Computational Chemistry
Vol.04 No.03(2016), Article ID:67044,8 pages
10.4236/cc.2016.43006
Protonation Sites in Benzimidazolyl-Chalcones Molecules: An ab Initio and DFT Investigation
Mamadou Guy-Richard Kone, Sopi Thomas Affi, Nahossé Ziao*, Kafoumba Bamba, Edja Florentin Assanvo
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, Abidjan, République de Côte-d’Ivoire

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 March 2016; accepted 30 May 2016; published 2 June 2016
ABSTRACT
In this work, we have focused our investigations on the protonation sites predilection in the benzi- midazolyl-chalcones (BZC) derivatives. Particularly, we are interested in the study of geometrical and energetical parameters. BZC are well known for their particularly nematicidal activity. Ten (10) BZC derivatives coded BZC-1 to BZC-10, with various larvicidal concentrations, have been selected for this work. They all are different one from another by the phenyl ring which is substi- tuted by electron modulators such as alkyl, hydroxyl, alkoxy, aminoalkyl, halogen and nitro or replaced by the furan. Quantum chemical methods, namely HF/6-311 + G(d,p) and MPW1PW91/6- 311 + G(d,p) theory levels have been used to determine the geometrical and energetical parame- ters by the protonation on each heteroatom of the BZC derivative. An accuracy results with rela- tively less time consuming has been obtained using Hartree-Fock (HF) and Density Functional Theory methods (DFT/MPW1PW91). The calculations results allow identifying the sp2 nitrogen as the preferential site of protonation in BZC derivative compounds.
Keywords:
Benzimidazolyl-Chalcone, Quantum Chemistry, Protonation, Proton Affinity, Gas Phase Basicity

1. Introduction
The derivatives of the benzimidazolyl present a pharmacological interest significantly due to their therapeutic virtues in many diseases. Several studies have demonstrated that the derivatives of benzimidazolyl are antihistaminic [1] , antifungal [2] , antiallergic [3] , antibacterial [4] - [6] , analgesic [7] , antiplasmodial [8] and antiviral [9] . In recent years, it has been documented and reported that the main cause of gastro-intestinal infections of small ruminants as well as loss and reduction in productivity of the livestock is due to the effect of nematodes [10] [11] . Nematodes (worm from hearth, freshwater or sea) are also being the origin of most human parasitic diseases [12] [13] . The development of an “ideal” anthelmintic seems to be possible with the benzimidazoles, imidazothiazoles, tetrahydropyrimidines and organophosphate compounds. Such ananthelmintic should possess a broad spectrum of action, a high degree of efficiency, a good safety margin and aflexibility of use. However, all reported studies related to benzimidazolyl-chalcones are limited to the synthesis, structural characterization and the investigation of the activity properties. Till date, researches have led to synthesize several hundred of compound. A few have been selected for their effective anthelmintic activity at broad spectrum, and among them, the benzimidazolyl-chalcones (BZC) kernel.
These different molecules have been synthesized by Ouattara et al. [14] . The numerous therapeutic properties of BZC could be related to the conformation of molecules and the interactions they can establish with other molecules. Among the different properties of a biological molecule, the proton-transfer reactions play a very important role in the molecular interactionsand biologicalsystems [15] [16] . The BZC ability to protonate is likely to affect its fate in the environment, both as regards its transport, his stay, its reactivity in the surrounding environment until the target molecule and as regards its recognition by the receiver. Protonation or deprotonation is the first step in many fundamental chemical rearrangements and in most of the enzymatic reactions [16] . Two quantities are used to characterize the ability of a molecule in the gas phase or phase condensed to accept a proton. The gas phase basicity (GB) which is the opposite of the variation of free energy associated with the protonation reaction (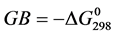 ) and the proton affinity (
) and the proton affinity ( ) [17] - [19] . The gas phase basicity and the proton affinity (PA) can inform us on the capacity of a site to accept a proton. A recently work [20] has tried to determine the preferential site of protonation between nitrogen atoms and oxygen. The aim of our present work is to characterize the preferential site of protonation in benzimidazolyl-chalcones by using different quantum chemical methods.
) [17] - [19] . The gas phase basicity and the proton affinity (PA) can inform us on the capacity of a site to accept a proton. A recently work [20] has tried to determine the preferential site of protonation between nitrogen atoms and oxygen. The aim of our present work is to characterize the preferential site of protonation in benzimidazolyl-chalcones by using different quantum chemical methods.
2. Computational Details
2.1. The Calculation Level
All the calculations have been carried out, on ten BZC compounds (Figure 1), with the software GAUSSIAN 03 [21] , in vacuo, at the HF/6-311 + G(d,p) and MPW1PW91/6-311 + G(d,p) theories levels. Choosing Hartree- Fock (HF) and Density Functional Theory methods (DFT/MPW1PW91) allows accuracy results with relatively

Figure 1. Structure of the benzimidazolyl-chalcones (BZC) studied.
less time consuming. Usually, it’s highly recommended to use at least two different methods in quantum mecha- nical calcultions, so that consistency of results can be, in some way, verified. The choice of split-valence and triple-dzeta basis sets is justified by the need of sufficiently extended levels. Diffuse and polarization functions are important, whenever the matter is intermolecular interactions. Each of the protonation complexes has been fully optimized, with a frequencies calculation at the same levels of theories.
2.2. Geometry Optimization
Geometry initialization of the protonated molecules has been carried out by utilizing the valence angles around the concerned heteroatoms (carbonyl oxygen, sp2 and sp3 nitrogens) of the benzimidazolyl-chalcones (BZC) kernel. According to Gillespie method or V.S.E.P.R (Valence Schell Electron Pair Repulsion) method, the average values of valence angles around sp2 and sp3 atoms equal 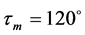 and
and  respectively. Figure 2 shows the initial geometries of protonation.
respectively. Figure 2 shows the initial geometries of protonation.
2.3. Energetic Parameters
The protonation is a process in which a Lewis base B fixed a proton to give a protonated molecule BH+ according Equation (1):
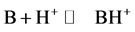 (1)
(1)
Knowing the variations of energy contributions to the internal energy at 0 K and at 298.15 K between the products and reactants contributes to the energy characterization of a chemical reaction. For a given energy parameter X, its variation is determined according Equation (2):
 (2)
(2)
The geometrical optimization and the calculation of the frequencies of the free molecules allow us to determine the variation of the internal energy at 0 K (ΔE0K) and at 298.15 K (ΔE298K) with respect to the reaction studied. The variation of the internal energy to 298.15 K, ΔE298K, constitutes a sum of different electronic, translation, rotation and vibration contributions and the internal energy at 0 K given in Equation (3):
 (3)
(3)
ZPVE (Zero Point Vibrational Energy) contribution, i.e. lowest vibrational level energy, due to 3N − 6 normal vibrational modes (3N − 5 for the linear molecules), each with frequency ni, up to N kernels at 0 K, is defined according Equation (4):
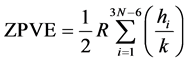 (4)
(4)
k is the Boltzmann constant; h Planck’s constant; R the constant of perfect gases. To obtain the corresponding energy at 298.15 K, it is necessary to take into account the extra energy due to vibrational levels population during temperature rising from 0 to 298.15 K. Thus, Equation (4) becomes Equation (5):
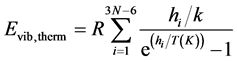 (5)
(5)

Figure 2. Definition of geometrical parameters describing the protonation sites.
As regards the contributions of rotation and translation, they are drawn from the approximation of perfect gases according relationship (6):
 (6)
(6)
As a result, internal energy variation at 298.15 K is given by Equation (7):
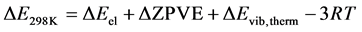 (7)
(7)
From this relationship, it is deducted the enthalpy of the reaction at 298.15 K. It corresponds to the variation of the internal energy corrected by the term ∆(PV), either 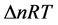 (∆n being the variation in the number of gaseous moles of the reaction):
(∆n being the variation in the number of gaseous moles of the reaction):
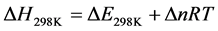 (8)
(8)
The entropic contributions of translation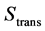 , of rotation
, of rotation  and of vibration
and of vibration  of given species at 298.15 K are regrouped in the total entropy term S and the Gibbs energy, at 298.15 K, linked to the reaction is simply obtained by the relationship (9):
of given species at 298.15 K are regrouped in the total entropy term S and the Gibbs energy, at 298.15 K, linked to the reaction is simply obtained by the relationship (9):
 (9)
(9)
The electronic energy of an isolated proton equals zero, therefore it doesn’t appear in the calculation of the variation of the electronic energy. Again, for the proton, the translational energy is different from zero
( at 298.15 K and the entropy of translation equals
at 298.15 K and the entropy of translation equals
 . Therefore, the proton affinity in the gas phase (PA) and the gas phase basicity
. Therefore, the proton affinity in the gas phase (PA) and the gas phase basicity
(GB), are easily determined according the above equations.
3. Results and Discussion
3.1. Geometrical Parameters
The descriptors selected for this study are related to the average valence angles around the potential protonated sites ,
,  and
and . It is assumed that in sp2 hybridization state, each of the lone pairs of the oxygen atom forms with the carbonyl bond
. It is assumed that in sp2 hybridization state, each of the lone pairs of the oxygen atom forms with the carbonyl bond  an angle of 120.0˚, and that the lone pair of the nitrogen atom also points out with an angle of 120.0˚ with each adjacent bond. In the case of sp3 nitrogen, the optimal angle is assumed to be 109.5˚. It is clear from these observations that the binding geometry, the more probable, which can be formed between hydrogen and each basic site, will be the one measuring the average valence angles θm, which better approaches the optimal angles. The values measured of the valence angles θm are shown in Table 1. In this table, θ1Nsp2 equals the average of the angles
an angle of 120.0˚, and that the lone pair of the nitrogen atom also points out with an angle of 120.0˚ with each adjacent bond. In the case of sp3 nitrogen, the optimal angle is assumed to be 109.5˚. It is clear from these observations that the binding geometry, the more probable, which can be formed between hydrogen and each basic site, will be the one measuring the average valence angles θm, which better approaches the optimal angles. The values measured of the valence angles θm are shown in Table 1. In this table, θ1Nsp2 equals the average of the angles  and
and , when θ3Nsp3 equals the average of the angles
, when θ3Nsp3 equals the average of the angles

Table 1. Average angles of protonated BZC on the sp2(θ1Nsp2), sp3 (θ3Nsp3) nitrogenatoms and carbonyl oxygen (θ2Osp2) calculated at the HF/6-311 + G(d,p) and MPW1PW91/6-311 + G(d,p) levels (Values expressed in ˚).
 ,
,  and
and . θ2Osp2 stands for
. θ2Osp2 stands for  describing the angle
describing the angle  (Figure 2).
(Figure 2).
The review of the values in Table 1 shows that respectively at HF/6-311+G (d,p) and MPW1PW91/6-311 + G(d,p) levels, the average values of the angles extend for θ1Nsp2, from 125.37˚ to 125.40˚ and from 125.13˚ to 125.14˚; for θ2Osp2, from 114.70˚ to 115.56˚ and from 112.36˚ to 113.18˚ and for θ3Nsp3, from 109.50˚ to 110.16˚ and from 109.91˚ to 110.06˚ respectivelyat the HF/6-311 + G(d,p) and MPW1PW91/6-311 + G(d,p) levels. Now, let’s examine which calculated angles are closer to the theoretical optimal angles according to Figure 2. For bothlevels of theories, the average θ1Nsp2 angles vary from 125.13˚ to 125.40˚ corresponding to a maximum gap of 5.40˚comparing with the ideal angle of 120.0˚. Samely, the maximum gaps obtained for θ2Osp2 and θ3Nsp3 are respectively 7.64˚ and 0.66˚. And finally, the form  corresponding to the angle θ3Nsp3, shows theclosestvalue to the theoretical one. Which confirms the tetragonalisation of sp3 nitrogen under the effect of protonation. It is thus established, following the criterion to the optimality of the valence angles, that sp3 nitrogen atom is the major protonation site in benzimidazole-chalcone kernels. Now, we’re going to examine energitical criterion to confirm or not the above conclusion.
corresponding to the angle θ3Nsp3, shows theclosestvalue to the theoretical one. Which confirms the tetragonalisation of sp3 nitrogen under the effect of protonation. It is thus established, following the criterion to the optimality of the valence angles, that sp3 nitrogen atom is the major protonation site in benzimidazole-chalcone kernels. Now, we’re going to examine energitical criterion to confirm or not the above conclusion.
3.2. Energetic Parameters
The values of the proton affinityand those of the gas phase basicity calculated at HF/6-311 + G(d,p) and MPW1PW91/6-311 + G(d,p) levelsare reported respectively in the Table 2 and Table 3.
All the values, of proton affinity and gas phase basicity of the different sites, reported in Table 2 and Table 3, are positive, indicating that protonation reactions on the different sites are exothermic and spontaneous. Further-
Table 2. Protonation energies calculated for heteroatoms at level HF/6-311 + G(d,p) in kcal/mol.
Table 3. Protonation energies calculated for heteroatoms at level MPW1PW91/6-311 + G(d,p) in kcal/mol.
more, at level HF/6-311 + G(d,p), the average values (PA and GB) for ,
,  and
and  are respectively 221.724 and 214.401 kcal/mol, 207.426 and 199.773 kcal/mol and 188.496 and 180.930 kcal/mol. At MPW1PW91/6-311 + G(d,p), the average values (PA and GB) are respectively 222.063 and 214.236 kcal/mol, 212.991 and 205.061 kcal/moland 186.658 and 178.953 kcal/mol We note greater energy values on the Nsp2 nitrogen atom compared to Osp2 and Nsp3. This means that oxygen atom and sp3 nitrogen atoms are bothlessbasic sitesandthereforehave thelowestproton affinities. All the values trend to show that the sp2 nitrogen is the major protonation site in BZC. Besides, according the energetic values, the following ascending sequence can be made:
are respectively 221.724 and 214.401 kcal/mol, 207.426 and 199.773 kcal/mol and 188.496 and 180.930 kcal/mol. At MPW1PW91/6-311 + G(d,p), the average values (PA and GB) are respectively 222.063 and 214.236 kcal/mol, 212.991 and 205.061 kcal/moland 186.658 and 178.953 kcal/mol We note greater energy values on the Nsp2 nitrogen atom compared to Osp2 and Nsp3. This means that oxygen atom and sp3 nitrogen atoms are bothlessbasic sitesandthereforehave thelowestproton affinities. All the values trend to show that the sp2 nitrogen is the major protonation site in BZC. Besides, according the energetic values, the following ascending sequence can be made: . Furthermore, the percentage of protonation on each site, can be calculated. The process was the following way, depending on the below protonation reactions ((10)-(12)), respectively, with Nsp2, Ocarbonyl and Nsp3 leading to the complexes
. Furthermore, the percentage of protonation on each site, can be calculated. The process was the following way, depending on the below protonation reactions ((10)-(12)), respectively, with Nsp2, Ocarbonyl and Nsp3 leading to the complexes ,
,  and
and , corresponding respectively to thermodynamic equilibrium constants
, corresponding respectively to thermodynamic equilibrium constants ,
,  and
and :
:
 (10)
(10)
 (11)
(11)
 (12)
(12)
If  is the fraction of the amount of BZC protonated with Nsp2, the below calculation process can lead to it:
is the fraction of the amount of BZC protonated with Nsp2, the below calculation process can lead to it:
 (13)
(13)
The different equilibrium constants are calculated from variations of free enthalpies according to the relationship (14):
 (14)
(14)
In the same way, the fraction of the amount of BZC protonated with Ocarbonyl is given by the below relation (15) and finally, the fraction of the amount of BZC protonated Nsp3 is drawn according relation (16).
 (15)
(15)
and
 (16)
(16)
From the results in Table 2 and Table 3, one can state that BZC kernel substituents have, in practice, no effet on protonation properties of the BZC kernel, since, for a kind of heteroatom, energetic values do not really vary. So, to simplify, we’ll use the average values of gas phase basicity,  , to calculate the average values of equi-
, to calculate the average values of equi-
Table 4. Average gas phase basicity and protonation percentages on sp2 nitrogen atom, carbonyl oxygen and sp3 nitrogen atom.
librium constants according Equation (14). Further, we’ll get the average protonation percentages,  of each site using Equations ((13), (15) and (16)). Results are given in Table 4.
of each site using Equations ((13), (15) and (16)). Results are given in Table 4.
With the above average values of  reported in Table 4, all the fractions
reported in Table 4, all the fractions ,
,  and
and 
tend toward 0, whereas  tends toward
tends toward . Therefore, at level HF/6-311 + G(d,p) as well as at level MPW1PW91/6-311 + G(d,p), the average protonation percentage on sp2 surrounds 100% when it surrounds 0% on both carbonyl oxygen and sp3 nitrogen. Thus, according the energetic analysis, the major protonation site is strongly the sp2 nitogen.
. Therefore, at level HF/6-311 + G(d,p) as well as at level MPW1PW91/6-311 + G(d,p), the average protonation percentage on sp2 surrounds 100% when it surrounds 0% on both carbonyl oxygen and sp3 nitrogen. Thus, according the energetic analysis, the major protonation site is strongly the sp2 nitogen.
4. Conclusion
The aim of this work was to determine the protonation major site in benzimidazolyl-chalcone (BZC) kernel as well as its energetic characteristics. Interpreting the valence average angles around each of the three heteroatoms, it has been noticed that the sp3 nitrogen atom is slightly the major site since the gap from ideal valence angle is the lowest. In the contrary, interpretation of energetic parameters, meaning proton affinity (PA) and gas phase basicity (GB), leads to design sp2 nitrogen as, strongly, the major site. These conclusions are available whatever the calculation level, meaning HF/6-311 + G(d,p) or MPW1PW91/6-311 + G(d,p). Therefore, the protonation percentage on each site has been calculated. Results show that the protonation percentage surrounds 100% on sp2 nitrogen. The conclusion is that, taking into account thermodynamic analysis, the sp2 nitrogen is the unique protonation site in BZC. From the whole results, one can also state that BZC kernel substituents have no effect on protonation properties of the kernel, since, for a kind of heteroatom, geometric or energetic values do not really vary, although substituents vary.
Cite this paper
Mamadou Guy-Richard Kone,Sopi Thomas Affi,Nahossé Ziao,Kafoumba Bamba,Edja Florentin Assanvo, (2016) Protonation Sites in Benzimidazolyl-Chalcones Molecules: An ab Initio and DFT Investigation. Computational Chemistry,04,65-72. doi: 10.4236/cc.2016.43006
References
- 1. Souness, E., Aldous, D. and Sargent, C. (2004) Immunosuppressive and Anti-Inflammatory Effects of Cyclic AMP Phosphodiesterase (PDE) Type 4 Inhibitors. Immunopharmacology, 47, 127-162. http://dx.doi.org/10.1016/S0162-3109(00)00185-5
- 2. Sanja, O., Podunavac, K. and Dragoljub, D.C. (2011) Lipophilicity and Antifungal Activity of Some 2-Substituted Benzimidazole Derivatives. Chemical Industry and Chemical Engineering Quarterly, 17, 9-15. http://dx.doi.org/10.2298/CICEQ100329044P
- 3. Kilcigil, G.A., Kus, C., Coban, T.B., Can-Eke and Iscan, M. (2004) Synthesis and. Antioxidant Properties of Novel Benzimidazole Derivatives. Journal of Enzyme Inhibition and Medicinal Chemistry, 19, 129-135.
- 4. Carta, A., Loriga, M., Zanetti, S. and Sechi, L.A. (2003) Quinoxalin-2-Ones. Part 5. Synthesis and Antimicrobial Evaluation of 3-Alkyl-, 3-Halomethyl- and 3-Carboxyethylquinoxaline-2-Ones Variously Substituted on the Benzo- Moiety. Farmaco, 58, 1251-1255. http://dx.doi.org/10.1016/s0014-827x(03)00198-8
- 5. Andriole, V.T. (2000) The Quinolones. 3rd Edition, Elsevier Academic Press, 517. http://dx.doi.org/10.1016/b978-012059517-4/50017-9
- 6. Emami, S., Shafiee, A. and Foroumadi, A. (2005) Quinolones: Recent Structural and Clinical Developments. Iranian Journal of Pharmaceutical Research, 3, 123-136.
- 7. Selvakumar, S., Sudeer Babu, I. and Chidambaranathan, N. (2012) Pharmacological Evaluation of Some Potent 2-Sub- stituted Benzimidazolylchalcones for Analgesic, Anti-Inflammatory, Anthelmintic and Central Nervous System Acti- vities. International Journal of Phytopharmacology, 3, 163-172.
- 8. Ouattara, M., Sissouma, D., Yavo, W. and Kone, M.W. (2015) Synthèse et criblage antiplasmodial de quelques benzi- midazolyl-chalcones. International Journal of Biological Sciences, 9, 1697-1710.
- 9. Mertens, A., Muller-Beckmann, B., Kampe, W., Holck, J.P. and Von der Saal, W. (1987) Synthesis of Benzimida- zol-2-Thiones from Dimedone. Journal of Medicinal Chemistry, 30, 1279-1287. http://dx.doi.org/10.1021/jm00391a004
- 10. Geerts, S. and Gryseels, B. (2000) La résistance aux médicamentschezles helminthesdroits: situation actuelle et les leconsdu bétail. Clinical Microbiology Reviews, 13, 207-222.
- 11. Waller, P.J. (2003) L’avenir devermifugesdans les programmesde lutte antiparasitairedurablespour le bétail. Helmin- thologia, 40, 97-102.
- 12. Horton, J. (2003) Programme mondialde chimiothérapie anthelminthique: l’Apprentissagede l’histoire. Tendances Parasitol, 9, 403-409.
- 13. Hotez, P.J., Brindley, P.J., Bethony, J.M., Le Roi, C.H. and Pearce, E.J. (2008) Infections Jacobson J. helminthiases: Lesgrandesmaladies Tropicales Négligées. Journal of Clinical Investigation, 118, 1311-1321. http://dx.doi.org/10.1172/JCI34261
- 14. Ouattara, M., Sissouma, D., Koné, M.W., Menan, H.E., Touré, S.A. and Ouattara, L. (2011) Synthesis and Anthelmin- tic Activity of Some Hybrid Benzimidazolyl-Chalcone Derivatives. Tropical Journal of Pharmaceutical Research, 10, 767-775. http://dx.doi.org/10.4314/tjpr.v10i6.10
- 15. Caldin, E. and Gold, V., Eds. (1975) Proton-Transfer Reactions. Chapman & Hall, London.
- 16. Reinchardt, C. (2002) Solvents and Solvent Effects in Organic Chemistry. Wiley-VCH, Weinheim. http://dx.doi.org/10.1002/3527601791
- 17. Gawinecki, R., Raczyńska, E.D., Rasala, D. and Styrcz, S. (1997) Tautomeric and Conformational Preferences in Nitraminopyridines: Comparison of Theoretical and Experimental Data. Tetrahedron, 53, 17211-17220. http://dx.doi.org/10.1016/S0040-4020(97)10142-9
- 18. Raczyńska, E. D., Darowska, M., Rudka, T. and Górnicka, E. (2002) Acid-Base Equilibria in Polyfunctional Com- pounds. Current Topics in Analytical Chemistry, 3, 125.
- 19. Raczyńska, E.D., Darowska, M., Dabkowska, I., Decouzon, J.M., Gal, F., Maria, P.C. and Dubin-Poliart, C. (2004) Experimental and Theoretical Evidence of Basic Site Preference in Polyfunctional Superbasic Amidinazine: N1,N1- Dimethyl-N2-β-(2-pyridylethyl)formamidine. Journal of Organic Chemistry, 69, 4023-4030. http://dx.doi.org/10.1021/jo030308j
- 20. Affi, T.S., Ziao, N. and Bamba, K. (2015) Détermination, par des méthodes ab initio et DFT, des sites et énergies de protonation d’une série de molécules d’imidazopyridinyl-chalcones substituées. European Scientific Journal, 11, 148- 158.
- 21. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr., J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, A.D., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C. and Pople, J.A. (2004) Gaussian, Inc., Wallingford.
NOTES

*Corresponding author.