Computational Chemistry
Vol.2 No.3(2014), Article
ID:48208,7
pages
DOI:10.4236/cc.2014.23006
DFT Study of Monochlorinated Pyrene Compounds
Ehab AlShamaileh
Department of Chemistry, Faculty of Science, The University of Jordan, Amman 11942, Jordan
Email: ehab@ju.edu.jo, ehabju@gmail.com
Copyright © 2014 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/
![]()
![]()

Received 11 May 2014; revised 10 June 2014; accepted 2 July 2014
ABSTRACT
The structure, electronic properties, and chemical reactivity of pyrene and its three monochlorinated derivatives have been theoretically studied by using density functional theory (DFT). Several methods and basis sets are tested for the optimization of the geometrical structure and the B3LYP/6-311G** method/basis set is found to be the most suitable in predicting the heat of formation of pyrene and the minimum energy optimized structure of pyrene and its monochlorinated derivatives. The geometrical parameters and the heat of formation of pyrene are in good agreement with available experimental data. The chemical reactivity and stability of the four compounds are investigated. The computed results agree well with experimental results of monochlorination of the pyrene molecule. The relative stability of the monochlorinated pyrene compounds is in the order: 1-chloropyrene > 4-chloropyrene > 2-chloropyrene.
Keywords:DFT, Pyrene, Chloropyrene, Heat of Formation, Chemical Reactivity

1. Introduction
Pyrene (C16H10) is a polycyclic aromatic hydrocarbon (PAH) consisting of four fused benzene rings. Pyrene is a flat aromatic compound and forms during incomplete combustion of organic compounds.
Pyrene and its derivatives are very important industrial materials. They are used commercially to make dyes and dye precursors. A good example is the fluorescent derivative of pyrene (sulfonated hydroxy pyrene trisodium salt) or pyranine [1] . Due to their possible use as molecular probes via fluorescence spectroscopy, several researchers investigated the theoretical and experimental electronic structure, UV-Vis absorption and fluorescence emission spectrum of pyrene and its derivatives [2] [3] .
The molecular design and the organic synthesis of light emitting materials are of great interest in the fundamental research and commercial applications [4] . Monochlorinated pyrene derivatives have been synthesized by organic chemistry and find applications as organic light emitting diodes (OLEDs) [5] .
Pyrene has strong π electron delocalization energy and hence may be substituted with many functional groups of interest. The derivatives then become potential candidate electronic materials. These pyrene derivatives can then be substituted with one or more atoms or groups with electron donor and acceptor properties. Their geometrical structures and electronic energies are therefore influenced by the substitution positions in the pyrene ring.
One of the promising pyrene derivatives are chlorinated pyrene compounds. They have been studied for many different properties and applications. For example, they have been recommended as standards in environmental chemistry and biochemical applications [6] . Chlorinated pyrene derivatives are promising materials in the fabrication of light emitting organics and as fluorescent probes in biochemistry.
The chlorination of pyrene falls in the category of electrophilic chlorination which is the combination of a carbon-centered nucleophile with an electrophilic source of chlorine to produce a chlorinated organic compound. Chlorinating agents include molecular chlorine, reagents incorporating an oxygen-chlorine bond and reagents containing a nitrogen-chlorine bond. The most common reagents for organic synthesis is N-chlorosuccinimide (NCS). The literature contains thorough experimental details of the process [7] .
A previous experimental study for the chlorination of pyrene with NCS employed HPLC fractionating followed by 1H-NMR characterization and GC-MS determination gave >95% of the substituent 1-chloropyrene [8] .
A recent study by Muff et al. aimed at studying the extent of formation of chlorinated pyrene during electrochemical oxidation processes detected, using mass spectroscopy, very small amounts of 1-chloropyrene while 2- and 4-chloropyrenes were not detected [9] . DFT calculations have been successfully applied to the study of polyaromatic hydrocarbons [10] .
In the present paper, the monochlorinated pyrene derivatives were chosen to be studied by DFT methods. Their structure, electronic properties, and chemical reactivity will be investigated and compared to available experimental findings.
2. Computational Details
The optimization of the geometries and the frequencies was carried out employing the basis set 6-311G** using the following DFT levels of theory: B3LYP, B3PW91, MPW1PW91, and PBE1PBE. All single point energy (SPE) and vibrational frequency calculations were performed at the level of theory B3LYP/6-311G**. The calculations for the heat of formation were performed using the methodology of a combination of isodesmic reactions with DFT methods. All the calculations used the Gaussian 03 program package [11] . Calculations were performed on a DELL T3500 Workstation.
3. Results and Discussion
3.1. Structural and Electronic Properties of Pyrene and Its Monochlorinated Derivatives
The ground state geometry optimization of the pyrene molecule and its monochlorinated derivatives: 1-chloropyrene, 2-chloropyrene, and 4-chloropyrene were performed by using the DFT/B3LYP/6-311G** method and shown in Figure 1. All the optimized geometries of pyrene and its derivatives kept good planarity. Details of the resulted calculated structural parameters for all four molecules are presented in Table1
The calculated bond lengths and bond angles of pyrene are in good agreement with the experimental values obtained by X-ray crystal diffraction [12] . This result is a good indication for the validity of the computational method. The similarity of the derivatives to the parent molecule explains the similarity in bond lengths and bond angles in all the molecules especially as we move farther away from the substituent (Cl). The carbon-chlorine bond length values average at around 1.763 Å and this is very close to the mean Caromatic-Cl bond length of 1.76 Å.
The computed dipole moments are in the order of 4-chloropyrene (2.04) < 1-chloropyrene (2.11) < 2-chloropyrene (2.49). This predicts that 4-chloropyrene is the most stable compound in organic chlorination reaction media due to its lowest dipole moment value followed by 1-chloropyrene. On the other hand, the computed minimum energy (a.u.) is in the order of 2-chloropyrene (−1075.53213120) < 1-chloropyrene (−1075.53096880) < 4-chloropyrene (−1075.53084833). This result assumes that 2-chloropyrene exhibits the
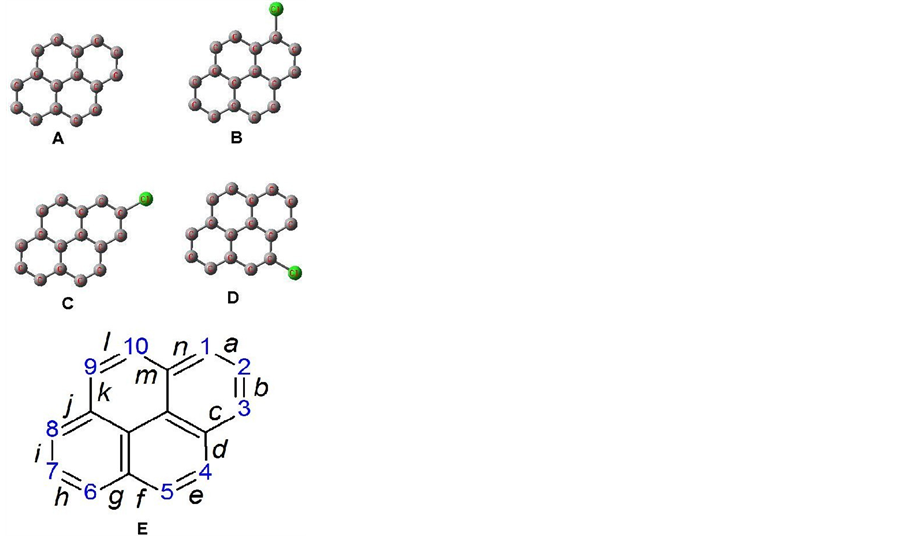
Figure 1. Optimized ground state geometry of pyrene (A) and its monochlorinated derivatives: 1-chloropyrene (B), 2-chloropyrene (C), and 4-chloropyrene (D). The numbering of the pyrene molecule and the bond labeling (derived from the IUPAC nomenclature of organic chemistry) is shown in the structure (E) and is used for all the molecules.

Table 1. Calculated bond lengths (Å), dipole moments (Debye) and energies (hartree) for the studies molecules using the DFT/B3LYP/6-311G** method. Bond designations are shown in Figure 1(E). Experimental values were taken from reference [12] .
lowest thermodynamic heat of formation. Since 2-chloropyrene is very polar, the conditions of the chlorination reaction will dictate the stability of the product. Experimentally, when pyrene is chlorinated with NCS, the main product was monochloropyrene [8] . Therefore, it can be concluded that the product is a mixture of the three derivatives: 1-chloro, 2-chloro and 4-chloropyrene. A study by Jensen and Berg [13] suggested that the 1-chloropyrene and the 2-chloropyrene have good reactivity in the organic medium ether. Moreover, 1-chloropyrene was one of the main products of the chlorination of pyrene in aqueous solution [14] .
The appropriateness of the DFT method in predicting the geometrical values was tested by applying different levels of the DFT theory. They include: B3PW91, MPW1PW91, and PBE1PBE in addition to B3LYP. The same methods were also used in the vibrational frequency calculations.
The energies of frontier orbitals: the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were computed. Figure 2 illustrates the energies (in hartrees) of frontier molecular orbitals and their gaps (ΔE) at the B3LYP/6-311G** level. It can be seen that the values of EHOMO increase in the order pyrene < 1-chloropyrene < 4-chloropyrene < 2-chloropyrene. The same trend is seen for the ELUMO. We notice that ΔE values are very similar within the monosubstituted derivatives. The trend indicates that 1-chloropyrene is less involved in chemical or photochemical processes with electron transfer and hence it is the least reactive in agreement with previous results. Our calculated value for ΔE for pyrene (3.84 eV = 0.141 a.u.) is in excellent agreement with the experimental value of 3.85 eV [15] .
3.2. Chemical Reactivity
The energies of frontier orbitals: the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are used to determine several chemical reactivity parameters as a measure of relative stability and reactivity. These include total energy (E0), chemical hardness (η), electronic chemical potentials (μ) and electrophilicity (ω). The energies of the HOMO and LUMO obtained by the B3LYP/6-311G** method (Figure 2) are listed in Table2
Chemical hardness is linked to the stability and reactivity of a chemical system. It measures the resistance of a compound to changes in the electron density distribution or to electron charge transfer. According to the frontier molecular orbital approach, chemical hardness is directly proportional to the energy gap between the HOMO and the LUMO and is estimated as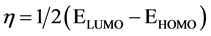 . The larger the HOMO-LUMO energy gap, the harder and more stable (less reactive) the compound [16] .
. The larger the HOMO-LUMO energy gap, the harder and more stable (less reactive) the compound [16] .

Figure 2. The frontier orbitals of the molecules of pyrene (A) and its monochlorinated derivatives: 1-chloropyrene (B), 2-chloropyrene (C), and 4-chloropyrene (D). The highest occupied molecular orbital (HOMO) are shown at the bottom and the lowest unoccupied molecular orbital (LUMO) are shown at the top. The values for EHOMO (a.u.) and ELUMO (a.u.) are shown on the graph.
Table 2. Calculated values for the energies of frontier orbitals: HOMO and LUMO, chemical hardness (η), electronic chemical potentials (μ) and electrophilicity (ω) of the molecules of pyrene (A) and its monochlorinated derivatives: 1-chloropyrene (B), 2-chloropyrene (C), and 4-chloropyrene (D).
Table 2 lists the calculated chemical hardness (η) values for the four studied compounds. Among the monochlorinated pyrene derivatives, 1-chloropyrene has the lowest η value (0.0690) and therefore it is expected to be the most stable. 2-chloropyrene seems to be the least stable with the highest value of η (0.0705).
The electronic chemical potential is defined as the negative of electronegativity of a molecule [17] and from DFT computations, it is calculated using the equation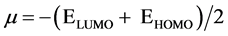 .
.
The smaller the electronic chemical potential value (μ), the more stable is the compound. Table 2 lists the calculated electronic chemical potential (μ) values for the four studied compounds. It is clear that since 1-chloropyrene has the lowest μ value (0.141 a.u.), it is expected to be the most stable while 2-chloropyrene with the highest μ value (0.1445 a.u.) is the least stable. The results for the electronic chemical potential (μ) agree well with the experimental chlorination of pyrene [8] .
The electrophilicity parameter (ω), is calculated using the electronic chemical potential (μ) and the chemical hardness (η) using the equation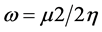 . In a chemical process, it measures the capacity of a species to accept electrons and therefore measures the stabilization in energy after a system accepts additional electronic charge.
. In a chemical process, it measures the capacity of a species to accept electrons and therefore measures the stabilization in energy after a system accepts additional electronic charge.
The electrophilicity values (ω) for the studied compounds are listed in Table2 They are 0.1481 for 2-chloro, 0.1461 eV for 4-chloro and 0.1441 for 1-chloro. Among the compounds, and having the lowest ω value, 1-chloro is the strongest nucleophile while 2-chloro is the strongest electrophile with the lowest ω value. The result suggests some sort of selectivity in the mechanistic chemistry of pyrene chlorination. Experimentally, more stable compounds correspond to smaller electrophilicity values and this is the case with 1-chloropyrene.
3.3. Heats of Formation
Standard Heat of formation (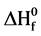 ) is a basic thermodynamic property of a chemical compound. Computational methods are increasingly being used for predicting and verifying the heat of formation of gas-phase systems at 298 K. Many studies focused on computing
) is a basic thermodynamic property of a chemical compound. Computational methods are increasingly being used for predicting and verifying the heat of formation of gas-phase systems at 298 K. Many studies focused on computing 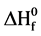 using DFT methods (such as B3LYP, MPW1PW91, PBE1PBE) for a large number of organic or inorganic compounds [18] .
using DFT methods (such as B3LYP, MPW1PW91, PBE1PBE) for a large number of organic or inorganic compounds [18] .
To our knowledge, the literature contains no attempts to calculate 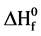 for chlorinated pyrene compounds. Here, we apply DFT calculations in predicting the gas phase heat of formation for pyrene and its three monochlorinated derivatives at 298 K.
for chlorinated pyrene compounds. Here, we apply DFT calculations in predicting the gas phase heat of formation for pyrene and its three monochlorinated derivatives at 298 K.
The calculations for the heat of formation were performed using the methodology of computing the total electronic energy of molecules and their individual atomic constituents. Utilizing the properties of isodesmic reactions and the high similarity between reactants and products, 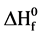 of the studied compounds is evaluated [16] .
of the studied compounds is evaluated [16] .
Table 3 lists the electronic energy along with the calculated heats of formation for pyrene and its three monochlorinated derivatives. Our calculations for the heats of formation predict that the most stable monochloropyrene is 4-chloropyrene and 1-chloropyrene while the least stable is 2-chloropyrene. Experimentally, when pyrene is chlorinated, the main product was 1-chloropyrene [8] .
Other monochloropyrenes were not detected in the products indicating their high heat of formation in agree ment with our computational results. In the literature, the closest chlorinated polyaromatic molecules with experimental  values are the 1-chloro and the 2-chloronapthalene [19] . The heat of formation of 1-chloronapthalene (115 kJ/mol at 298 K) is considerably less than that for the 2-chloronapthalene (137 kJ/mol). Our findings follow a similar trend in which 1-chloropyrene exhibits a smaller heat of formation at 298 K (103.13 kJ/mol). However, we notice that the heat of formation of 4-chloropyrene is slightly lower than that for the 1-chloropyrene
values are the 1-chloro and the 2-chloronapthalene [19] . The heat of formation of 1-chloronapthalene (115 kJ/mol at 298 K) is considerably less than that for the 2-chloronapthalene (137 kJ/mol). Our findings follow a similar trend in which 1-chloropyrene exhibits a smaller heat of formation at 298 K (103.13 kJ/mol). However, we notice that the heat of formation of 4-chloropyrene is slightly lower than that for the 1-chloropyrene
Table 3 . Electronic energy, and calculated heats of formation for pyrene and its three monochlorinated derivatives. Experimental value was taken from reference [21] .
indicating that the 4-chloropyrene derivative is the most stable thermodynamically. This result is in agreement with a study conducted by Nilsson et al. that found 1-chloro and 4-chloropyrene to be the most abundant molecule upon continuous heating of a mixture of pyrene derivatives [20] .
4. Conclusion
Based on DFT computations for pyrene and its monochlorinated derivatives, B3LYP/6-311G** method/basis set is found to be the most suitable in predicting the minimum energy optimized structure and the heat of formation of pyrene and its monochlorinated derivatives. The geometrical parameters and the heat of formation of pyrene are in good agreement with available experimental data. The chemical reactivity and the relative stability of the monochlorinated pyrene compounds are in the order: 1-chloropyrene > 4-chloropyrene > 2-chloropyrene in agreement with available experimental data.
Acknowledgements
This work has been carried out during a sabbatical leave granted to the author from the University of Jordan in the year 2012-2013.
References
- Pagnoni, A., Kligman, A.M. and Stoudenayer, T. (1998) Pyranine, a Fluorescent Dye, Detects Subclinical Injury to Sodium Lauryl Sulfate. Journal of Cosmetic Science, 49, 33-38.
- Clar, E., Roberston, J.M., Schloegl, R. and Schmidt, W. (1981) Photoelectron Spectra of Polynuclear Aromatics. 6. Applications to Structural Elucidation: “Circumanthracene”. Journal of the American Chemical Society, 103, 1320-1328. http://dx.doi.org/10.1021/ja00396a003
- Goodpaster, J.V., Harrison, J.F. and McGuffin, V.L. (1998) Ab Initio Study of Polycyclic Aromatic Hydrocarbons in Their Ground and Excited States. The Journal of Physical Chemistry A, 102, 3372-3381. http://dx.doi.org/10.1021/jp980467k
- Kerr, C.E., Mitchell, C.D., Headrick, J., Eaton, B.E. and Netzel, T.L. (2000) Synthesis and Photophysics of a 1-Pyrenyl Substituted 2’-Deoxyuridine-5-carboxamide Nucleoside: Electron Transfer Products as CISINDO/S Excited States. The Journal of Physical Chemistry B, 104, 1637-1650. http://dx.doi.org/10.1021/jp992773j
- Charles Li, X.-C., Okamura, Y., Ueno, K., Tashiro, M. and Prakash, G.K. (2005) Organic Electroluminescent Device Based on Pyrene Derivatives. US Patent No. 6852429.
- Luthe, G.M., Ariese, F., Udo, A. and Brinkman, T. (2002) Monochlorinated Polycyclic Aromatic Hydrocarbons: Standards in Environmental Chemistry and Biochemical Applications. The Handbook of Environmental Chemistry, 3N, 249-275.
- Dewhurst, F. and Kitchen, D.A. (1972) Synthesis and Properties of 6-Substituted Benzo[a]pyrene Derivatives. Journal of the Chemical Society, Perkin Transactions 1, 2,710-712.
- Ohura, T., Kitazawa, A. and Amagai, T. (2004) Seasonal Variability of 1-Chloropyrene on Atmospheric Particles and Photostability in Toluene. Chemosphere, 57, 831-837. http://dx.doi.org/10.1016/j.chemosphere.2004.08.069
- Muff, J. and Sogaard, E.G. (2011) Identification and Fate of Halogenated PAHs Formed during Electrochemical Treatment of Saline Aqueous Solutions. Journal of Hazardous Materials, 186, 1993-2000. http://dx.doi.org/10.1016/j.jhazmat.2010.12.110
- Takeuchi, H. (2013) Structures, Stability, and Growth Sequence Patterns of Small Homoclusters of Naphthalene, Anthracene, Phenanthrene, Phenalene, Naphthacene, and Pyrene. Computational and Theoretical Chemistry, 1021, 84-90. http://dx.doi.org/10.1016/j.comptc.2013.06.029
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr., J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C., Pople, J.A. (2004) Gaussian 03, Revision B.03. Gaussian, Inc., Wallingford CT.
- Robertson, J.M. and White, J.G. (1947) The Crystal Structure of Pyrene. A Quantitative X-Ray Investigation. Journal of the Chemical Society, 1, 358-368. http://dx.doi.org/10.1039/jr9470000358
- Jensen, A. and Berg, A. (1965) Reactions of Halopyrenes with Phenyllithium in Ether. Acta Chemica Scandinavica, 19, 1838-1842. http://dx.doi.org/10.3891/acta.chem.scand.19-1838
- Hu, J., Jin, X., Kunikane, S., Terao, Y. and Aizawa, T. (2006) Transformation of Pyrene in Aqueous Chlorination in the Presence and Absence of Bromide Ion:? Kinetics, Products, and Their Aryl Hydrocarbon Receptor-Mediated Activities. Environmental Science Technology, 40, 487-493. http://dx.doi.org/10.1021/es0516108�
- Fabina, J. and Hartmann, H. (1980) Theoretical Treatment and Empirical Rules. Springer, Berlin, 117.
- Vektariene, A, Vektaris, G. and Svoboda, J. (2009) A Theoretical Approach to the Nucleophilic Behavior of Benzofused Thieno[3,2-b]furans Using DFT and HF Based Reactivity Descriptors. ARKIVOC, 7, 311-329. http://dx.doi.org/10.3998/ark.5550190.0010.730
- Parr, R.G. and Pearson, R.G. (1983) Absolute Hardness: Companion Parameter to Absolute Electronegativity. Journal of the American Chemical Society, 105, 7512. http://dx.doi.org/10.1021/ja00364a005
- Li, Y.-F., Fan, X.-W., Wang, Z.-Y. and Ju, X.-H. (2009) A Density Functional Study of Substituted Pyrazole Derivatives. Journal of Molecular Structure: THEOCHEM, 896, 96-102. http://dx.doi.org/10.1016/j.theochem.2008.11.004
- Smith, L., Bjellerup, L., Krook, S. and Westermark, H. (1953) Heats of Combustion of Organic Chloro Compounds Determined by the “Quartz Wool” Method. Acta Chemica Scandinavica, 7, 65-86. http://dx.doi.org/10.3891/acta.chem.scand.07-0065
- Nilsson, U.L. and Ostman, C.E. (1983) Chlorinated Polycyclic Aromatic Hydrocarbons: Method of Analysis and Their Occurrence in Urban Air. Environmental Science Technology, 27, 1826-1831. http://dx.doi.org/10.1021/es00046a010
- Roux, M.V., Temprado, M., Chickos, J.S. and Nagano, Y. (2008) Critically Evaluated Thermochemical Properties of Polycyclic Aromatic Hydrocarbons. Journal of Physical and Chemical Reference Data, 37, 1855-1996. http://dx.doi.org/10.1063/1.2955570