Computational Chemistry
Vol.05 No.03(2017), Article ID:77430,12 pages
10.4236/cc.2017.53008
Quantum Chemical Characterization of Hydrogen Bonding Sites in Three 4-(4-Halo-Phenyl)-6-(Furan-2-yl) Pyrimidin-2-Amine Derivatives
Yafigui Traore, Kafoumba Bamba, Nahossé Ziao*, Sopi Thomas Affi, Mamadou Guy-Richard Kone
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université NanguiAbrogoua, Abidjan, Côte-d’Ivoire
![]()
Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: May 25, 2017; Accepted: July 2, 2017; Published: July 5, 2017
ABSTRACT
Hydrogen bonding (HB) sites in three pyrimidine compounds derivatives (DP), namely 4-(4-fluorophenyl)-6-(furan-2-yl) pyrimidin-2-amine (DP-1), 4-(4-ch- lorophenyl)-6-(furan-2-yl) pyrimidin-2-amine (DP-2) and 4-(4-bromophenyl)- 6-(furan-2-yl) pyrimidin-2-amine (DP-3), have been investigated by quantum chemistry methods, especially at HF/6-311+G(d,p) and B3PW91/6-311+G(d,p) levels. Hydrogenfluori deserved as probe for hydrogen bonding complexes. Molecular electrostatic potential maps, geometricparameters of HB com- plexes, as well as energetic parameters of the complexation reactions have been computed. Finally, one out of two nitrogen atoms of pyrimidine nucleus has been identified as the major hydrogen bonding site in the three pyrimidine derivatives, with respective percentages of around 83.0% and 93.2% at HF/6-311+G(d,p) and B3PW91/6-311+G(d,p) levels.
Keywords:
Analgesic, Hydrogen Bonding, Pyrimidine Derivatives, Quantum Chemistry

1. Introduction
Pain accounts for more than 90% of medical consultations because it is present in most diseases [1] [2] . It is widely spread and has a very high social and economic cost [3] . Thus, fight against pain mobilizes medical corps, researchers and policy makers for its management [4] as well as the discovery of new molecules capable of treating it more efficiently. Pyrimidine derivatives offer a good access to new varieties as evidenced by numerous works for this purpose [5] [6] . The three derivatives referred to in this work belong to this group and have analgesic properties. They were synthesized by Vishal et al. [7] , their 2D structures are shown in Figure 1.
2. Computational Details
2.1. Calculation Level
2.2. Molecular Electrostatic Potential (MEP)
Electrostatic potential is the interaction energy between a molecule and a positive unit charge (proton) placed at a point of its environment. It is by the electrostatic potential that a molecule is first “seen” or “estimated” by another chemical species that approaches it [24] . It is a real physical property that can be determined theoretically as well as experimentally by X-ray diffraction methods [24] [25] . It is defined by the expression 1 below in atomic unit [24] [25] [26] .
ZA is the charge of the nucleus A, located at  A and
A and 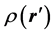 the electron density function of the molecule. Molecular electrostatic potential is an important tool for predicting the reactive properties of a molecule and it has been widely used, especially in the case of electrostatic reactions [24] such as hydrogen bonding interactions. Thus an electrophilic attack will tend to occur at the region where the potential
the electron density function of the molecule. Molecular electrostatic potential is an important tool for predicting the reactive properties of a molecule and it has been widely used, especially in the case of electrostatic reactions [24] such as hydrogen bonding interactions. Thus an electrophilic attack will tend to occur at the region where the potential 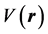 is the most negative [27] , the local minima [24] .
is the most negative [27] , the local minima [24] .
2.3. Geometry Optimization
Each of the DP derivatives has four heteroatoms. Four hydrogen bonding complexes were constructed, with the hydrogen fluoride molecule (HF) as the hydrogen bond donor (HBD). The characteristic geometric parameters of an hydrogen bond [28] are presented in Figure 2.
α is the HB linearity angle, θ is the directionality angle, d length of Hydrogen bond. Thus, before starting a computation on a complex, the initial values below have been assigned (Figure 3): α = 180˚, θ1 = θ2 = 120˚ for sp2 nitrogen and oxygen, 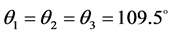 for the sp3nitrogen and d = 2 Å for all the complexes.
for the sp3nitrogen and d = 2 Å for all the complexes.
2.4. Energetic Parameters
Energetic parameters of the hydrogen bond were calculated considering the

Figure 2. Geometric parameters α, θ and d describing an Hydrogen bond.

Figure 3. Definition of geometric parameters characteristic of hydrogen bond.
formation of complexes on each heteroatom according Reaction (2):
Hydrogen bonding energy is calculated by subtracting the energies of the isolated fragments from that of the complex [23] [29] [30] [31] according to Equation (3):
![]() stands for BSSE. Calculations are carried out at a temperature of 298.15 K under normal atmospheric pressure (P = 1 atm). Moreover, the total energy E of an isolated molecule is obtained by summing the energies of the electronic movements, translation, rotation and vibration [32] according to Equation (5):
stands for BSSE. Calculations are carried out at a temperature of 298.15 K under normal atmospheric pressure (P = 1 atm). Moreover, the total energy E of an isolated molecule is obtained by summing the energies of the electronic movements, translation, rotation and vibration [32] according to Equation (5):
In the perfect gas approximation, the translation and rotation contributions are given by the Relations (6), (7) and (8). For nonlinear molecule A and A ・・・ D:
For the diatomic molecule D (HF):
The contribution of the vibration motion to the total energy is given by Equation (9):
The Zero Point Vibrational Energy (ZPVE) contribution is the lowest vibrational level energy due to the 3N-6 normal vibration modes (3N-5 for linear molecules) of frequencies νi of the N kernels at 0 K, and defined by relation 10:
The contribution 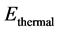 is the additional energy due to the population of vibration levels during the temperature rise from 0 to T = 298.15 K, and defined by Equation (11):
is the additional energy due to the population of vibration levels during the temperature rise from 0 to T = 298.15 K, and defined by Equation (11):
Finally, the change in the internal energy E upon Reaction (2) to 298 K is given by expression 12:
Frequencies calculations provide these different electronic, vibrational and thermal contributions, as well as entropy. The enthalpy variation at 298.15 K is given by the expression 13:
Free Enthalpy variation, at 298.15 K, is given by Equation (14):
Variation in entropy, 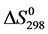 , is calculated according to expression 15:
, is calculated according to expression 15:
3. Results and Discussion
3.1. Molecular Electrostatic Potentials
Molecular electrostatic potential maps of studied DP were designed after optimization at B3PW91/6-311+G(d,p). Potential surfaces (Figure 4) are associated

Figure 4. Molecular electrostatic potential maps of the three pyrimidine derivatives.
with a color code evolving continuously from red indicating the most negative potentials to blue indicating the most positive potentials [33] . These maps indicate that the heteroatom N6 is favorable to an electrophilic attack given its approaching zone which is the redder. As for the other heteroatoms, O and N8 are disadvantaged for electrophilic attacks because of their approach zones which are in the green whereas N2, whose approach zone is orange, is less favorable than N6. The results are similar for the three molecules. It is thus apparent that N2 and N6 nitrogen atoms of pyrimidine nucleus are the most nucleophilic, thus favorable to hydrogen bonding in these three derivatives.
3.2. Geometric Parameters
Geometry optimization of the hydrogen bond complexes on the oxygen atom did not succeed. Indeed, after optimization, migration of the probe from the oxygen atom to the N6 nitrogen atom of the pyrimidine nucleus is observed, whatever the DP molecule.
This phenomenon may be explained by the participation of one of the lone pairs of oxygen atom in the aromatic character of the furan. The commitment of this doublet in electronic delocalization depletes the oxygen atom. Probe migrations have recently been observed in studies conducted by Ziao et al. [34] [35] , both in the case of two imidazopyridinyl-chalcone derivatives and four benzimidazolyl-chalcone derivatives using water molecule as probe. Thus, only energetic parameters of the complexes on the nitrogen atoms of these three DP molecules have been calculated. Geometric parameters of the complexes formed on the three nitrogen atoms are recorded in Table 1 below.
Results of Table 1 show that for the same level of calculation and hydrogen bonding site, values obtained for each parameter are virtually the same from one molecule to another. Thus for the linearity angles α of the complexes formed on the N2 and N6 atoms with sp2 hybridization, the mean values are respectively 165.7˚ and 160.5˚ at the DFT level and 165.3˚ and 156.8˚ at the HF level for the

Table 1. Geometrical parameters of hydrogen bond complexes on N2, N6 and N8 atoms.
3.3. Energetic Parameters
All values of the enthalpies changes are negative (Table 2), expressing exother- mic reactions whereas some values of the free enthalpies changes are negative translating spontaneous complexations, while others are positive, showing non- spontaneous reactions in the conditions of the study. Thus, the two levels of theory predict spontaneous reactions on N6 for the three derivatives and on N2 for the DP-1 and DP-2 derivatives; for DP-3, the HF method predicts a spon- taneous reaction and DFT provides a non-spontaneous reaction as on nitrogen N8 for the two theories levels. A classification in ascending order of the free enthalpy change of complexation gives the following result for each derivative, and also for each computation level:  . It appears from this energetic study that on N6 nitrogen atom, the variation of the free enthalpy of complexation is the smallest, values being −4.649 kcal/mol, −4.500 kcal/mol and −0.951 kcal/mol with DFT method and −1.840 kcal/mol, −1.758 kcal/mol and −1.740 kcal/mol with HF method respectively for DP-1, DP-2 and DP-3. Therefore N6 is the preferred hydrogen bonding site in these pyrimidine derivatives.
. It appears from this energetic study that on N6 nitrogen atom, the variation of the free enthalpy of complexation is the smallest, values being −4.649 kcal/mol, −4.500 kcal/mol and −0.951 kcal/mol with DFT method and −1.840 kcal/mol, −1.758 kcal/mol and −1.740 kcal/mol with HF method respectively for DP-1, DP-2 and DP-3. Therefore N6 is the preferred hydrogen bonding site in these pyrimidine derivatives.
Table 3. Percentages of N2 and N6 nitrogen H-bond complexes.
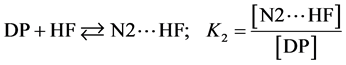
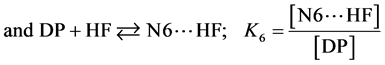
If we consider 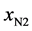 and
and 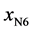 the fractions of the complexes formed on N2 and N6 respectively, then:
the fractions of the complexes formed on N2 and N6 respectively, then:

The various values of  and
and  are summarized in Table 3.
are summarized in Table 3.
Values in Table 3 show that, whatever the calculation method is, the complexes formed on N2 nitrogen are in a minority compared to those formed on N6 nitrogen. This result indicates that the N6 nitrogen atom in the pyrimidine is the major hydrogen bonding site with the average percentages of 82.98% at the B3PW91/6-311+G(d,p) level and 93.19% at the HF/6-311+G(d,p) level.
4. Conclusion
Cite this paper
Traore, Y., Bamba, K., Ziao, N., Affi, S.T. and Kone, M.G.-R. (2017) Quantum Chemical Characteriza- tion of Hydrogen Bonding Sites in Three 4- (4-Halo-Phenyl)-6-(Furan-2-yl) Pyrimidin- 2-Amine Derivatives. Computational Che- mistry, 5, 91-102. https://doi.org/10.4236/cc.2017.53008
References
- 1. Le Bars, D. and Willer, J.-C. (2004) EMC-Anesthésie Réanimation. Physiologie de la douleur, 1, 227-266.
- 2. Jitendra, K.G., Pramod, K.S., Rupesh, D., Sambhu, C.M., Anshu, C. and Prabhakar, K.V. (2011) Synthesis and Analgesic Activity of Novel Pyrimidine Derivatives of Coumarin Moiety. Acta Poloniae Pharmaceutica-Drug Research, 68, 785-793.
- 3. Harald, B., Beverly, C., Vittorio, V., Rob, C. and Derek, G. (2006) Survey of Chronic Pain in Europe: Prevalence, Impact on Daily Life, and Treatment. European Journal of Pain, 10, 287-333.
https://doi.org/10.1016/j.ejpain.2005.06.009 - 4. Clément Cousin (2012) Du droit du patient de recevoir des soins antalgiques à l’obligation du médecin de prendre en charge la douleur. Médecine & Droit, 2012, 158-160.
https://doi.org/10.1016/j.meddro.2012.05.001 - 5. Anshu, C., Pramod, K.S., Prabhakar, V. and Rupesh, D. (2011) Synthesis of Novel Pyrimidine Derivative and Its Biological Evaluation. Analele Universitatii din Bucuresti-Chimie (serie noua), 20, 123-140.
- 6. Mohamed, M.S., Awad, S.M. and Sayed, A.I. (2010) Synthesis of Certain Pyrimidine Derivatives as Antimicrobial Agents and Anti-Inflammatory Agents. Molecules, 15, 1882-1890.
https://doi.org/10.3390/molecules15031882 - 7. Vishal, D.J., Kshirsagar, M.D. and Singhal, S. (2012) Synthesis and Pharmacological Study of Some Novel Pyrimidines. Der Pharmacia Sinica, 3, 343-348.
- 8. Naik, T.A. and Chikhalia, K.H. (2007) Studies on Synthesis of Pyrimidine Derivatives and Their Pharmacological Evaluation. E-Journal of Chemistry, 4, 60-66.
https://doi.org/10.1155/2007/507590 - 9. Salman, A., Tamara, A., Naji, S. and Farooq, I.M. (2013) Synthesis, Characterization and Cytotoxic Activity of Some Pyrimidine Derivatives. Journal of Al-Nahrain University, 16, 84-92.
- 10. Sun, L., Wu, J., Zhang, L.Z., Luo, M. and Sun, D.Q. (2011) Synthesis and Antifungal Activities of Some Novel Pyrimidine Derivatives. Molecules, 16, 5618-5628.
https://doi.org/10.3390/molecules16075618 - 11. Asmaa, E., Ehab, K. and Gedawy, M. (2013) Synthesis and Anticancer Activity of Novel 2-Pyridyl Hexahyrocyclooctathieno [2,3-d]Pyrimidine Derivatives. European Journal of Medicinal Chemistry, 63, 224-230.
- 12. Bansal, S., Chaudhary, A.N. and Kothiyal, P. (2013) Microwave Assisted Synthesis and Antibacterial Activity of Pyrimidine Derivatives. International Journal of Pharmacy and Pharmaceutical Sciences, 5, 346-348.
- 13. Sharma, O.P., Rajeev, K.S., Birendra, S., Varadaraj, B.G., Gautham, G.S., Jayashree, B.S. and Sreenivasan, K.K. (2012) Synthesis, Spectral Characterization & Antimicrobial Evaluation of Some Novel Pyrimidine-2,4(1H,3H)-Diones. Indo Global Journal of Pharmaceutical Sciences, 2, 70-75.
- 14. Bhargava, S. and Rajwanshi, L.K. (2013) Synthesis of Some Novel Pyrido[2,3-d]Pyrimidine Derivatives and Their Antimicrobial Investigations. Indian Journal of Chémistry, 52B, 448-452.
- 15. Sonia, D., Arikkatt, B., Mathew, V., Chandran, J., Joseph, M., Bhat, A.R. and Krishnakumar, K. (2014) Highly-Efficient Conversion of Primary Amides to Nitriles Using Indium(III) Triflate as the Catalyst. International Journal of Organic and Bioorganic Chemistry, 4, 1-5.
https://doi.org/10.4236/ijoc.2014.41001 - 16. Madhu, R., Mahetaa, N.M., Pashab, T.Y. and Patel, S. (2013) Synthesis and Antitubercular Activity of Some Novel {1[(1phenylethylidene) Amino] Naphtho [2,1-B] Furan-2-yl}4-Substituted Pyrimidin-2-Amine Derivatives. International Journal of Pharmaceutical Sciences Review and Research, 23, 72-76.
- 17. Chaydhary, A., Singh, A., Kumarverma, P. and Walailak, J. (2016) Novel Series of Pyrimidine Derivatives as Anti-inflammatory Agents. Science & Technology, 13, 789-802.
- 18. Dansena, H., Hj, D. and Chandrakar, K. (2015) Pharmacological Potentials of Pyrimidine Derivative: A Review. Asian Journal of Pharmaceutical and Clinical Research, 8, 171-177.
- 19. Sharma, V., Chitranshi, N. and Agarwal, A.K. (2014) Significance and Biological Importance of Pyrimidine in the Microbial World. International Journal of Medicinal Chemistry, 2014, 1-31.
- 20. Arunan, E., Desiraju, G.R., Klein, R.A., Sadlej, J., Scheiner, S., Alkorta I., Clary, D.C., Crabtree, R.H., Dannenberg,J.J., Hobza, P., Kjaergaard, H.G., Legon, A.C., Mennucci, B. and Nesbitt, D.J. (2011) Definition of the Hydrogen Bond. Pure and Applied Chemistry, 83, 1637-1641.
https://doi.org/10.1351/PAC-REC-10-01-02 - 21. Ziao, N., Questel, J.Y.L. and N’guessan, T. (2005) Caractérisation Energétique Des Sites De Fixation De Liaisons Hydrogène Dans Les Aminonitriles Par La Méthode De La Fonctionnelle De La Densité. Journal de la Société Ouest-Africaine de Chimie, 020, 101-118.
- 22. Gaussian 03, Revision C.01, Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K.,Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C. and Pople, J.A. (2004) Gaussian Incorporation, Wallingford.
- 23. Daza, M.C., Dobado, J.A. and Molinaa, J.M. (1999) Basis Set Superposition Error-Counterpoise corrected Potential Energy Surfaces. Application to Hydrogen Peroxide...X (X=F-, Cl-, (X=F-, Cl-, Br-, Li+,Na+)Br-,Li+, Na+) Complexes. Journal of Chemical Physics, 110, 11806-11813.
https://doi.org/10.1063/1.479166 - 24. Murray, J.S., Seminario, J.M., Concha, M.C. and Politzer, P. (1992) An Analysis of Molecular Electrostatic Potentials Obtained by a Local Density Functional Approach. International Journal of Quantum Chemistry, 44, 113-122.
https://doi.org/10.1002/qua.560440204 - 25. Riley, K.E., Tran, K.A., Lane, P., Murray, J.S. and Politzer, P. (2016) Comparative Analysis of Electrostatic Potential Maxima and Minima on Molecular Surfaces, as Determined by Three Methods and a Variety of Basis Sets. Journal of Scientific Computing, 17, 273-284.
https://doi.org/10.1016/j.jocs.2016.03.010 - 26. Drissi, M., Benhalima, N., Megrouss, Y., Rachida, R., Abdelkader, C. and Hamzaoui, F. (2015) Theoretical and Experimental Electrostatic Potential around the M-Nitrophenol Molecule. Molecules, 20, 4042-4054.
https://doi.org/10.3390/molecules20034042 - 27. Balachandran, V. and Karunakaran, V. (2014) Molecular Structure, Vibrational Spectroscopic, Hyperpolarizability, Natural Bond Orbital Analysis, Frontier Molecular Orbital Analysis and Thermodynamic Properties of 2, 3, 4, 5, 6-Pentafluoro-phenylacetic Acid. Spectrochimica Acta Part A, 127, 473-483.
https://doi.org/10.1016/j.saa.2014.02.129 - 28. Desiraju, G.R. and Steiner, T. (1999) The Weak Hydrogen Bond in Structural Chemistry and Biology. Oxford University Press, Chichester.
- 29. Richard, R.M., Lao, K.U. and Herbert, J.M. (2013) Achieving the CCSD (T) Basis-Set Limit in Sizable molecular Clusters: Counterpoises Corrections for the Many-Bady Expansion. The Journal of Physical Chemistry, 4, 2674-2680.
- 30. Halkier, A., Klopper, W., Helgaker, T., Jorgensen, P. and Taylor, P.R. (1999) Basis Set Convergence of the Interaction Energy of Hydrogen-Bonded Complexes. Journal of Chemical Physics, 111, 9157-9167.
https://doi.org/10.1063/1.479830 - 31. Frans, B., Duijneveldt, V., Jeanne, G.C.M., Rijdt, D. and Lenth, J.H. (1994) State of the Art in Counterpoise Theory. Chemical Reviews, 94, 1873-1885.
https://doi.org/10.1021/cr00031a007 - 32. Ochterski, J.W. (2016) Thermochemistry. Gaussian Incorporation, Wallingford.
- 33. Abdel, B.H.M., Hanan, G.E., Mohamed, M.E.O. and Ibrahim, M.A. (2015) Molecular Electrostatic Potential Analysis of Nano-Scale Fullerene (C60) Crystals and Some Specifc Derivatives: DFT Approach. Journal of Nanomaterials & Molecular Nanotechnology, 4, 2.
- 34. Affi, S.T., Nahossé, Z., Mahama, O., Drissa, S. and Kicho, Y. (2014) Caractérisation Théorique des Sites D’interaction par Liaison Hydrogène de 3-(4-Isopropylphenyl)-1-(2-ethylimidazopyridin-3-yl) Prop-2-en-1-One et de 3-(2-Methoxyphenyl)-1-(2-Methylimidazopyridin-3-yl) Prop-2-en-1-One. European Journal of Scientific Research, 123, 340-347.
- 35. Kone M.G.R., Affi, S.T., Nahossé, Z., Kafoumba, B. and Assanvo, E.F. (2015) Hydrogenbonding Sites in Benzimidazolyl-Chalconesmolecules: An Ab Initio and DFT Investigation. Journal of Chemical and Pharmaceutical Research, 7, 805-8012.
- 36. Bagheri, S., Masoodi, H.R. and Yousofvand, A. (2016) Exploring the Role of Substituents on Cooperativity between N...HF and CH...F Hydrogen Bonds in Ternary Systems Involving Aromatic Azine: Substituted Complexes of S-Triazine: HF: S-Triazine as a Working Model. Computational and Theoretical Chemistry, 1092, 12-18.
https://doi.org/10.1016/j.comptc.2016.07.002 - 37. Silva, D.S. and Oliveira, B.G. (2017) New Insights about the Hydrogen Bonds Formed between Acetylene and Hydrogen Fluoride: π ... H, C ... H and F ... H. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 173, 160-169.
https://doi.org/10.1016/j.saa.2016.08.054 - 38. Nathalia, B., Lima, D., Victor, H., Rusu, M. and Ramos, N. (2011) Hydrogen Bonds between Phthalimide and Hydrogen Fluoride: A Theoretical Study. International Journal of Quantum Chemistry, 111, 1387-1394.
https://doi.org/10.1002/qua.22648 - 39. Batsanov, S.S. (2001) Van der Waals Radii of Elements. Inorganic Materials, 37, 871-885.
https://doi.org/10.1023/A:1011625728803