International Journal of Analytical Mass Spectrometry and Chromatography
Vol.05 No.01(2017), Article ID:74088,16 pages
10.4236/ijamsc.2017.51001
Reactivity of N-Methylidenemalonates of 3-Arylaminoindoles and p-Dimethylamino-N-Phenylaniline in the Course of Their Analysis by Electrospray Ionization Mass Spectrometry
Yuri S. Nekrasov*, Nikolai S. Ikonnikov, Yuri A. Borisov, Sergey S. Kiselev, Albert G. Kornienko, Valeriya S. Velezheva, Yury I. Lyakhovetsky
A.N. Nesmeyanov Institute of Organoelement Compounds, The Russian Academy of Sciences, Moscow, Russia
![]()
Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: November 26, 2016; Accepted: February 10, 2017; Published: February 13, 2017
ABSTRACT
The behavior of N-methylidenemalonates of 3-arylaminoindoles and p-di- metylamino-N-phenylanyline (M = ANa) was studied during their analysis with ESI mass spectrometer operated in negative (NI) and positive (PI) ion modes. Anions [A]- and both [M + H]+ and [M + Na]+ were recorded under conditions of the NI-ESI and PI-ESI, respectively. The fragmentation processes of [A]- and [M + H]+ were found that probably occurred as “in- source collusion induced dissociation”. The main paths for [A]- proved to be elimination of CO2 and breakage of the N-methylidenemalonate bond. A route [A]- − CO2 − ROH (R = Me or Et) was less expressed and occurred for the indolyl-containing compounds with the NH bond only. Experiments employing heavy water demonstrated the isotope exchange to occur involving the hydrogen atom of this bond. This and other facts evidenced that the last fragmentation included abstraction of just this atom. Quantum-chemical calculations allowed picking out a structure for the product ion from the possible ones. The calculations also indicated that the protonation of M occurred at the anionic oxygen atom of the malonate moiety. The fragmentation of [M + H]+ ions included elimination of two water molecules that was supported by their MS2 spectra. A common feature of the NI- and PI-ESI mass spectra was the presence of oligomeric ions, up to tetramers and trimers for the NI- and PI-ESI ones, respectively. The oligomers were formed by interaction of the corresponding ions with neutral molecules. When ions contained extra hydrogen atoms, they were introduced by hydrolysis.
Keywords:
N-Methylidenemalonates of 3-Aminoarylindoles, Electrospray Ionization, Fragmentation Processes, Quantum-Chemical Calculations, Ion Structures

1. Introduction
3-Aminoindoles along with their derivatives and heterocycles with the embedded 3-aminoindole scaffold were found in a number of natural products and represented a group of pharmacologically promising compounds that show diverse biological activities, such as antimalarial, antimuscarinic, antibacterial, antiviral, antihypoglicemic, antiplasmodium, and PARP-inhibiting activities [1] [2] . Earlier, some of us reported a new reaction for the synthesis of 3-amino- indoles and their aniline analogs tethered to methylidenemalonate fragments. They were prepared from easily available indole-3-carbaldehyde/p-dimetyl- aminobenzaldehyde aryl nitrones and sodium malonates [3] . Mass spectrometry (MS) seems to be a good tool for their analysis and identification. Since they are sodium salts, a MS method with the desorption type ion source should be employed. Such instruments include, for example, one with the ion sources of field desorption (FD) [4] , secondary ion mass spectrometry (SIMS) [5] , those with desorption chemical ionization (DCI) [6] , laser desorption/ionization (LDI) [7] , fast atom bombardment (FAB) [8] , and some other types [9] [10] . For the analysis of low- and non-volatile organic, bioorganic, and organoelement compound, however, the most applied mass spectrometers are now the matrix assisted laser desorption/ionization (MALDI), and electrospray ionization (ESI) instruments [11] [12] [13] [14] [15] . These ionization methods belong to the group of so-called “mild methods”, for which the fragmentation of the precursor ions (molecular radical anions and cations, deprotonated, protonated, and cationated molecules) either does not occur or occurs to a small extent. In those cases when the fragmentation is of interest, the tandem technique (MS2) or generally, MSn is employed. For the analysis of the aforementioned sodium malonates, the ESI method appears to be more appropriate since negative ions are already present in analyte solutions. Moreover, the construction peculiarities of the ESI instruments involve ionization at the atmospheric pressure (with the formation of positive ions in the case of the malonates), while ion filtration and their scanning over the ratio of mass to charge (m/z) occur in high vacuum. As a result, the ion sources have the region of a medium pressure where a background gas (often, N2) is present. The electrodes designed for focusing ions toward the skimmer, for example, tube lens, are built in this zone. An additional potential between 0 and ± 200 V (for cations and anions, respectively) can be applied to the lens to focus and accelerate ions. Collisions of the accelerated ions with the background gas favor desolvation of the ions, thus increasing the sensitivity of the instrument. However, if the module of the potential is sufficiently high, the internal energy of ions may increase to such a level that the fragmentation of the ions occurs. Thus, in some cases, the fragmentation called “in-source collision-in- duced dissociation (in-source CID or source-CID)” may come into being under conditions of ESI [16] .
Fragmentation patterns and the structures of fragment ions formed can help chemists to establish the analyte structure. Moreover, they provide valuable information explaining the behavior of the species studied in chemical or biochemical processes.
In the present work, we have studied the behavior of a number of sodium methylidenemalonates of 3-arylaminoindoles or p-dimethylamino-N-phenyla- niline, obtained by the above mentioned reaction, under conditions of ESI in the positive and negative ion modes when in-source CID occurred. As it was told before, the results of the investigation could provide important information on the reactivity and the reaction mechanisms of these compounds in drug synthesis processes.
2. Materials and Methods
The syntheses of species studied were carried out by the method described in reference [3] .
The ESI mass spectra were taken on a Finnigan LCQ Advantage mass spectrometer with ion trap mass analyzer (USA) and equipped with a Surveyor MS pump, and a Schmidlin-Lab nitrogen generator (Germany). Nitrogen served as a sheath gas, while no auxiliary gas was used. A flow of acetonitrile (purchased from the Merk company) of 25 µL∙min−1 was maintained. The temperature of the ion transfer capillary was 150˚C; the electric voltages between the needle and the counter electrode were ±4.5 kV and ±55 V for tube lens (for the positive and negative ion polarity modes, respectively). The samples with the concentration of 10−4 mol∙L−1 in acetonitrile:water 9:1 were introduced into the ion source through the Reodyne injector with a 5 µL loop. The data acquisition and treatment were fulfilled using the program X Calibur, version 1.3. The removal of the contributions of the 13C- and other natural heavy-isotope-containing ions from the peak intensities of the deuterium labeled compounds was performed iteratively using the program SCIPE [17] .
The quantum chemical calculations were made in the framework of the density functional theory (DFT) [18] . The Becke-Lee-Yang-Parr hybrid method (B3LYP/6-31G*) was employed [19] [20] . All calculations with total optimization of the geometry of molecules and computation of normal vibrational frequencies were carried out with the GAUSSIAN 09 program operated under LINUX [21] .
3. Results and Discussion
The N-methylidenmalonates of tertiary 3-arylaminoindoles and p-dimethyla- mino-N-phenylaniline the ESI mass spectra of which have been studied are presented in Figure 1 (compounds 1 - 5).
All Figure 1 compounds formed both negative and positive ions (NI and PI, respectively) under condition of the NI-ESI and PI-ESI, respectively.
3.1. ESI Mass Spectra in the Negative Ion Polarity Mode
The NI mass spectra of all compounds examined showed peaks of anions [ArN(C6H4R2)-CH=C(COOR3)COO]− ([A]−) (Ar = R2indolyl or Me2NPh) and products of their dissociation: [A − CO2]− (except 5) and [ArN(C6H4R2)]−. Protonated and cationated dimers, [A2H]− and [A2Na]−, trimers [A3НnNam]- (n + m = 2) and tetramers [A4НnNam]− (n + m = 3) were also recorded (Table 1 and Figure 2(a)).
Besides the above ion peaks, the NI-ESI mass spectra of compounds 1 and 2 displayed small intensity peaks of ions at m/z 259 and 273, respectively. It was reasonable to assign them to the ions formed due to the elimination of the CO2 and R3OH molecules from [A]- ([A]- and [A2]-, respectively). Moreover, the

Figure 1. The species subjected to this study.

Table 1. M/z values and peak abundances with respect to the maximal peaks (Irel, %) in NI-ESI mass spectra of compounds 1 - 5a.
aThe Table lists the averaged over scans values.


Figure 2. NI-ESI mass spectra of compound 1: (a) mass spectrum obtained by averaging single-scan spectra over 30 scans; (b) MS2 spectrum of anion [A]-.
corresponding peaks at 273 and 263 m/z were absent in the spectra of compounds 3, 4 and 5. Since 3 and 4 lack the hydrogen atoms at the nitrogen one of the indole ring and 5 hasn’t this ring at all, the first impulse was to consider that for 1 and 2 the elimination process discussed involved the hydrogen atom at the indolyl nitrogen atom. However, it was rather surprising to find that, while in the case of compound 4, the MS2 spectrum of the similar ion [A4]- lacked a peak at m/z 273 which would correspond to ion [A4 − CO2 − EtOH]-, the MS2 spectrum of [A3]- in the case of compound 3 showed an abundant peak of the [A3 − CO2 − MeOH]- ion. It may be caused by the point that, though the compound lacks the hydrogen atom at the indole nitrogen one, but under the more drastic conditions of MS2 CID in comparison with those of in-source CID, the fragmentation pathway with the participation of another and less mobile hydrogen atom occurred. Nevertheless, this MS2 spectrum of 3 cased some doubts on the validity of the above suggestion that the hydrogen atoms at the indole nitrogen atoms were involved in these fragmentations of ions [A]- and [A2]-.
To solve this problem and for the elucidation of other reactions peculiarities, experiments with compounds 1 and 4 were performed in the presence of heavy water. Four modes were employed; in the first variant, D2O was introduced in the ion source as a solvent for the analytes (method A). Protocols C and D involved additional introduction in the ion source of heavy water and water via a syringe pump, respectively. By application of method B, the analytes were introduced in the source in water solutions, while heavy water was admitted through the syringe pump. In the case of compound 1, ion [A]- and its fragment ions save one contained deuterium-labeled components thus indicating hydrogen-deuterium exchange to have occurred (Table 2). The comparison of the data for method A with those for method B demonstrates that the H,D-exchange proceeded inside the ion source to a noticeable extent. With that, however, the percentage of deuterium containing ions was greater in each deuterated entry of this ionic group when method A was employed. It can be explained by the fact that under the protocol A conditions, the exchange occurred in pure D2O, while a mixture of D2O and H2O was present in the ion source when method B was employed. This finds support in the points that the percentages of deuterated components in the ions increased or decreased when methods C or D were used, respectively. It reveals especially clear by the example of ions [A]- with m/z 335 and 336 (Table 2). However, the results obtained can’t exclude the possibility that the exchange could partly proceed in the solution of the analyte in D2O before its introduction into the ion source.
Since ion [A − CO2 − MeOH]- in the case of compound 1 and all ions listed in Table 2 for compound 4 did not contain deuterium atoms, these experiments strictly supported two points: 1) the hydrogen-deuterium exchange in compound 1 occurred with the hydrogen atom located at the nitrogen atom of the indole ring; 2) the fragmentation of [A]- to give the above mentioned first ion involved this ion exactly. The latter is apparently valid for ion [A2]- of compound 2.
In the cases of compounds 1, 2, and 4 the dissociation patterns of [A]-, [A2]-, and [A4]- ions, respectively, occurred due to in-source CID turned out to be virtually the same as in MS2 experiments differing in the intensities of the corresponding peaks only (cf. Figure 2(a) and Figure 2(b)). At the same time, besides
Table 2. Number of the deuterium atoms (nd) in the ionic structures and the relative abundances of the corresponding ions (%) in these structures in the NI-ESI mass spectra from the experiments with the use of heavy watera.
aThe contributions from ions containing 13C and the other natural heavy isotopes were removed as described afore (see Section 2).
the coincided ions (305 and 221 Da), other fragment ions of [A3]- were present in its MS2 spectrum (331, 323, 317, 291, and 273 Da). This difference again seems to be caused by the fact that the collision energies were different in these CID modes.
Ions [A2Na]- were formed most likely due to interaction of ions [A]- with neutral molecules of analytes (reaction 1).
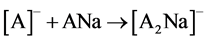 (1)
(1)
Two possible ways for the formation of ions [A2H]- could be anticipated. The first path involved hydrolysis of ions [A2Na]- (reaction 2). The second implied the hydrolysis of neutral ANa or protonation in the ion source of [A]- by H2O and even CH3CN (a solvent in many experiments). AH formed then added [A]- (reaction 3)1.
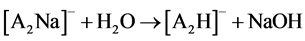 (2)
(2)
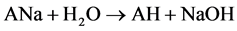
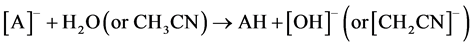
![]() (3)
(3)
Trimeric and tetrameric ions appear to be formed by the analogous schemes involving addition the corresponding dimeric or trimeric ions to neutral molecules and hydrolysis processes.
3.2. ESI Mass Spectra in the Positive Ion Polarity Mode
Peaks of the protonated and cationated molecules [M + H]+ and [M + Na]+of compounds 1 - 5 (M = ANa) were present in their PI-ESI mass spectra. Besides them, the mass spectra showed peaks of fragment ions of the former ions, ions [AH + H]+ (with significant peak abundances of such ion and its fragments for 1 only), and also ions of dimers and trimers (Table 3, Figure 3(a)).
With that, the peak abundances of protonated ions [M + H]+ significantly exceeded those of cationated ions [M + Na]+, while the relative abundances of ion peaks of dimers at m/z 695, 751, 723, 751 and 703 for compounds 1 - 5, respectively, were great, being even 100% for 1 - 3, and 5. This suggests [MAH + H]+ structure of these ions and the reaction 4 for their formation.
![]()
![]() (4)
(4)
Two other theoretically possible schemes involving the participations of [AH + H]+ or [M + Na]+ ions, and AH neutral molecules seem less likely for compounds 2 - 5. Actually, the peak abundances of [AH + H]+ were extremely small in their spectra (see Table 3). This means that the amounts of the neutral AH molecules were also small. Further, as we underlined above, the peak abundances
Table 3. Values m/z and relative peak abundances of PI (Irel, %) with respect to the maximal peaks in the PI-ESI mass spectra of compounds 1 - 5a.
aSee footnote “a” to Table 1.
of [M + Na]+ ions were small as compared with those of [M + H]+ ones that speaks in favor of the fact that the reaction mostly occurred via the latter ions.
As relates to compound 1, the peak of [AH + H]+ possessed a pronounced abundance in the mass spectrum. Thus, the reactions 5 could contribute to the process of the [MAH + H]+ cation formation.
![]()
![]()
![]()
![]() (5)
(5)
The participation of [M + Na]+ is unlikely in this case by the same reason as above.
The origin of fragment ions of [M + H]+ with m/z 319, 347, 333, 347, and 323 in the spectra of 1 - 5, respectively, is worth discussion. The ions can be described as [M + H ? NaOH]+, and this fragmentation seems quite reasonable. However, the MS2 spectra of the precursor ions showed these products ions either to be present with negligible abundances, or to be absent. An alternative fragmentation process is [AH + H]+ − H2O (see reaction 5 for a possible way of formation of this precursor ion; it could also be generated via hydrolysis of the [M + H]+ ion.) The above MS2 argument that would indirectly speak in favor of the latter process has a hint of doubt for compounds 2 - 4, since the abundances of the precursor-product pair were rather small in these cases, while ions [M + H]+ were abundant. Moreover, ion [A5H + H]+ was virtually absent on the spectrum of 5, whereas a small abundance ion with m/z 323 was present and again
![]()
Figure 3. PI-ESI mass spectra of compound 1: (a) the spectrum obtained by summarizing 40 single-scan spectra and (b) MS2 spectrum of cation [M + H]+.
[M5 + H]+ was abundant. Thus, we cannot exclude possibility of contributions of [M + H − NaOH]+ to the abundances of the ion discussed in the cases of species 2 ? 5. At the same time, the peak abundances of the ions of the corresponding pair, 337 and 319 Da, were relatively great in the case of 1 (Table 3). Hence, the aforementioned MS2 data for the protonated molecule of 1 indirectly support the fact that the 319 Da ion was formed mostly owing to elimination of water from the [AH + H]+ cation.
Another fragmentation of [M + H]+ cations is of interest. It involved the loss of two water molecules, was expressed for compounds 1, 3, and 5, while minor for 2 and 4 (Table 3). It seemed rather unexpected, but was supported by MS2 spectra in the cases of the first above group of the compounds, these product ions being dominant in the spectra (see, e.g., Figure 3(b)).
The 297 Da ions in the spectra of 2 and 4 can be described as either [M + H − EtOH − CO2]+ or [M + H − CO2 − EtOH]+. Though both ions [M + H − EtOH]+ and [M + H − CO2]+ were absent in the spectra, the MS2 spectrum of [M4 + H]+ ion of 4 showed the former ion rather than the latter. Also, analogous ion [M5 + H − MeOH]+ and not [M5 + H − CO2]+ was present in the MS2 spectrum of [M5 + H]+ in the case of compound 5. Based on all this, we believe that the 297 Da ions discussed were formed due to the sequential elimination of the alcohol and carbon dioxide from the protonated molecules of 2 and 4. We also believe that this path took place in the cases of compounds 1, 3, and 5 producing ions 283, 297, and 287 Da, respectively.
An alternative fragmentation that could also have provided the above m/z ions in the mass spectra of these compounds seemed to be [M + H]+ − H2O − H2O − NaOH. However, the mass spectra of 2 and 4, and the MS2 spectrum of ion [M4 + H]+ of 4 lacked the corresponding product ions (311 Da). It allowed us to exclude such fragmentation from the consideration.
When 1 was introduced in D2O solution, the treatment of the mass spectrum as mentioned in section 2 showed ions of analyte 1 formed owing to addition of a proton or deuteron to contain between zero and two deuterium atoms [359 Da (12%), 360 Da (43%) and 361 Da (45%)]. This means that the deuterium atoms were introduced into the analyte molecule by both H,D-exchange and the addition of a deuteron. At the same time, the corresponding ions of compound 4 obtained and registered under the same conditions contained maximum 1 deuterium atom [387 Da (25%) and 388 Da (75%)]. Contrary to 1, this compound has no hydrogen atom at the nitrogen one of the indole ring. Thus, all of this is consisted with the conclusion drawn before when negative ions were examined that the H,D-ex- change in compound 1 and its ions involved just this hydrogen atom. It is essential to note, that protonation (or deuteron addition) could hardly proceed in the solutions of analytes before introduction in the ion source, since the analytes are salts of weak acids and a strong base and thus provide basic mediums. Hence, the addition of proton or deuteron most likely occurred immediately in the ion source.
3.3. Quantum-Chemical Calculations
As we mentioned above, the abundant peaks of fragment ions [A − CO2]- and [ArN(C6H4R2)]- are present in the NI-ESI mass spectra of compounds 1 - 4 (Table 1). At the same time, peaks of ions [A − CO2 − R3OH]-, though of low intensities, were detected reliably for compounds 1 and 2. Their formation from ions [A]- ([A]- and [A2]-, respectively) was supported by the corresponding MS2 spectra. Quantum-chemical calculation performed in the case of compound 1 provided three possible structures of [A − CO2 − MeOH]− (Figure 4). However, the ion with structure 1 turned out to be the most stable and even more stable than that with a cumulene fragment (such cumulene motif was reported, e.g., as propadienone [22] ). The formation of this ion seems to be kinetically rather than thermodynamically controlled. However, during the period when this ion was present in the ion source, isomerization could occur for it to acquire the most stable structure 1. Moreover, according to the extended Hammond-Leffler postulate, [23] the more the reaction is endothermic the more the transition state for generating the [A − CO2 − MeOH]− ion should be shifted to the reaction products
![]()
Figure 4. Three possible structures of the [A − CO2 − MeOH]- ion of compound 1 and their parameters obtained from the quantum-chemical calculations.
along the reaction coordinate. If the process is exothermic, the shift has to occur in the opposite direction, to the reagents. In both cases, the activation barrier is minimal most likely for the most stable product. From all of this, anion [A − CO2 − MeOH]− appears to have structure 1.
Its formation apparently included the nucleophilic attack of the C(2) atom of the indole ring upon the C-atom of a carbonyl group followed by elimination of MeOH involving abstraction of the hydrogen atom from the indole nitrogen one. Then 1,3-hydrogen shift occurred to give ion 1. The other formally possible structures that could be suggested a priori proved to be either impossible sterically, or the optimization of their geometry resulted in structure 2.
When a compound lacked the hydrogen atom at the indole nitrogen one and the collision energy was high enough, an ion with the structure like 3 (e.g., with N-Me bond instead N-H one) might be formed as a result of the abstraction of the hydrogen located at the α-carbon atom of the indole ring. It seems to be the case of compound 3, for which fragmentation [A3]- − CO2 − MeOH was observed under MS2 conditions.
As indicated above, molecules of 1 - 5 added a proton or sodium cation under conditions of PI-ESI (reactions 6 an 7, respectively). The abundances of ions [M + H]+ were always higher than those of the [M + Na]+ ions (see Table 3). As above, in the framework of the extended Hammond-Leffler postulate, this is consistent with the calculated values of proton affinity (PA) and the affinity to cation Na+ for compound 1, the former being 3.5 times greater than the later.
 (6)
(6)
 (7)
(7)
 (8)
(8)
It is worthy of noting that the calculated PA of molecule 1 is very high prevailing that for ammonia (852.6 kJ∙mol−1) [24] .
There is little likelihood that the reaction of hydrolysis of [M + Na]+ ion (reaction 8) could to any noticeable extent contribute to the process of the formation of [M + H]+ ion, since it is highly endothermic.
A priori, eight atoms in molecule 1 are amenable to protonation. Those are C(2), C(3), N(10), C(18), O(20), O(22), O(23), and O(25) (see Figure 5). The calculations demonstrated that there are two positions in the molecule with the lowest and relatively close PA, the protonation leading to structures A and B. However, the PA value for structure A is nevertheless greater, and protonation seems to predominantly occur at O(25) position. As aforementioned, ions [M2 + H]+ were formed most probably by interaction of molecules M with their protonoted forms (the first scheme of reaction 4). The heat effect of this interaction was calculated to be −261.8 kJ∙mol−1 for compound 1. An alternative path could

Figure 5. The protonated molecules of 1 with two lowest DН of protonation (the protons added are marked by the asterisks).
comprise the interaction of the molecules with their cationated forms and the hydrolysis of the dimers obtained (reaction 9).

 (9)
(9)
However, the abundances of [M + Na]+ ions in the spectra were considerably less than those of [M + H]+ ions, the ΔН of the interaction between M and [M + H]+ was less than that of M and [M + Na]+ exemplified by the case of 1. Besides, the formation of [M2 + H]+ ions involving the latter interaction would be a two-step process, the second step being highly endothermic. All this supports that the [M2 + H]+ ions were formed predominantly according reaction 4.
4. Conclusions
Thus, both anions [A]- and cations [M + H]+ of compounds 1 - 5 (M = ANa) were detected under NI-ESI and PI-ESI conditions, respectively. These ions underwent fragmentation processes that occurred probably as in-source CID.
In the case of NI-ESI, the main fragmentation paths proved to be the loss of CO2 and the elimination of methylidenmalonate moiety from [A]- (the former with the exception of compound 5). The less expressed process found for compounds 1 and 2 was the successive elimination of CO2 and an alcohol molecule (MeOH or EtOH). The absence of the corresponding product ions in the case of compound 4 lacking a hydrogen atom at the nitrogen atom of the indolyl moiety and for compound 5 containing no indolyl group at all suggested that just this atom was abstracted during this fragmentation. Experiments with 1 and 4 with the use of heavy water demonstrated the isotope exchange to occur, at least partly, in the ion source that involved the above hydrogen atom of 1. Also, these experiments supported the fact of the abstraction of this atom during the formation of [A − CO2 − MeOH]-. Quantum-chemical calculations showed that the ion formed had to be of a cyclic structure rather than a linear one.
Apart from ions [A]− and their fragment ions, protonated and cationated oligomeric ions [A2H]-, [A2Na]-, [A3НnNam]- (n + m = 2) and [A4НnNam]- (n + m = 3) were detected. The schemes of their formation include interaction of ions with neutral molecules, e.g., of [A]- with ANa, and hydrolysis to get species containing extra hydrogen atoms.
In the case of PI-ESI, ions [M + Na]+ were recorded along with the [M + H]+ ones, peaks of the former being considerably less abundant than those of the latter. Ion [AH + H]+ was also present, but pronounced only in the case of 1.
Based on the MS2 spectra of [M + H]+ of 2 and 4, we believe that this ion successively lost alcohol and CO2 molecules, contrary to the analogous fragmentation of A- that seems to occur in the opposite consequence.
A fragmentation of ions [M + H]+ with elimination of two water molecules appeared to be rather surprising, but was supported by their MS2 spectra for compounds 1, 3, and 5.
By the example of compound 1, quantum-chemical calculation indicated that protonation of molecule M occurred at the oxygen atom adjacent to the sodium one in Figure 1.
As in the case of NI-ESI mass spectra, peaks of oligomeric cations were rather greatly abundant in the PI-ESI spectra. Again, they were formed because of the interaction of the monomeric cations [M +H]+ and their oligomeric cations with neutral molecules M. Those ions, where extra hydrogen atoms were present, acquired these atoms owing to hydrolysis processes. As an example, quantum- chemical calculation made for 1 along with point that the abundance of ion [M + Na]+ was rather small proved to be consistent with the proposal that ions [M2 + H]+ were formed via interaction of [M + H]+ with M rather than the alternative route involving the reaction of [M + Na]+ with M followed by the hydrolysis of the dimer obtained.
Completing the paragraph, it is worth to note that the depicted results can be of interest to solution chemists who work with compounds 1 - 5 and similar species, since fragmentation and oligomerization processes observed in the ion source in this study and processes occurring in flasks can be akin. Moreover, the possibility of formation of oligomeric ions during ESI-MS analysis of similar compounds should be taken into consideration in order not to make identification mistakes.
Acknowledgements
The work was made under the financial support from the Russian Foundation for Basic Research (grant No. 09-03-00535). It is a great sadness for us to record the death of our friend and co-author, Professor Yuri Nekrasov, occurred before the completion of this paper. He worked with us untiringly until the end. May his memory live forever.
Cite this paper
Nekrasov, Y.S., Ikonnikov, N.S., Borisov, Y.A., Kiselev, S.S., Kornienko, A.G., Velezheva, V.S. and Lyakhovetsky, Y.I. (2017) Reactivity of N- Methylidenemalonates of 3-Arylaminoin- doles and p-Dimethylamino-N-Phenylani- line in the Course of Their Analysis by Electrospray Ionization Mass Spectrometry. International Journal of Analytical Mass Spectrometry and Chromatography, 5, 1- 16. https://doi.org/10.4236/ijamsc.2017.51001
References
- 1. Yao, B., Wang, Q. and Zhu, J. (2012) Paladium(II)-Catalyzed Intramolecular Diamination of Alkynes under Aerobic Oxidative Conditions: Catalytic Turnover of an Iodide Ion. Angewandte Chemie International Edition, 51, 5170-5174.
https://doi.org/10.1002/anie.201201640 - 2. Pews-Davtyan, A. and Beller, M. (2011) Efficient and Simple Zinc-Mediated Synthesis of 3-Amidoindoles. Organic & Biomolecular Chemistry, 9, 6331-6334.
https://doi.org/10.1039/c1ob05576c - 3. Velezheva, V.S., Azev, V.N., Kornienko, A.G., Peregudov, A.S., Godovikov, I.A. and Sebyakin, Y.L. (2010) The Stereoselective Synthesis of Highly Functionalized Tertiary 3-Aminoindoles/Anilines or Dihydropyrroles from C-(3-Indolyl)-N-aryl and C,N-diaryl Nitrones. Tetrahedron Letters, 51, 6594-6597.
https://doi.org/10.1016/j.tetlet.2010.10.045 - 4. Reynolds, W.D. (1979) Field Desorption Mass Spectrometry. Analytical Chemistry, 51, 283A-293A.
https://doi.org/10.1021/ac50038a857 - 5. Benninghoven, A., Rudenauer F.G. and Werner, H.W. (1987) Secondary Ion Mass Spectrometry: Basic Concepts, Instrumental Aspects, Applications and Trends. John Wiley & Sons, New York.
- 6. Esmans, E.L., Alderweireldt, F.C., Marescau, B.A. and Lowenthal, A.A. (1984) Desorption Chemical Ionization of Guanidino Compounds. Analytical Chemistry, 56, 693-695.
https://doi.org/10.1021/ac00268a024 - 7. Hercules, D.M., Day, R.J., Balasanmugam, K., Dang, T.A. and Li, C.P. (1982) Laser Microprobe Mass Spectrometry 2. Applications to Structural Analysis. Analytical Chemistry, 54, 280A-305A.
https://doi.org/10.1021/ac00239a001 - 8. Barber, M., Bordoli, R.S., Donald Sedgwick, R. and Tyler, A.N. (1981) Fast Atom Bombardment of Solids (F.A.B.): A New Ion Source for Mass Spectrometry. Journal of Chemical Society, Chemical Communications, 1981, 325-327.
https://doi.org/10.1039/c39810000325 - 9. McEwen, C.N. and Larsen, B.S. (2015) Fifty Years of Desorption Ionization of Nonvolatile Compounds. International Journal of Mass Spectrometry, 377, 515-531.
https://doi.org/10.1016/j.ijms.2014.07.018 - 10. Trimpin, S., Wang, B., Lutomski, C.A., EI-Baba, T.J. and Harless, B.N. (2016) A Convenient Alternative to MALDI and ESI. Spectroscopy, 31, 53-61.
- 11. Karas, M., Bachman, D., Bahr, U. and Hillenkamp, F. (1987) Matrix-Assisted Ultraviolet Laser Desorption of Non-Volatile Compounds. International Journal of Mass Spectrometry and Ion Processes, 78, 53-68.
https://doi.org/10.1016/0168-1176(87)87041-6 - 12. Tanaka, K., Waki, H., Ido, Y., Akita, S. Yoshida, Y. and Yoshida, T. (1988) Protein and Polymer Analyses up to m/z 100000 by Laser Ionization Time-of-Flight Mass Spectrometry. Rapid Communications in Mass Spectrometry, 2, 151-153.
https://doi.org/10.1002/rcm.1290020802 - 13. Alexandrov, M.L., Gal’, L.N. Krasnov, N.V., Nikolaev., V.I. Pavlenko, V.A. and Shkurov, V.A. (1984) Extraction of Ions from Solutions under Atmospheric Pressure—A Method for Mass Spectral Analysis of Bioorganic Species. Doklady Akademii Nauk SSSR, 277, 379-383.
- 14. Yamashita, M. and Fenn, J.B. (1984) Electrospray Ion Source. Another Variation on the Free-Jet Theme. The Journal of Physical Chemistry, 88, 4451-4459.
https://doi.org/10.1021/j150664a002 - 15. Whitehouse, C.M., Dreyer, R.N., Yamashita, M. and Fenn, J.B. (1985) Electrospray Interface for Liquid Chromatographs and Mass Spectrometers. Analytical Chemistry, 57, 675-679.
https://doi.org/10.1021/ac00280a023 - 16. Gabelica V. and De Pauw, E. (2005) Internal Energy and Fragmentation of Ions Produced in Electrospray Sources. Mass Spectrometry Reviews, 24, 566-587.
https://doi.org/10.1002/mas.20027 - 17. Sukharev, Y.N., Sizoi, V.F. and Nekrasov, Y.S. (1981) The Computer Processing and Interpretation of Mass Spectral Information. VI—Computing the Isotopic Spectrum of Assumed Composition. Organic Mass Spectrometry, 16, 23-25.
https://doi.org/10.1002/oms.1210160107 - 18. Parr, R.G. and Yang, W. (1989) Density-Functional Theory of Atoms and Molecules (International Series of Monographs on Chemistry: 16). Oxford University Press, New York.
- 19. Becke, A.D. (1988) Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Physical Review A, 38, 3098-3100.
https://doi.org/10.1103/PhysRevA.38.3098 - 20. Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Physical Review B, 37, 785-789.
https://doi.org/10.1103/PhysRevB.37.785 - 21. Gaussian 09. www.gaussian.com
- 22. Burgers, P.C., Holmes, J.L., Lossing, F.P., Mommers, A.A., Povel, F.R. and Terlouw, J.K. (1982) Isomeric and Tautomeric [C4H4O]+ Ions; Their Thermochemistry and Collisionally Induced Fragmentation Characteristics. Canadian Journal of Chemistry, 60, 2246-2255.
https://doi.org/10.1139/v82-319 - 23. Murdoch, J.R. (1972) Rate-Equilibria Relationships and Proton-Transfer Reactions. Journal of the American Chemical Society, 94, 4410-4418.
https://doi.org/10.1021/ja00768a002 - 24. Ruusuvuori, K., Kurtén, T., Ortega, I.K., Faust, J. and Vehkamäki, H. (2013) Proton Affinities of Candidates for Positively Charged Ambient Ions in Boreal Forests. Atmospheric Chemistry and Physics, 13, 10397-10404.
https://doi.org/10.5194/acp-13-10397-2013
NOTES

*Died on 22 June 2012.
![]()
1These AH compounds could also be formed directly in solution via hydrolysis of salts M occurred directly in solution before introduction of the samples into the ion source.