Open Journal of Clinical Diagnostics
Vol.2 No.3(2012), Article ID:22505,3 pages DOI:10.4236/ojcd.2012.23009
Sensitivity assay of polymerase chain reaction for detection of Canine Parvo Virus infection in dogs
![]()
Central Military Veterinary Laboratory, Meerut, Uttarpradesh, India
Email: *Prashant.bio@gmail.com
Received 10 June 2012; revised 13 July 2012; accepted 12 August 2012
Keywords: CPV; Canine Parvo Virus; PCR; Polymerase Chain Reaction; Sensitivity
ABSTRACT
A polymerase chain reaction was performed using reported primers for detection of Canine Parvo Virus (CPV) in the stool sample obtained from repository. The PCR primers were specific to VP1/VP2 gene of CPV. Sensitivity assay of PCR detection was performed by making dilutions of CPV positive DNA sample and running each dilution in a PCR and visualizing amplicons in ethidium bromide stained agarose gel under UV radiation. Study was valuable in determining the efficiency of PCR. The sensitivity of PCR in present study showed 20 ng/µl. The study was conducted to analyze the variation and sensitivity.
1. INTRODUCTION
CPV is a single stranded DNA virus and is a major pathogen of dogs. The virus is known to cause myocarditis in young puppies and hemorrhagic gastroenteritis in older animals. Over a period of time enteric form of disease has predominated and it persists as a major problem in breeding kennels, or where vaccination is widely practiced [1]. The presence of CPV in India has been confirmed by Ramadass and Khader [2]. Strain of CPV present in India has been documented to be CPV 2a [3]. Althogh the disease is vaccinated against; there is still a chance of vaccine failure amongst pups. If neglected the same may result in an episode of full blown disease. In an organized kennel, an outbreak may result in an epidemic and hence it is essential that presence of infectious agent is detected in shortest possible time, before organism can cause disease. DNA based detection using PCR is one of the most precise and rapid method for detection of CPV. Present study was undertaken to estimate the minimum detection limit of CPV PCR.
2. MATERIALS AND METHODS
2.1. Sample
Sample of Pup No. 18 CPV positive stool was taken from repository. DNA was extracted and PCR was run to check Pup No. 18 positive DNA for CPV. DNA concentration for sample extracted DNA was found to be 8.3 ng/µl by spectrophotometric analysis as reported by AIIMS.
2.2. Primers for PCR
Already reported primers C4A (5’-CAAATAGAGCATTGGGCTTACC-3’) and C4B (5’-CAATCTCCTTCTGGATATCTTC-3’) [4] amplifying VP1/VP2 gene of CPV were used.
2.3. Amplification
DNA was extracted using Qiagen DNA Extraction kit according to the protocol supplied with it. The PCR assay mixture (25 µl) contained primers (2 µM/L each), 1X-PCR buffer [750 mM/L Tris HCL (pH 8.8 at 25˚C), 200 mM (NH4)2SO4, 0.1% Tween-20], 1.5 mM/L MgCl2, 160 µM/L dNTPs and 1.5 units Taq DNA Polymerase. Thermal cycling parameters were 94˚C for 5 minutes and 35 cycles of each 1 minute at 94˚C, 1 minute at 55˚C and 1 minute at 72˚C followed by final extension at 72˚C for 10 minutes. Amplified product obtained in PCR assay is 400 bp.
2.4. Detection of PCR Amplified Products
For gel based PCR detection amplicons were detected by standard ethidium bromide staining and UV illumination of 2% agarose gels.
2.5. Sensitivity of Assay
Limit of sensitivity of PCR assays was determined by serial dilutions of the extracted DNA. Dilutions were made in nuclease free water up to 100–4. Each tube is vigorously mixed, vortex and sinned after each dilution. PCR was run for each dilution. Eight replicates were run on PCR for last dilution (Figure 1). Eight replicates were run on PCR for last dilution (Figure 2).
3. RESULT
The positive sample of Pup No. 18 CPV DNA was properly amplified by the PCR protocol followed. We added 5 µL of template/25µL reaction. Since DNA concentration as reported by AIIMS by spectrophotometric analysis was 8.3 ng/µL therefore DNA concentration in 5 µL template was (5 × 8.3) 41.50 ng (Figure 1).
As PCR is detecting up to 100–4 dilution of CPV DNA therefore the detection limit would be 41.50/100000000 i.e. 415 ng/5µL = 20 ng/µL (Figure 2).
4. DISCUSSION
PCR is primer directed enzymatic amplification of specific target DNA sequences. It has become an important part of modern diagnostic methods and also used for basic research. When used with proper caution PCR can
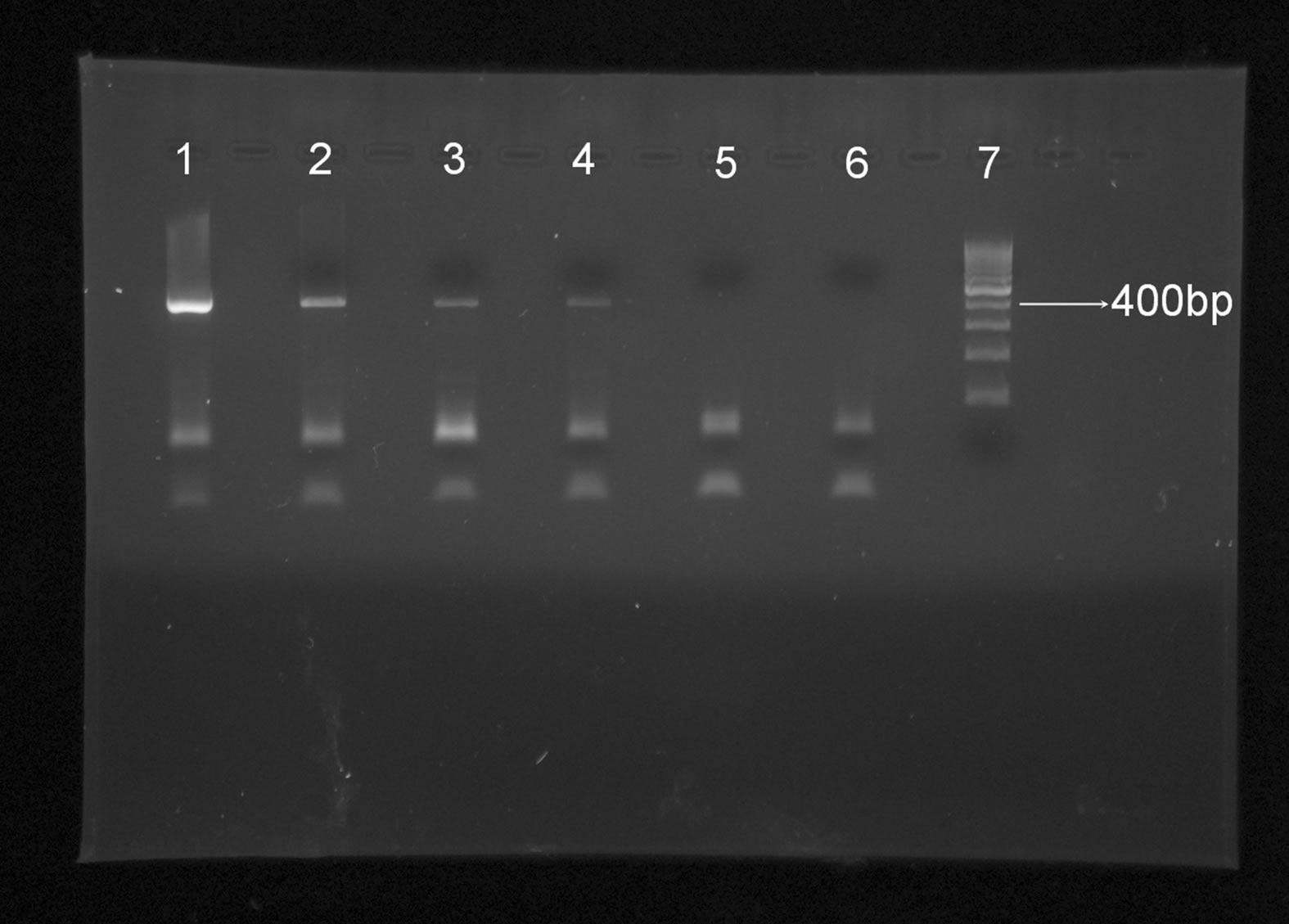
Figure 1. Agarose gel electrophoresis with dilutions PCR products, Lane 1—100–1, Lane2—100–2, Lane 3—100–3, Lane 4—100–4, Lane 5—100–5, Lane 6—100–6, Lane 7— 100 bp marker.
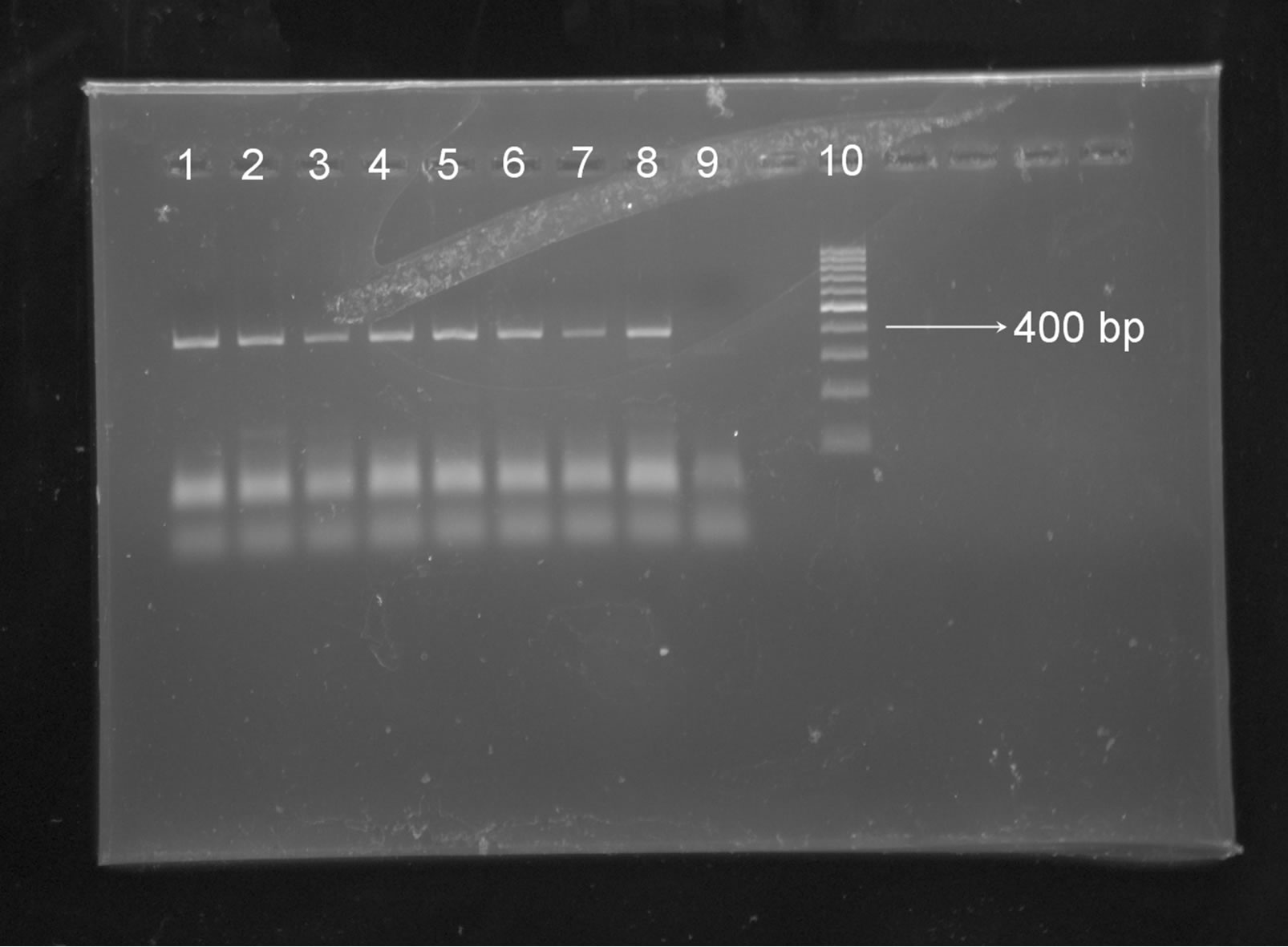
Figure 2. Agarose gel electrophoresis of PCR products of replicates of dilution of 100–4 of sample DNA, Lane 1-8 —100–4 dilution of sample DNA, Lane 8—100 bp marker, Lane 9—negative control, Lane 10—100 bp marker.
provide invaluable help in rapid identification of specific pathogens which are difficult to grow. PCR has been applied for detection of several viruses. Currently, the standard method for diagnosing the presence of viral pathogens in clinical samples relies on culture and other techniques which are time consuming and cumbersome. However, active research is under way using new molecular methods to decrease detection time and increase assay sensitivity. PCR has emerged as the molecular method of choice in achieving objectives. A PCR-based method cannot be given diagnostic status, until it includes methods to determine minimum detection limit, positive control, negative control and a reagent control (blank). The minimum detection limit and thereby the diagnostic sensitivity of a PCR assay, particularly on sub clinical samples with low target pathogens, depends on an effective sample treatment procedure. Thus by estimating minimum detection limit of a diagnostic PCR one can diagnose a disease condition where infection is in subclinical phase and sample volume is less. In this study we developed a PCR using reported primers to detect CPV infection. PCR showed good concordance with virus isolation “gold standard” for diagnostic virology, demonstrates specificity and sensitivity of this method. In addition PCR is more economical than virus isolation because it requires neither cell culture facilities nor highly trained specialists.
We got DNA concentration in sample DNA quantified from AIIMS by spectrophotometric analysis. We made dilutions of concentration of the DNA and used these dilutions as template for PCR to calculate the minimum detection limit for the viral infection. We calculated the minimum amount of DNA detected by PCR to be 0.20 ng which is very less compared to other methods. When replicates of the minimum detected dilution were run PCR was able to detect the DNA every time demonstrates, specificity, sensitivity and reproducibility of this method. PCR therefore is found to be of immense applicability in diagnosing virus in apparently healthy animals in latent stage or very early stage long before it shows any symptoms of disease in them thus controlling infection and formulating policies on prevention and control of disease at a very early stage at right time.
5. ACKNOWLEDGMENTS Authors are grateful to Directorate General Remount Veterinary Services for providing facilities for conduct of this work and to Dr H. K. Prasad, Professor of Department of Biotechnology AIIMS, for assistance provided.
REFERENCES
- Sagazio, P., Tempesta, M., Buonavoglia, D., Cirone, F. and Bounavoglia, C. (1998) Antigenic characterization of canine parvovirus strains isolated in Italy. Journal of Virological Methods, 73, 197-200. doi:10.1016/S0166-0934(98)00055-X
- Ramadass, P. and Khader, T.G.A. (1982) Diagnosis of canine parvovirus infection by agar gel precipitation test and fluorescent antibody techniques. Cherion, 11, 323-328.
- Kumar and Dharmadheeran (2008) Detection of CPV from fecal samples by PCR. Journal of Remount and Veterinary Corps, 47, 197-201.
- Narayanan, S., Amit, K., Chug, P.K., et al. (2001) Strain characterization of Indian isolates of Canine Parvo Virus by PCR RFLP. Journal of Remount and Veterinary Corps, 40, 67-74.
NOTES
*Corresponding author.