correction or gene insertion, should only be the first choice in germline gene therapy, but not in somatic gene therapy. Its application in somatic gene therapy can be beneficial in conditions with no or minimal immune rejection to the wild type protein from the normalized gene;
2) If intentional deletion of an extra exon can return the reading frame of the transcript and the deletion of an extra exon is tolerable in gene function, this strategy could be the first choice in somatic gene therapy in mutations that occurred in large genes with tens of exons;
3) Switching frameshift mutation to inframe mutation through gene disruption or gene correction should target downstream locations from the deletion, rather than the upstream sites;
4) When gene disruption is used in remedying mRNA reading frame, scheduled codon shifting from NHEJ must not create a novel stop codon before the reading frame is normalized. Additionally, if a frameshift mutation is caused by the addition of one base, simple disruption may be the first choice in gene therapy.
Based on these rules, four possible gene therapy strategies are available for the treatment of exon 44 deletion in dystrophin gene (Table 1). Actually, all of these four options are currently under investigation at cellular level in our group.
Although it is difficult to speculate which of the four therapeutic strategies would have the highest efficacy in restoring truncated dystrophin protein, the deletion of the extra exon is theoretically the most efficient approach in restoring the reading frame of the transcript of mutated
Table 1. Possible gene therapy strategies applicable to the treatment of DMD patients with exon 44 deleted.

dystrophin and some other mutations with one exon deletion. When the splice acceptor site at the 3’ end of the intron 42 is targeted with gene editing technology, nearly all edited products formed double-strands break by TALENs will lead to the deletion of exon 43 since most of the mutations from NHEJ disrupt the acceptor site for splicing. For genome failed to form DSB or acceptor site recovered from NHEJ, no novel protein translated as the transcript is still in frameshift, which is important in minimizing or avoiding toxic effects in gene therapy (Figure 2).
TALENs specifically targeting the acceptor site of exon 43 yield double-strand breaks at the acceptor site. Except for a small portion of DNA molecules with DSBs repaired by homologous recombination still transcribe mRNAs containing the exon 43, most of the DNA molecules with DSBs were fixed by NHEJ and were disrupted with their acceptor site. A represents the acceptor sited disrupted from single base substitution; B from single base deletion; C from single base insertion; and D represents acceptor site eliminated by all other types of mutations such as 2 basepair deletion or insertion and 3 or more basepairs insertion or deletion. When exon 43 is deleted, the transcripts are then restored in frame again.
Exon 44 of dystrophin gene is pretty short, a feature which suggests the technical possibility of gene normalization by adding the exon through gene editing. For future germline gene therapy, the addition of exon 44 is an ideal or even perfect therapy for fertilized eggs. However, several factors are to be considered when this approach is applied in somatic gene therapy. First, gene addition has a lower efficiency as compared to gene disruption. Secondly, the wild type of the intact dystrophin protein may cause an immune response or rejection. Therefore, clinical trials testing the addition of an exon should pay extra attention about the possible immune rejection in a larger population.
As listed in Table 1, the third available strategy is gene disruption at the coding region of exon 45. This strategy is less efficient as compared to the first strategy of disrupting the acceptor site of intron 42. All disrupted
Gene Disruption Gene Correction Gene Addition
Figure 2. Illustration of the exon 43 skipping with gene editing technology.
acceptor site of intron 43 may lead to the deletion of exon 43, whereas only certain types of NHEJ, two bases deletion and one base insertion, are able to restore the reading frame of the transcripts as illustrated in Figure 3. The third strategy requires the target site downstream of the deletion, not upstream of the deletion. In the case of exon 44 deletion, the simple gene disruption in exon 45 is the correct choice. However, if in situations when novel stop codon(s) formed right after the deletion junction in the transcript, upstream locations could be the target of gene editing. Elimination of stop codon(s) from exon deletion and avoidance of creating novel stop codon(s) from the scheduled codon shift are crucial in the application of gene disruption in gene therapy.
A represents the mRNA from wild type sequence of either exon 43 or 45 in DMD patient with exon 44 deleted. B represents the shifted codons in case of deleting 2 nucleotides (green highlighted), and C represents the mRNA fragment in case of adding 1 nucleotide (pink highlighted). Since the exon 43 has 3n + 2 and exon 44 has 3n + 1 nucleotides, only selected gene editing effectssuch as adding extra 1 nucleotide or deleting 2 nucleo-

Figure 3. Representative examples of remedying the reading frame of transcripts by gene disruption.
tides are able to restore the reading frame. As illustrated, relevant gene changes created new stop codon in exon 43 but not in exon 45.
Considering that the three leading types of mutations are single base substitution, single base deletion, and single base insertion [27,28], simple gene disruption by TALENs could be the first gene therapy strategy in diseases caused by mutation with one base added. However, for diseases caused by mutations with single base substitution or deletion, gene correction in the presence of homologous oligonucleotides is the more efficient strategy. Gene correction has a low efficiency and may cause immune rejection when applied in somatic gene therapy, although it has a potential in germline gene therapy.
4. Concluding Remarks
Genetically engineered nucleases, particular the TALENs, have been widely used in genome modifications. This gene editing ability presents great potential in gene therapy for human diseases. Single base substitution, small deletion, and small insertion are the three leading types of mutations causing human diseases. As these three types of mutations are good targets for gene editing by TALENs, it is not too optimistic to expect more and more clinically effective gene therapies available in the coming years.
We took DMD as an example in discussing the possible gene therapy strategies using gene editing technology. One of the algorithms in switching DMD to BMD was discussed in more detail, which highlights a practical therapy strategy in remedying the reading frame of transcripts from frameshift mutations such as some deletions and insertions. Considering the application of TALENs is in its early stage, some general rules governing gene therapy for human diseases with gene editing technology were suggested based on the analysis of DMD. These general rules may also be applicable in gene therapy of some other human diseases such as hemophilia, thalassemia, neuronal deafness, Stargardt’s syndrome, essential hypertension, TP53 gene mutation as well as lung, breast, cervix, prostate, colon, rectum cancers and leukemia. There is no doubt that TALENs are a new type of “drugable” macromolecules that will be invaluable for future gene therapy in the era of individualized medicine.
5. Acknowledgements
This study is partially supported by National Natural Science Foundation of China (No. 30970877), Chinese National 863 Major Grant (No. 2012AA020905), the Priority Academic Program Development of Jiangsu Higher Education Institutions and Innovation Project of Jiangsu Graduate Education (No. CXZZ11_0117).
REFERENCES
- Natalie de Souza, “Zinc-Finger Nucleases,” Nature Methods, Vol. 8, No. 1, 2011, p. 43.
- M. J. Moscou and A. J. Bogdanove, “A Simple Cipher Governs DNA Recognition by TAL Effectors,” Science, Vol. 326, No. 5959, 2009, p. 1051. doi:10.1126/science.1178817
- J. Boch, H. Scholze and S. Schornack, “Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors,” Science, Vol. 326, No. 5959, 2009, pp. 10509- 10512. doi:10.1126/science.1178811
- R. Morbitzer, P. Römer and J. Boch, “Regulation of Selected Genome Loci Using de Novo-Engineered Transcription Activator-Like Effector (TALE)-Type Transcription Factor,” Proceedings of the National Academy of Sciences of the United States, Vol. 107, No. 50, 2010, pp. 21617-21622. doi:10.1073/pnas.1013133107
- R. Geissler, H. Scholze and S. Hahn, “Transcriptional Activators of Human Genes with Programmable DNASpecificity,” PLoS One, Vol. 6, No. 5, 2011, Article ID: e19509. doi:10.1371/journal.pone.0019509
- M Christian, T Cermak and EL Doyle, “Targeting DNA Double-Strand Breaks with TAL Effector Nucleases,” Genetics, Vol. 186, No. 2, 2010, pp. 757-61. doi:10.1534/genetics.110.120717
- M. M. Mahfouz, L. Li and M. Shamimuzzaman, “De Novo-Engineered Transcription Activator-Like Effector (TALE) Hybrid Nuclease with novel DNA Binding Specificity Creates Double-Strand Breaks,” Proceedings of the National Academy of Sciences of the United States, Vol. 108, No. 6, 2011, pp. 2623-2628. doi:10.1073/pnas.1019533108
- J. C. Miller, S. Tan and G. Qiao, “A TALE Nuclease Architecture for Efficient Genome Editing,” Nature Biotechnology, Vol. 29, No. 2, 2011, pp. 143-148. doi:10.1038/nbt.1755
- P. Huang, A. Xiao and M. Zhou, “Heritable Gene Targeting in Zebrafish Using Customized TALENs,” Nature Biotechnology, Vol. 29, No. 8, 2011, pp. 699-700. doi:10.1038/nbt.1939
- G. Silva, L. Poirot and R. Galetto, “Meganucleases and Other Tools for Targeted Genome Engineering: Perspectives and Challenges for Gene Therapy,” Current Gene Therapy, Vol. 11, No. 1, 2011, pp. 11-27. doi:10.2174/156652311794520111
- D Hockemeyer, H Wang and S Kiani, “Genetic Engineering of Human Pluripotent Cells Using TALE Nucleases,” Nature Biotechnology, Vol. 29, No. 8, 2011, pp. 731-734. doi:10.1038/nbt.1927
- H. Li, V. Haurigot and Y. Doyon, “In Vivo Genome Editing Restores Haemostasis in a Mouse Model of Haemophilia,” Nature, Vol. 475, No. 7355, 2011, pp. 217-221. doi:10.1038/nature10177
- J. A. Townsend, D. A. Wright and R. J. Winfrey, “HighFrequency Modification of Plant Genes Using Engineered Zinc-Finger Nucleases,” Nature, Vol. 459, No. 7245, 2011, pp. 442-445. doi:10.1038/nature07845
- K. Osakabe, Y. Osakabe and S. Toki, “Site-Directed Mutagenesis in Arabidopsis Using Custom-Designed Zinc Finger Nucleases,” Proceedings of the National Academy of Sciences of the United States,Vol. 107, No. 26, 2010, pp. 12034-12039. doi:10.1073/pnas.1000234107
- A. D. Goldberg, L. A. Banaszynski and K. M. Noh, “Distinct Factors Control Histone Variant H3.3 Localization at specific Genomic Regions,” Cell, Vol. 140, No. 5, 2010, pp. 678-691. doi:10.1016/j.cell.2010.01.003
- J. R. Mendell, C. Shilling and N. D. Leslie, “EvidenceBased Path to Newborn Screening for Duchenne Muscular Dystrophy,” Annals of Neurology, Vol. 73, No. 4, 2012, pp. 304-313. doi:10.1002/ana.23528
- J. Oshima, D. B. Magner and J. A. Lee, “Regional Genomic Instability Predisposes to Complex Dystrophin Gene Rearrangements,” Human Genetics Vol. 126, No. 3, 2009, pp. 411-423. doi:10.1007/s00439-009-0679-9
- L. L. Baumbach, J. S. Chamberlain and P. A. Ward, “Molecular and Clinical Correlation of Deletion Leading to Duchenne and Becker Muscular Dystrophies,” Neurology, Vol. 39, No. 4, 1989, pp. 465-474. doi:10.1212/WNL.39.4.465
- A. Pizzuti, M. Pieretti and R. G. Fenwick, “A Transposon-Like Element in the Deletion-Prone Region of the Dystrophin Gene,” Genomics, Vol. 13, No. 3, 1992, pp. 594-600. doi:10.1016/0888-7543(92)90129-G
- Z. Lu, “Interaction of Nonsense Suppressor tRNAs and Codon Nonsense Mutations or Termination Codons,” Advances in Biological Chemistry, Vol. 2, No. 3, 2012, pp. 301-314. doi:10.4236/abc.2012.23038
- J. F. Burke and A. E. Mogg, “Suppression of a Nonsense Mutation in Mammalian Cells in Vivo by the Aminoglycoside Antibiotics G-418 and Paromomycin,” Nucleic Acids Research, Vol. 13, No. 17, 1985, pp. 6265-6272. doi:10.1093/nar/13.17.6265
- C. Bertoni, C. Lau and T. A. Rando, “Restoration of Dystrophin Expression in Mdx Muscle Cells by Chimeraplast-Mediated Exon Skipping,” Human Molecular Genetics, Vol. 12, No. 10, 2003, pp. 1087-1099. doi:10.1093/hmg/ddg133
- J. Alter, F. Lou and A. Rabinowitz, “Systemic Delivery of Morpholino Oligonucleotide Restores Dystrophin Expression Bodywide and Improves Dystrophic Pathology,” Nature Medicine, Vol. 12, No. 2, 2006, pp. 175-177. doi:10.1038/nm1345
- T. Yokota, Q. L. Lu and T. Partridge, “Efficacy of Systemic Morpholino Exon-Skipping in Duchenne Dystrophy Dogs,” Annals of Neurology, Vol. 65, No. 6, 2009, pp. 667-676. doi:10.1002/ana.21627
- C. J. Klein, D. D. Coovert and D. E. Bulman, “Somatic Reversion/Suppression in Duchenne Muscular Dystrophy (DMD): Evidence Supporting a Frame-Restoring Mechanism in Rare Dystrophin-Positive Fibers,” The American Journal of Human Genetics, Vol. 50, No. 5, 1992, pp. 950- 959.
- J. R. Mendell, L. Rodino-Klapac and Z. Sahenk, “Gene Therapy for Muscular Dystrophy: Lessons Learned and Path Forward,” Neuroscience Letters, Vol. 527, No. 2, 1992, pp. 90-99. doi:10.1016/j.neulet.2012.04.078
- J. Zhang, L. Xiao and Y. Yin, “A Law of Mutation: Power Decay of Small Insertions and Small Deletions Associated with Human Diseases,” Applied Biochemistry and Biotechnology, Vol. 162, No. 2, 2010, pp. 321-328. doi:10.1007/s12010-009-8793-7
- L. Xiao, W. Sun and J. Zhang, “An Excess of G over C Nucleotides in Mutagenesis of Human Genetic Diseases,” Molecular Biotechnology, Vol. 48, No. 1, 2011, pp. 1-6. doi:10.1007/s12033-010-9341-y
NOTES
*These authors contributed equally to this paper.
#Corresponding author.