Open Journal of Immunology
Vol.2 No.3(2012), Article ID:22534,13 pages DOI:10.4236/oji.2012.23011
Single doses of p38 MAP kinase inhibitors or prednisolone affect CRP and IL-6 in patients with active Rheumatoid Arthritis (RA)
![]()
GlaxoSmithKline R&D, Medicines Research Centre, Stevenage, UK; *Corresponding Author: Pauline.t.lukey@gsk.com
Received 10 December 2011; revise 20 January 2012; accepted 3 February 2012
Keywords: p38 MAPK; Prednisolone; RA; Biomarkers; Proof of Mechanism; Randomised Controlled Trial
ABSTRACT
Purpose: To investigate the effects of single doses of losmapimod, dilmapimod (inhibitors of p38 MAPK) and prednisolone on biomarkers of systemic inflammation (serum C-reactive protein (CRP) and interleukin (IL)-6) in subjects with active rheumatoid arthritis (RA). Methods: Two randomized, double blind, placebo controlled, parallel group, single dose studies investigated the CRP and IL-6 dose-response relationships for losmapimod (study A) and dilmapimod and prednisolone (study B) in patients with active RA on stable weekly doses of methotrexate. CRP, IL-6 and other exploratory biomarkers were measured in blood at baseline and up to 72 hours post-dosing. Results: In study A, 51 subjects were randomized to receive losmapimod (7.5, 20 and 60 mg) or placebo and in study B, 77 subjects were randomized to receive dilmapimod (7.5, 15 and 25 mg) or prednisolone (10, 20 and 50 mg) or placebo. Single doses of prednisolone caused a decrease in circulating IL-6 detectable at 3 and 24 hours post-dose as well as a CRP single-dose relationship at 48 hours post-dose in patients with active RA. All doses of losmapimod produced a statistically significant reduction in serum IL-6, 3 hours post-dose but had no effect on CRP. Dilmapimod had no effect on IL-6 or CRP at any dose or time. Conclusion: This single-dose response clinical study design could be useful in the early clinical development of anti-inflammatory agents for the treatment of rheumatoid arthritis. Inhibition of both circulating IL-6 and CRP would increase confidence that a new chemical entity may have the potential to deliver clinical benefit in RA. Trial registration: [clinicaltrials.gov: NCT00256919 and NCT00134693].
1. INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterised by over-production of inflammatory cytokines (e.g. TNFa, IL-6, IL-1) [1] which result in increased levels of acute phase proteins (e.g. C-Reactive Protein (CRP)) in the blood [2]. CRP is a well-validated surrogate marker of disease activity and correlates with functional outcome in RA [2,3]. CRP has a half-life measured in days [4] and is not subject to circadian rhythms. These factors reduce the variability in measuring CRP and contribute to its success as a surrogate marker. Treatments that affect CRP could reasonably be expected to translate into clinical efficacy in RA [3]. In fact, CRP is reduced significantly by a single dose of infliximab in RA patients with maximum inhibition observed 3 days postdose [5]. Transcription of CRP is regulated by pro-inflammatory cytokines [6] including IL-6 which may be derived directly from the inflamed synovium [4,7] and therefore a reduction in IL-6 should precede a decrease in CRP. However, given the relatively short half-life of IL-6 [4] and the known circadian variability of this cytokine [8], IL-6 is more variable and difficult to correlate with outcome measures. Thus, observing an early effect on IL-6 and a subsequent effect on CRP in the same subjects with RA could provide confidence that a novel therapy has the potential to deliver clinical efficacy.
p38 mitogen-activated protein kinase (MAPK) is a serine/threonine-directed protein kinase which phosphorylates a number of intracellular signal transduction molecules that are involved in regulating the biosynthesis of inflammatory cytokines [9]. Consequently, inhibitors of this kinase, were expected to be effective in diseases where the pro-inflammatory mechanisms are cytokinedependent such as rheumatoid arthritis. It is now evident that clinical studies with p38 MAPK inhibitors have generally been disappointing in RA. Recently, RA data for pamapimod (Roche) [10] and VX-702 (Vertex) [11] have failed to show efficacy in RA patients. There is a growing body of evidence that some initial activity in RA patients may be observed but that for reasons unknown this effect may not be sustained [12]. Losmapimod [13] and dilmapimod (potent and selective p38 MAPK inhibitors) were safe and well-tolerated in proof of concept studies in RA but did not provide significant benefit in this disease (manuscript in preparation). Recently, however, BMS-582949 has been reported to provide efficacy in RA patients [14], and this is attributed to its apparent dual activity which inhibits p38 MAPK activity and activation [15]. Thus, there remains a need to understand this mechanism and the single-dose response studies described here add to the body of knowledge in this significant pathway.
Prior to investing in large proof of concept studies, p38 MAP kinase inhibitors have been tested in smaller proof of mechanism studies. Healthy subjects have been challenged systemically with bacterial lipopolysaccharide (LPS) and inhibition of resultant inflammatory cytokines was observed in the presence of p38 mitogen activated protein kinase (MAPK) inhibitors [16]. In addition, VX-702, another p38 MAPK inhibitor, was dosed for 5 days in patients with acute coronary syndrome undergoing percutaneous coronary intervention (PCI) and showed a detectable reduction in CRP 24 hours post-dose [17,18]. These results indicate the potential of p38 MAPK inhibitors to deliver an anti-inflammatory effect in an acute inflammatory setting. This apparent disconnect between the early experimental medicine studies and the RA PoC studies may be due to a number of factors. There may be differences in the mechanisms driving an acute inflammatory response and those driving chronic inflammation in RA [19]. Furthermore, there is a history of p38 MAPK inhibitors and a risk of hepatotoxicity which is likely to have limited administration of sufficiently high doses for extended periods [20]. There is also a recent discussion in the literature regarding transient effects of p38 MAPK inhibitors in RA which tend to wane with duration of dosing [12]. Further understanding of these observations requires ongoing investigation into the anti-inflammatory response in RA and the role of p38 MAPK.
In the early development of two p38 MAPK inhibitors (losmapimod and dilmapimod, previously known as GW856553 and SB-681323 respectively) the novel study design described here was tested in order to show antiinflammatory effects and to indicate an appropriate dose range. These studies were conducted in patients with RA in order to overcome the shortcomings of testing these molecules in an acute inflammatory model. Prednisolone, which has proven efficacy in RA was included as a positive control [21-23]. These clinical studies provide additional information about p38 MAPK inhibitors in RA and describe a new study design which may be used in the early clinical development of other anti-inflammatory mechanisms in this disease.
2. PATIENTS AND METHODS
2.1. Ethical Conduct of the Studies
These studies were reviewed and approved by national, regional, or investigational centre ethics committees or institutional review boards as required in the relevant countries.
These studies were conducted in accordance with Good Clinical Practice and all applicable regulatory requirements, and, the guiding principles of the Declaration of Helsinki. Written informed consent was obtained from each subject prior to the performance of any studyspecific procedures. Electronic case report forms (eCRFs) were provided for each subject’s data to be recorded. Study A (GSK protocol number: RA3103730) can be found on ClinicalTrials.gov under the study identifier NCT00256919 and the study B (GSK protocol number RA1104046) identifier is NCT00134693.
2.2. Patients
Male and female subjects aged ³18 years, with a diagnosis of RA according to revised 1987 American College of Rheumatology criteria were included. Subjects had serum CRP ³ 10 mg/L and ³ 3/66 swollen or ³ 3/68 tender/painful joints and were receiving stable weekly doses of methotrexate (2.5 mg to 25 mg). Other antirheumatic therapy, including disease modifying antirheumatic drugs [DMARDs], non-steroidal anti-inflammatory drugs [NSAIDs], cyclo-oxygenase-2 (COX-2) inhibitors and, in Study A prednisolone (≤10 mg/day), was permitted providing the doses had been stable for 4 weeks prior to the study. Patients receiving biological anti-rheumatic therapies were excluded. Any history of liver disease or elevated liver transaminases (>1.5 times the upper limit of normal) were also exclusion criteria.
2.3. Study Design
The two studies were multi-centre, double blind, double dummy (study B only), randomized, placebo controlled and parallel group in design.
In Study A, eligible subjects were randomized to receive single doses of losmapimod (7.5, 20, and 60 mg) or placebo. In Study B, subjects were randomized to receive single doses of dilmapimod (7.5, 15 and 25 mg), or prednisolone (10, 20 and 50 mg) or placebo. Prednisolone was over-encapsulated in order to maintain the blinding. Dissolution testing showed no significant effect of the over-encapsulation against the compendial specification for prednisolone tablets. Subjects received the study treatment on a day between the two weekly doses of methotrexate and at least 1 hour after a light breakfast and two hours before the next meal.
Screening took place within 10 days prior to the first dose and the 120-h post-dose visit was used as a followup visit.
2.4. Endpoints
The primary endpoint was the single-dose response for serum CRP at 72 h post-dose (using placebo as the zero dose). Secondary endpoints were the single-dose response for serum CRP at 24 and 48 h and IL-6 singledose response (measured at 1, 3, 24 and 72 h post-dose).
CRP was measured using a high sensitivity assay and IL-6 using an ELISA (Quest diagnostics). Other exploratory biomarkers were assayed either at Pathway Diagnostics or at GSK R&D, Stevenage as previously described [24].
Other endpoints included estimations of previously described serum biomarkers of inflammation A-SAA, BCL/CXCL13, MPIF-1, TNFSFR14, calprotectin, sIL- 6R, and TNFα [24] measured at 1, 3, 24 and 72 hrs post dose. In addition, blood cytokine messenger ribonucleic acid (mRNA) levels were measured at 45, 90 and 180 minutes after dosing. These mRNAs were previously shown to be reduced by p38 MAPK inhibition (IL-6, TNF-a, IL-1b, COX2, IL-8, MMP9, iNOS manuscript in preparation), modulated by prednisolone (FKBP5, GILZ, IFI30, IL-1R2, DDIT4, TXNIP and ZNF145 manuscript in preparation) or modulated by both (DUSP1).
GAPDH mRNA, cyclophilin mRNA and β-Actin mRNA were used in the statistical analysis to control for variability between samples caused by confounding technical factors.
2.5. Dose Rationale
Doses of 15 - 20 mg/day of oral prednisolone are administered to control a flare of RA activity but this is rapidly tapered to 5 - 10 mg/day within the first few weeks of dosing. In addition doses of 60 mg/day have been administered for short periods to RA patients with significant benefit [25]. Therefore, a dose range of 10 - 50 mg oral prednisolone was considered reasonable for this single dose study.
The IC50 for inhibition of TNFa production by losmapimod in a whole blood assay of LPS-induced TNFa production, was 25 nM (9.5 ng/ml) [13]. Dilmapimod had a similar IC50 in the same assay (not shown).
Single doses of losmapimod administered to healthy subjects resulted in 30% (7 mg), 48% (20 mg) and 59% (60 mg) weighted mean reduction in ex vivo whole blood LPS-induced TNF-a production. Single doses of dilmapimod (7.5 mg and 25 mg) were administered to patients with COPD and shown to be active in an ex vivo whole blood assay of LPS-stimulated production of TNFa (33.4% weighted mean inhibition by 7.5 mg and 40% inhibition by 25 mg) [26].
All dosing in the studies described here was carried out between two weekly doses of methotrexate. This was based on a previous study which measured CRP levels in RA patients during weekly or daily administration of methotrexate [26,27]. This indicated that CRP levels remained stable with daily dosing of methotrexate, but, in subjects given weekly doses, CRP levels decreased after dosing and increased again prior to the subsequent dose [27]. Therefore, in the present studies, it was considered advantageous to dose subjects when CRP levels were at their lowest i.e. between two weekly doses of methotrexate. Theoretically, this would increase the probability of detecting a difference between active and placebo.
2.6. Pharmacokinetics
Plasma samples collected in study A were assayed for losmapimod using a validated analytical method based on protein precipitation followed by HPLC/MS/MS analysis. This method was validated over the range 0.2 - 200 ng/ml and the lower limit of quantification was 0.2 ng/ml using a 50 ml sample of human plasma. At all validation sample concentrations examined for losmapimod, the bias was less than 15%.
Pharmacokinetics samples were not collected in study B; however the PK characteristics of dilmapimod were studied in a separate study (not shown).
2.7. Statistical Methods
It was estimated that 12 evaluable subjects in each group were required to provide 90% power to detect a dose-response decrease in log CRP based on a test of significance at the two-sided 5% level [28]. For each dose and time separately, point estimates and corresponding 95% confidence intervals were constructed and back transformed and all pairwise treatment differences between doses of p38 MAPK inhibitors and placebo presented accordingly.
Principal components analysis was used to identify trends in the mRNA transcripts. A second principle component analysis was used as a covariate in a repeated measures analysis of mRNA transcripts to adjust for any technical factors.
The maximum plasma concentrations (Cmax) of losmapimod, the first time the plasma concentration reached maximum (tmax), and the area under the plasma concentration-time profiles by time t (t hours post dose) (AUC 0 - t) were calculated using a non-compartmental analysis method. The weighted mean concentration by time t was defined as AUC 0 - t divided by t.
The effect of systemic exposure of losmapimod on IL-6 reduction was studied by exploring the relationship between IL-6 ratio to baseline at time t and the weighted mean concentration by time t using a nonlinear mixed effects model that considered both the population and subject level effects.
3. RESULTS
3.1. Patient Population
The baseline and demographic characteristics of the subjects are shown in Table 1. Study A involved 51 and Study B involved 77 RA subjects receiving methotrexate. In study A approximately 50% in each group concomitantly received low dose glucocorticoids: prednisolone, prednisone or methylprednisone (Table 1). No subject in study B was receiving glucocorticoids. Baseline rheumatological assessments and serum levels of CRP and IL-6 were consistent with active rheumatoid arthritis (Table 1).
3.2. Pharmacokinetics of Losmapimod
Overall, 37 subjects provided a total of 268 samples for pharmacokinetic analysis of losmapimod in study A. The exploratory analysis of the losmapimod plasma concentration data obtained from these patients showed approximately dose proportional increase in exposure for doses of 7.5 mg and 20 mg. There was relatively less than proportional increase in the plasma concentration following the 60 mg dose. The elimination phase appeared slightly longer following the 60 mg dosing of losmapimod compared to the other two dose levels. The geometric mean observed maximum plasma concentration (Cmax) was 42 ng/mL (95% CI: 33; 55), 75 ng/mL (95% CI: 56; 102), and 126 ng/mL (95% CI: 101; 156) for 7.5 mg, 20 mg and 60 mg group, respectively. The Cmax was reached approximately 3 hours post dose. The area under the plasma concentration-time curve (AUC) up to 24 hours post dose was 333 ng·h/mL (95% CI: 281; 393), 714 ng·h/mL (95% CI: 573; 891) and 1377 ng·h/mL (95% CI: 1164; 1630) for 7.5 mg, 20 mg and 60 mg dose groups, respectively.
The pharmacokinetic data for dilmapimod was not collected in study B, but was measured in a previous study where dilmapimod was administered at 7.5 mg/day (2.5 mg in the morning and 5 mg in the evening) for 15 days to nine patients with RA on a background of stable doses of methotrexate. In this study the geometric mean Cmax (after morning dose of 5 mg) was 33 ng/ml (95% CI: 21; 51 ng/ml) with a Tmax of 0.75 hours and geometric mean AUC (0 - 24) was 125 ng·h/ml (95% CI: 82; 188).
3.3. CRP and IL-6
Prednisolone decreased circulating levels of serum CRP and baseline-adjusted ratios to placebo (Figure 1(c) and (f)). Single doses of prednisolone (10, 20 and 50 mg) produced a dose-dependent decrease in CRP at 48 hours only although the trend did not achieve statistical significance (slope = −0.01, p = 0.070) (Figure 3(c)). The percentage decrease in CRP compared to placebo at 48 hours was 41% (95% CI: 11%; 62%) for the 10 mg dose; 33% (95% CI: 0%; 55%) for the 20 mg dose and 39% (95% CI: 10%; 59%) for the 50 mg dose.
Prednisolone decreased circulating levels of serum IL-6 (Figure 2(c)) and baseline-adjusted ratios to placebo (Figure 2(f)). There was a statistically significant single dose response relationship between prednisolone and IL-6 at 3 hours (Figure 3(f)) (largely driven by the highest dose) and 24 hours post-dose. The percentage decrease in IL-6 compared to placebo at 3 hours was 40% (95% CI: −27%; 72%) for the 10mg dose; 11% (95% CI: −78%; 56%) for the 20 mg dose and 53% (95% CI: 12%; 75%) for the 50 mg dose, the percentage decrease in IL-6 compared to placebo at 24 hours was −6% (95% CI: −164%; 57%) for the 10 mg dose; 11% (95% CI: −166%; 57%) for the 20 mg dose and 56% (95% CI: −4%; 82%) for the 50 mg dose.
Losmapimod (7.5, 20, and 60 mg) showed no effect on CRP at any time point either compared with baseline or placebo (Figures 1(a), 1(d) and 3(a)).
All doses of losmapimod produced a statistically significant reduction in serum IL-6 at 3 hours post-dose although the differences were similar across the doses (Figures 2(a) and (d)). The percentage decrease in IL-6 compared to placebo at 3 hours was 55% (95% CI: 18%; 76%) for the 7.5 mg dose; 53% (95% CI: 12%; 75%) for the 20 mg dose and 59% (95% CI: 25%; 77%) for the 60 mg dose (Figure 3(d)).
Dilmapimod had no effect on CRP (Figures 1(b) and (e)) or IL-6 (Figures 2(b) and (e) and Figure 3(e)).
3.4. Relationship between Plasma Levels of Losmapimod and Serum IL-6
Figure 4 shows the relationship of IL-6 ratio to baseline versus the weighted mean concentration of losmapimod for each dose group (Figures 4(a)-(c)) and all dose groups including placebo (Figure 4(d)). Overall,

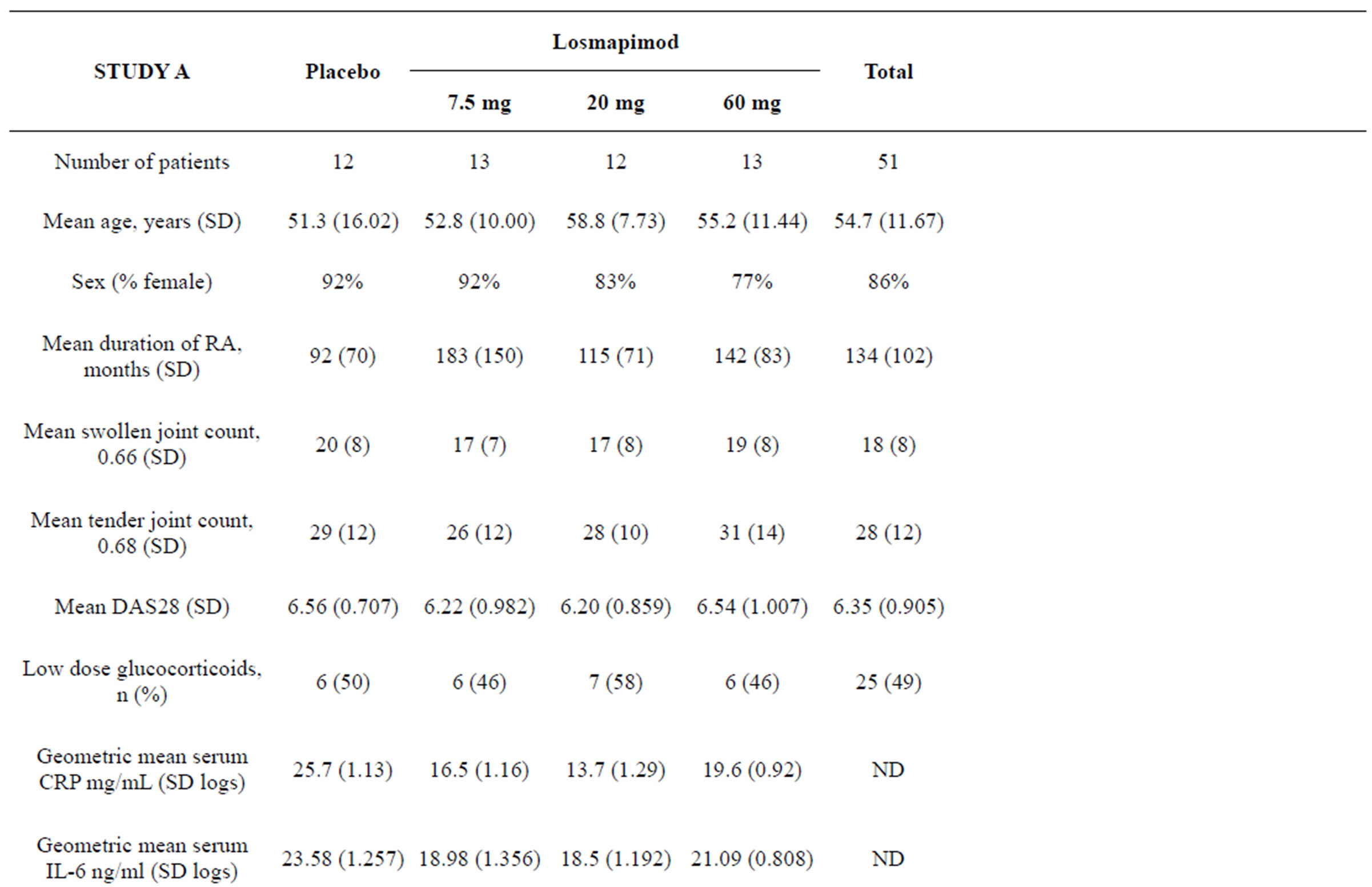
Table 1. Baseline characteristics.
mg/L Adjusted ratio to placebo

Figure 1. Serum CRP levels in patients with RA treated with single doses of losmapimod ((a) and (d)) dilmapimod ((b) and (e)) or prednisolone ((c) and (f)). Serum levels of CRP are presented as geometric mean mg/L and 95% confidence intervals ((a)-(c)) and as adjusted ratio to placebo and 95% confidence intervals (d)-(f).
there was a tendency towards an exposure-driven IL-6 response: an increase in response with an increase in exposure. A reduction in IL-6 was more likely to be observed when the weighted mean concentration of losmapimod was above 20 ng/mL. Compared to the two lower doses, more patients in the 60 mg dose group had IL-6 reductions. The nonlinear mixed effects model showed a potential non-linear trend over the concentration range, and the maximum reduction in IL-6 reached in this study was just under 60%. There was little evidence of further reduction in IL-6 with increases in the weighted mean concentration of losmapimod above 40 ng/mL.
3.5. Exploratory Protein Biomarkers
In Study A, single doses of losmapimod had no effect on circulating levels of serum TNFSFR14, MPIF-1, calprotectin, sIL-6R, or TNFα. However, there was a significant dose-response relationship with increasing dose of losmapimod and serum levels of both A-SAA (Figure 5(a)) and BLC/CXCL13 (Figure 5(b)) at 24 hours. Baseline serum levels of A-SAA were 179.6 (111.0; 290.3) μg/ml and of BLC/CXCL13 were 237.9 (68.8; 96.8) pg/ml; expressed as mean (95% CI).
In Study B, neither dilmapimod nor prednisolone had any effect on the exploratory serum biomarkers (data not shown). MPIF-1 was the only exception, which by comparison with placebo, was statistically significantly reduced in the 50 mg prednisolone group 24 hours posttreatment.
3.6. Exploratory mRNA biomarkers
In Study A, there was some evidence of an increase in TNF-α (Figure 5(g)), COX-2, IL-1b (Figure 5(h)) and MMP9 mRNAs following losmapimod at all doses at 90 and 180 minutes post-dose compared with placebo, but this was not statistically significant and may have been due to the reduction of these transcripts detected in the placebo group. There was no evidence of an effect of losmapimod on other biomarker mRNA levels at any time point.
In Study B, there was a downward trend of IL-1b (Figure 5(d)) and IL-8 which reached significance at 90 ng/L Adjusted ratio to placebo
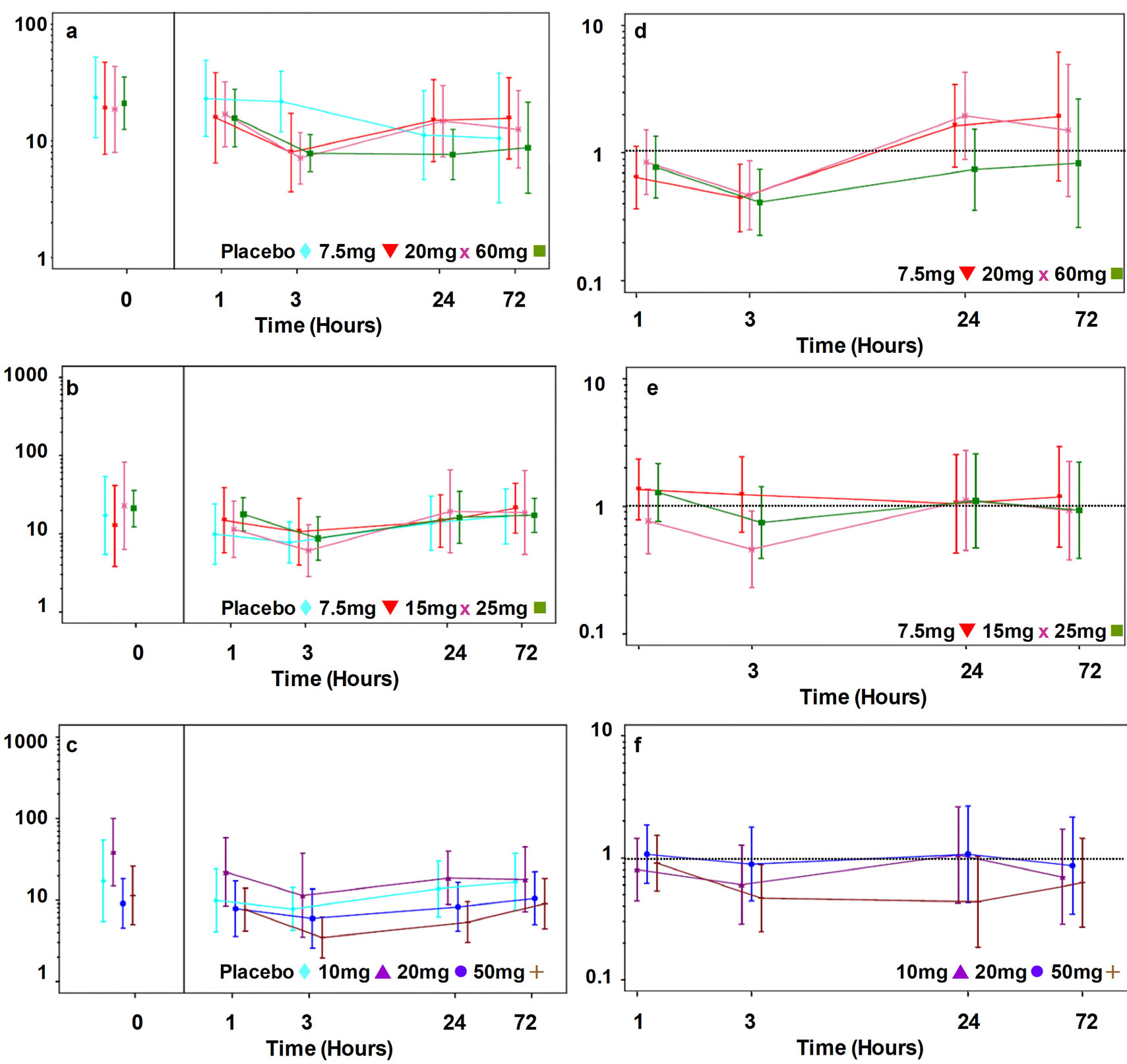
Figure 2. Serum IL-6 levels in patients with RA treated with single does of losmapimod ((a) and (d)), dilmapimod ((b) and (e)) or prednisolone ((c) and (f)). Serum levels are presented as geometric mean ng/L and 95% confidence intervals ((a)-(c)) and as adjusted ratio to placebo and 95% confidence intervals ((d)-(f)).
mins at 7.5 mg in the dilmapimod treated group. However, there was no other evidence of an effect of single doses of dilmapimod on mRNA biomarker levels. Single doses of prednisolone (20 mg or 50 mg) were associated with statistically significantly increased mRNA levels of GILZ, DUSP1 (Figure 5(f)), IL-1R2, TXNIP, ZNF145, FKBP5 and DDIT4 (Figure 5(e)), and statistically significantly decreased mRNA levels of IL-1b (Figure 5(d)), TNF-α (Figure 5(c)) and IFI30.
3.7. Safety
Both p38 MAPK inhibitors were well tolerated in subjects with RA. Due to liver toxicity associated with other compounds acting via this mechanism [20], liver function tests including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), gamma-glutamyl transferase (GGT) and bilirubin were monitored in both studies. In Study A, two subjects, (one receiving 20 mg losmapimod group, the other 60 mg losmapimod), had adverse events associated with liver function tests, which were judged by the Investigator to be possibly related to the investigational product. Both subjects had ALT and AST values more than two times the upper limit of normal values (of note both subjects were treated with methotrexate). In Study B, one subject had ALT levels considered to be of clinical concern (defined as > twice the upper limit of normal). However the subject had received prednisolone 20 mg and the raised ALT levels were present at screening.
4. DISCUSSION
Using the data generated in these studies it may be feasible to design a single dose cross-over study as an alternative to the parallel design used here. Crossover studies are not generally recommended for RA as the course of disease may be variable over time in individual subjects [29]. However, several crossover design studies have been conducted in RA over the past decade [30-32], but these have generally been studies of analgesics and devices rather than of DMARDs. The current observation that single doses of prednisolone decrease serum CRP for at least 48 hours with no significant effect at 72 hours CRP IL-6

Figure 3. Predicted dose response curves for CRP and IL-6. The CRP dose response is shown for the 48 hours post-dose timepoint and the IL-6 dose response for the 3 hours postdose timepoint. Predicted dose response is illustrated by the unbroken line and 95% confidence intervals by the dotted lines. Individual datapoints are shown with 95% confidence intervals for the different doses of losmapimod, dilmapimod and prednisolone.

Figure 4. Relationship between IL-6 ratio to baseline and weighted mean plasma concentration of losmapimod at doses of 7.5 mg (a), 20 mg (b) and 60 mg (c) and all doses together (d). The plasma levels of losmapimod are presented as weighted mean concentrations up to the time at which the IL-6 was measured. Each point represents an individual subject at one of the 4 timepoints (1 hour, 3 hours, 24 hours and 72 hours post-dose. The model-predicted IL-6 ratio to baseline is drawn in red (d).
could be used to define a reasonable between-period washout. Thus a crossover design with one week between single doses could deliver significant savings in the number of patients required to achieve a dose-response, while retaining the advantages of a relatively short study.
Such a single dose response study could be used during early clinical development of novel anti-inflammatories in patients with active RA. The advantages of this study design include: limited pre-clinical and clinical safety cover is required prior to study start; the short duration of study participation improves patient and physician acceptability; anti-inflammatory effects can be observed in the chronic inflammatory disease of interest, without any in vivo or ex vivo manipulations; provides an early indication of potential to deliver efficacy and provides an indication of possible therapeutic dose ranges.
Prednisolone (an effective anti-inflammatory agent with established repeat dose efficacy in RA) was used as a positive control to validate the study design. A single administration of prednisolone resulted in a dose-dependent decrease in levels of serum IL-6 for at least 24 hours in patients with active RA. This decrease in serum
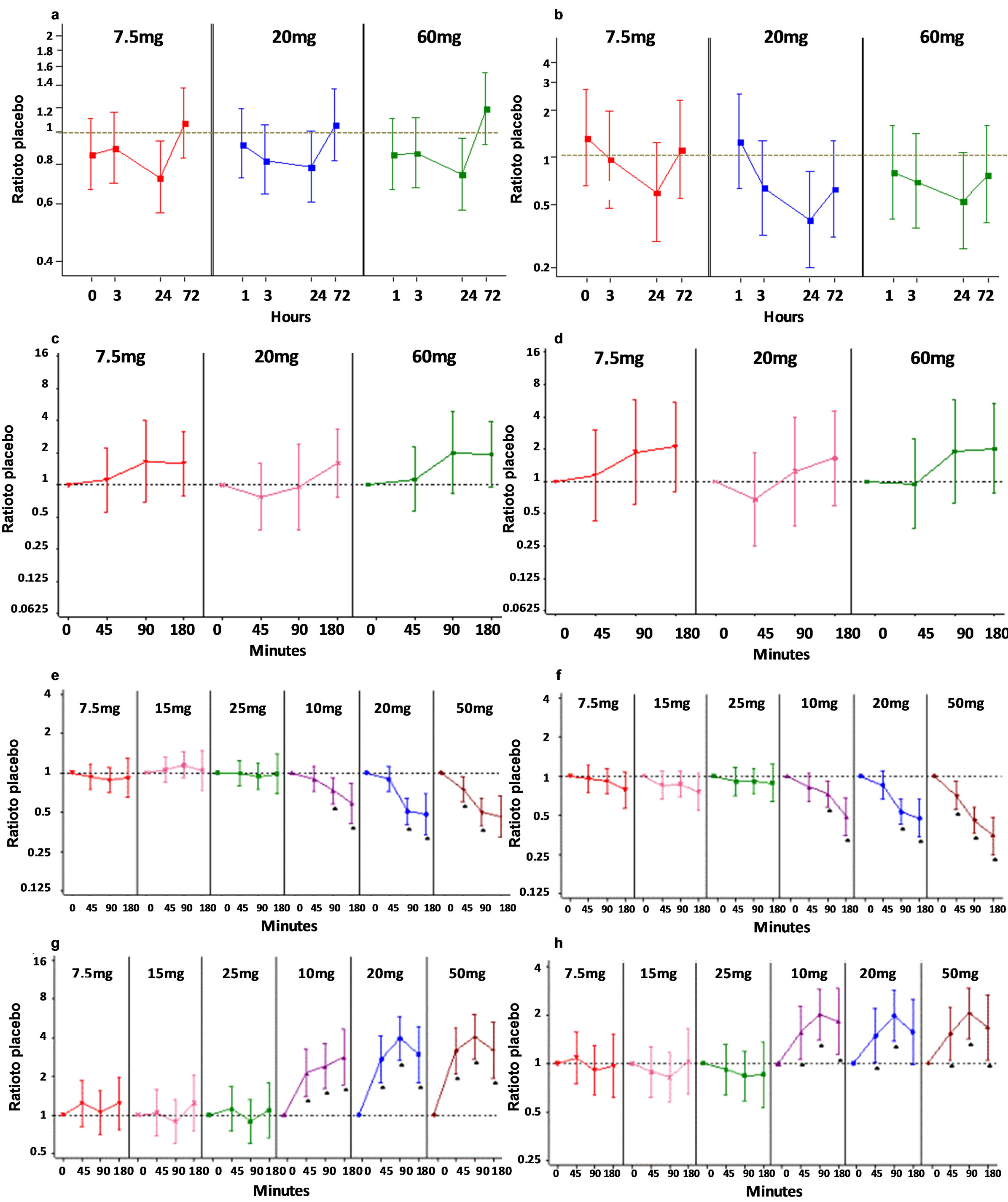
Figure 5. Exploratory biomarkers. Effects of single doses of losmapimod ((D 7.5 mg, ○ 20 mg, □ 60 mg) on serum protein levels of A-SAA (a) and BLC (b) are shown pre-dose and 1, 3, 24 and 72 hours post-dosing. Results are expressed as adjusted ratios to baseline compared to placebo (95% CIs). Effects of single doses of losmapimod on whole blood mRNA levels for TNF-a (c) and IL-1b (d) are shown pre-dose, 45, 90 minutes and 180 post-dose. Effects of single doses of dilmapimod (7.5 g, 15 mg and 25 mg) and prednisolone (10 mg, 20 mg, 50 mg) on whole blood mRNA levels for TNF-a (e), IL-1b (f), DDIT (g) and DUSP1 (h) are shown pre-dose and 45, 90 and 180 post-dosing. mRNA results are expressed and as logarithmic (base 2) ratios to placebo. The dotted grey line represents a ratio of 1 (where no difference is observed between baseline and placebo).
IL-6 for at least 24 hours resulted in a dose-dependent decrease in circulating levels of serum CRP observed 48 hours post-dose. This causative relationship between decreased serum IL-6 resulting in a subsequent decrease in CRP, is supported by the role of IL-6 in the transcriptional regulation of CRP [4]. These data would suggest that application of this study design to the development of a novel anti-inflammatory in RA, would require a dose-dependent decrease in IL-6 and a subsequent dose-dependent decrease in CRP in order to support progression to the next stage of development. Furthermore, the dose response curve would imply that the therapeutic dose of prednisolone would be in the range of the doses administered (10 - 50 mg, which is indeed the case) [25,33]. The two p38 MAPK inhibitors studied here had similar potency and selectivity in vitro; however, they delivered different effects in RA patients using this study design. Single administrations of dilmapimod at three doses had no effect on levels of serum CRP or IL-6 in patients with active RA. Whilst, losmapimod inhibited circulating serum levels of IL-6 but had no effect on CRP in a similar patient population. As plasma levels of both p38 MAP kinase inhibitors reached were above the in vitro IC50 at all doses, the differences in activity may be explained by the more rapid clearance of dilmapimod resulting in a shorter duration of exposure above the IC50 compared to losmapimod. Both compounds were safe and well tolerated in repeat dose clinical efficacy PoC studies in RA, however neither compound met the criteria for clinical progression in RA (manuscript in preparation). These data would imply that in order to meet the criteria to undertake a subsequent repeat dose study in RA, a novel anti-inflammatory molecule should provide a single dose response for both CRP and IL-6.
A complicating factor is the observation that about 50% of the subjects in Study A receiving losmapimod were taking concomitant glucocorticoids; whilst dilmapimod was administered to glucocorticoid naïve subjects. There is no basis to expect a pharmacokinetic interaction between glucocorticoids and losmapimod however, co-administration could potentially affect pharmacodynamics. There is evidence to suggest that administration of a p38 MAP kinase inhibitor to glucocorticoid-resistant asthmatics may reverse the resistant phenotype and restore glucocorticoid-sensitivity [34]. These data would suggest a possible beneficial effect of combining losmapimod and prednisolone in patients with relative resistance to glucocorticoids. On the other hand, glucocorticoids are reported to induce expression of a protein phosphatase (MKP-1) that is involved in negative regulation of MAP kinases including p38 [35,36]. It is possible that this mechanism could limit the potential for achieving efficacy benefits from a combination therapy (glucocorticoid plus p38 inhibitor). Therefore, the overall effect of co-administration is unclear and unlikely to significantly impact this single dose study.
In addition to the single dose response observed at 3 hours post-dose, a relationship was observed between the plasma concentration of losmapimod and the effect on serum IL-6 levels, such that increasing weighted mean plasma levels of losmapimod resulted in decreasing levels of serum IL-6 in patients with RA. This supports a causal relationship between inhibition of p38 MAPK and decreased levels of circulating IL-6 in patients with active RA. The effective range of weighted mean plasma concentration of losmapimod appears to be between 20 ng/ml and 40 ng/ml; where weighted mean concentrations below 20 ng/ml showed little effect, whilst the maximal effect (~60% decrease in serum IL-6 levels) was reached at 40 ng/ml with little additional benefit above this weighted mean concentration. It is noteworthy that a maximal decrease in IL-6 of ~60% was also observed with prednisolone; implying that 60% may be the maximal effect that can be delivered by p38MAPK inhibition or glucocorticoids in RA. However, the duration of the effect for 24 hours after prednisolone dosing may have resulted in the subsequent decrease in serum CRP at 48 hours.
In order to support the effects on IL-6 and CRP, exploratory pharmacodynamic markers were also measured. The pharmacodynamic effects of prednisolone were evidenced by the increased levels of mRNA markers (including amongst others GILZ, DUSP1 and DDIT4) observed at the early time point of 45 minutes post dosing. This effect has been examined in more detail [37]. Both A-SAA and BLC/CXCL13 exhibited a dose-response relationship with increasing doses of losmapimod whereas no such effect was evident with dilmapimod.
In conclusion, administration of single doses of a potent pro-inflammatory cytokine inhibitor to patients with ongoing chronic inflammation resulted in a detectable effect on circulating IL-6 and CRP. This study design may be usefully incorporated into the early clinical development of anti-inflammatories in order to increase confidence that the mechanism under study has the potential to deliver clinical efficacy.
5. ACKNOWLEDGEMENTS
These studies were GlaxoSmithKline R&D sponsored studies and were fully funded by GSK R&D.
We would like to acknowledge the contribution of the investigators and patients who participated in these studies and without whom none of this work could have been done. We also acknowledge the contribution of Peter Critchley (GSK) for his assistance in operationalising these multi-centre, multi-national studies and to Steve Mallett (GSK) for analyzing the qRT-PCR data. Sue Daly (self-employed medical writer) drafted the first, very early version of the manuscript based on the final study reports provided by GSK and was paid by GSK for this work. All subsequent versions were written by the in-house GSK team.
Investigators for study A: Dr. AZ Batalov, (Principal Investigator) Clinic of Rheumatology, Medical University, Plovdiv, Bulgaria; Prof. Folke Sjoberg, Berzelius Clinical Research Center, Linköping, S-582 25 Sweden; Dr. Anders Henriksson, Quintiles-Hermelinen, Lulea, SE- 972 33 Sweden; Dr. Eva Zaren, Quintiles AB, Uppsala, 753 23 Sweden; Dr. Bernhard Hellmich, Rheumaklinik Bad Bramstedt & Universitaetsklinikum Schleswig-Holstein, 24576 Bad Bramstedt, Germany; Dr. Florian Schuch, Moehrendorfer Str. 1c, 91056 Erlangen, Germany; Prof. Dr. Peter Herzner, Tal 6, 80331 Muenchen, Germany; Dr. Herbert Kellner, Romanstr. 9, 80639 Muenchen, Germany; Dr. Harald Tremel, Denickestr. 88, 21075 Hamburg, Germany; Dr. Federico Navarro, Hospital Virgen de la Macarena, 41009 Sevilla, Spain; Dr. Emilio Martin Mola, Hospital Universitario La Paz, 28046 Madrid, Spain; Dr. Juan Jesus Gomez Reino, Hospital de Santiago, 15703 Santiago de Compostela, Spain; Prof. Zlatimir Kolarov, MHAT “Sv.Ivan Rilski”, Sofia, 1621, Bulgaria; Prof. Yordan Sheytanov/Assoc. Prof. Rumen Stoilov, MHAT “Sv.Ivan Rilski”, Sofia, 1621, Bulgaria; Assoc. Prof. Antoaneta Toncheva, National Multiprofile Transport Hospital “Tsar Boris III”, Sofia, 1233, Bulgaria; Prof. Vladimir Kovalenko, Strazgesko Research Institute of Cardiology, Kyiv, 3680, Ukraine; Prof. Ekaterina Amosova, Kiev National Medical University, Kiev, 3680, Ukraine; Dr. Lyudmila Kononenko, City Clinical Hospital #27, Kharkiv, 61002, Ukraine; Dr. Andriy Gnylorybov, Donetsk Institute of Urgent and Restitution Surgery, Donetsk, 83045, Ukraine; Dr. Valentyna Chopyak, D. Galytsky Lviv National Medical University, Lviv, 79010, Ukraine.
Investigators for Study B: Dr. Wilson, Anthony G (Principal Investigator), Division of Genomic Medicine, University of Sheffield, Royal Hallamshire Hospital, Sheffield, UK; Dr. Day, Richard O, Clinical Trial Center. St. Vincents Hospital, DAW PARK SA 504, Australia; Dr. Vecchio, Phillip C, Dept of Rheumatology, Princess Alexandra Hospital, Woolloongabba, Australia; Dr. Fardellone, Patrice, Rhumatologie CHU-Hôpital Nord Service Rhumatologie, AMIENS, France; Dr. Lehmann, Reiner, Klinische Forschung Berlin-Buch, Berlin, Germany; Dr. Dreykluft, Tilman, Klinische Forschung Berlin, Berlin, Germany; Dr. Krause, Andreas, Immanuel-Krankenhaus, Berlin, Germany; Dr. Nischik, Ruth, Zentrum für Therapiestudien der Innomed Leipzig GmbH, Leipzig, Germany; Dr. Bodenschatz, Ralf, Pharmakologisches Studienzentrum Chemnitz, Chemnitz, Germany; Dr. Demary, Winfried, Bahnhofsallee 3 - 4, Hildesheim, Germany; Dr. Alten, Rieke, Schlosspark-Klinik Berlin, Abteilung Rheumatologie, Physikalische Therapie und Sportmedizin, Berlin Germany; Dr. Nasonov, Evgeny L, Rheumatology Research Institute, Moscow, Russian Federation; Dr. Lesnyak, Olga M, Sverdlovsk Regional Clinical Hospital, Ekaterinburg, Russia; Dr. Ershova, Olga B, Yaroslavl Soloviev’s Clinical Hospital, Yaroslavl, Russia; Dr. Moots, Robert J, Division of Infection and Immunity, Clinical Sciences Centre, University Hospital Aintree, Liverpool, UK; Dr. Chattopadhyay, Chandrabhusan, Wrightington Hospital, Wigan, UK; Dr. Luqmani, Raashid A, Rheumatology Department, Nuffield Orthopaedic Centre, Oxford, UK.
REFERENCES
- McInnes, I.B. and Schett, G. (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology, 7, 429-442. doi:10.1038/nri2094
- Van den Broek, T., Tesser, J.R.P. and Albani, S. (2008) The evolution of biomarkers in rheumatoid arthritis: From clinical research to clinical care. Expert Opinion on Biological Therapy, 8, 1773-1785. doi:10.1517/14712598.8.11.1773
- Devlin, J., Gough, A., Huisson, A., Perkins, P., Holder, R., Reece, R., et al. (1997) The acute phase and function in early rheumatoid arthritis, c-reactive protein levels correlate with functional outcome. Journal of Rheumatology, 24, 9-13.
- Pepys, M.B. and Hirschfield, G.M. (2003), C-reactive protein: A critical update. Journal of Clinical Investigation, 111, 1805-1812.
- Elliott, M.J, Maini, R.N., Feldmann, M., Kalden, J.R., Antoni, C., Smolen, J.S., et al. (1994) Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet, 344, 1105-1110. doi:10.1016/S0140-6736(94)90628-9
- Eklund, C.M. (2009) Proinflammatory cytokines in CRP baseline regulation. Advances in Clinical Chemistry, 48, 111-136. doi:10.1016/S0065-2423(09)48005-3
- Guerne, P.A., Zuraw, B.L., Vaughan, J.H., Carson, D.A. and Lotz, M. (1989) Synovium as a source of interleukin 6 in vitro. Contributions to local and systemic manifestations of arthritis. Journal of Clinical Investigation, 83, 585-592. doi:10.1172/JCI113921
- Perry, M.G., Kirwan, J.R., Jessop, D.S. and Hunt, L.P. (2009) Overnight variations in Coritisol, interleukin 6, tumour necrosis factor (alpha) and other cytokines in people with rheumatoid arthritis. Arthritis & Rheumatism, 68, 63-68. doi:10.1136/ard.2007.086561
- Campbell, J., Ciesielski, C.J., Hunt, A.E., Horwood, N.J., Beech, J.T., Hayes, L.A., et al. (2004) A novel mechanism for TNF-alpha regulation by p38 MAPK: Involvement of NF-B with implications for therapy in rheumatoid arthritis. Journal of Immunology, 173, 6928-6937.
- Cohen, S.B., Cheng., T.T., Chindalore, V., Damjanov, N., Burgos-Vargas, R., DeLora, P., et al. (2009) Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis & Rheumatism, 60, 335-344. doi:10.1002/art.24266
- Damjanov, N., Kauffman, R.S. and Spencer-Green, G.T. (2009) Efficacy, pharmacodynamics, and safety of VX- 702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: Results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis & Rheumatism, 60, 1232- 1241. doi:10.1002/art.24485
- Genovese, M.C. (2009) Inhibition of p38: Has the fat lady sung? Arthritis & Rheumatism, 60, 317-320. doi:10.1002/art.24264
- Aston, N.M., Bamborough, P., Buckton, J.B., Edwards, C.D., Holmes, D.S., Jones, K.L., et al. (2009) p38(alpha) mitogen-activated protein kinase inhibitors: Optimization of a series of biphenylamines to give a molecule suitable for clinical progression. Journal of Medicinal Chemistry, 52, 6257-6269. doi:10.1021/jm9004779
- Genovese, M.C., Gao, L., Yin, J., Smith, S., Weinblatt, M.E., Smolen, J.S., et al. (2010) Proof of concept study for a potent p38 MAPK dual action inhibitor BMS- 582949 in subjects with RA receiving comcomitant methotrexate. American College of Rheumatology (ACR).
- Schieven, G., Zhang, R., Pitt, S., Shen, D.-R., Cao, J., Sack, J., et al. (2010) BMS-582949 is a dual action p38 kinase inhibitor well suited to avoid resistance mechanisms that increase p38 activation in cells. American College of Rheumatology (ACR).
- Branger, J., Van den BB, Weijer, S., Madwed, J., Bos, C.L., Gupta, A., et al. (2002) Anti-inflammatory effects of a p38 mitogen-activate inhibitor during human endotoxemia. Journal of Immunology, 168, 4070-4077.
- Vertex Pharmaceuticals (2004) First demonstration of CRP reduction in cardiovascular patients using an oral cytokine inhibitor. PRNewswire-FirstCall, 18 October 2004.
- Ding, C. (2006) Drug evaluation: VX-702, a MAP kinase inhibitor for rheumatoid arthritis and acute coronary syndrome. Current Opinion in Investigational Drugs, 7, 1020-1025.
- Dallegri, F. and Ottonello, L. (2002) Pharmacological implications in the switch from acute to chronic inflammation. Inflammaphamacology, 10, 159-171. doi:10.1163/156856002321168187
- Dominguez, C., Powers, D.A. and Tamayo, N. (2005) p38 MAP kinase inhibitors: Many are made, but few are chosen. Current Opinion in Drug Discovery & Development, 8, 420-430.
- Kirwan, J.R., Hallgren, R., Mielants, H., Wollheim, F., Bjorck, E., Persson, T., et al. (2004) A randomized placebo controlled 12 week trial of budesonide and prednisolone in rheumatoid arthritis. Annals of the Rheumatic Diseases, 63, 688-695. doi:10.1136/ard.2003.008573
- Grisar, J.C., Kapral, T., Gonda, G., Stamm, T., Smolen, J.S. and Aletaha, D. (2006) Short term responsiveness of biomarkers to prednisolone in patients with active rheumatoid arthritis. Annals of the Rheumatic Diseases, 65, 171.
- Gerlag, D.M., Haringman, J.J., Smeets, T.J., Zwinderman, A.H., Kraan, M.C., Laud, P.J., et al. (2004) Effects of oral prednisolone on biomarkers in synovial tissue and clinical improvement in rheumatoid arthritis. Arthritis and Inflammation, 50, 3783-3791. doi:10.1002/art.20664
- Rioja, I., Hughes, F.J., Sharp, C.H., Warnock, L.C., Montgomery, D.S., Akil, M., et al. (2008) Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor (alpha), tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis & Rheumatism, 58, 2257-2267. doi:10.1002/art.23667
- Boers, M., Verhoeven, A.C., Markusse, H.M., Vandelaar, M.A.F.J., Westovens, R., Vendenderen, J.C., et al. (1997) Randomised comparison of combined step-down prednisolone, methotrexate and sulphasalazine with sulphasalazine alone in early rheumatoid arthritis. Lancet, 350, 309-318. doi:10.1016/S0140-6736(97)01300-7
- Singh, D., Smyth, L., Borrill, Z., Sweeney, L. and TalSinger, R. (2010) A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. Journal of Clinical Pharmacology, 50, 94-100. doi:10.1177/0091270009347873
- Seideman, P. (1993) Better effect of methotrexate on c-reactive protein during daily compared to weekly treatment in rheumatoid arthritis. Clinical Rheumatology, 12, 210-213. doi:10.1007/BF02231528
- Tukey, J.W., Ciminera, J.L. and Heyse, J.F. (1985) Testing the statistical certainty of a response to increasing doses of a drug. Biometrics, 41, 295-301. doi:10.2307/2530666
- Food and Drug Administration (1999) Clinical development programs for drugs devices and biological products for the treatment of Rheumatoid Arthritis (RA). Center for Drug Evaluation and Research (CDER), 1 February 1999.
- Pasero, G.P. and Di Munno, O. (2002) Analgesic dose range finding of lornoxicam compared to diclofenac. Crossover double blind study in rheumatoid arthritis. Reumatismo, 54, 238-242.
- Richmond, S.J. (2008) Magnet therapy for the relief of pain and inflammation in rheumatoid arthritis (CAMBRA): A randomized placebo-controlled crossover trial. Trials 9.
- Van Der Giesen, F.J., Van Lankveld, W.J., Kremers-Selten, C., Peeters, A.J., Stern, E.B., Le Cessie, S., et al. (2009) Effectiveness of two finger splints for swan neck deformity in patients with rheumatoid arthritis: A randomized, crossover trial. Arthritis Care & Research, 61, 1025-1031. doi:10.1002/art.24866
- Hafstrem, I., Albertsson, K., Boonen, A., Van Der Heijde, D., Landewe, R. and Svensson, B. (2009) Remission achieved after 2 years treatment with low-dose prednisolone in addition to disease-modifying ant-rheumatic drugs in early rheumatoid arthritis is associated with reduced joint destruction still present after 4 years: An open 2- year continuous study. Annals of the Rheumatic Diseases, 68, 508-513. doi:10.1136/ard.2008.087833
- Irusen, E., Matthews, J.G., Takahashi, A., Barnes, P.J., Chung, K.F. and Adcock, I.M. (2002) p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. Journal of Allergy & Clinical Immunology, 109, 649-657. doi:10.1067/mai.2002.122465
- Lasa, M., Abraham, S.M., Boucheron, C., Saklatvala, J. and Clark, A.R. (2002) Dexamethasone causes sustained expression of mitogen-activate protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Molecular & Cellular Biology, 22, 7802- 7811. doi:10.1128/MCB.22.22.7802-7811.2002
- Toh, M.L., Yang, Y., Leech, M., Santos, L. and Morand, E.F. (2004) Expression of mitogen-activated protein kinase phosphatase 1, a negative regulator of the mitogen-activated protein kinases, in rehmatoid arthritis: Upregulation by interleukin-1beta and glucocorticoids. Arthritis & Rheumatism, 50, 3118-3128. doi:10.1002/art.20580
- Dickson, M.C., Ellis, J., Genqian, C., Wilson, A.G., Bridges, L.S. Jr., Furst, D.E., et al. (2008) Corticosteroid-responsive pharmacodynamic transcriptional markers: Results from a randomized placebo-controlled study of rheumatoid arthritis patients treated with prednisolone. American College of Rheumatology 72nd Meeting, 24 October 2008.