Open Journal of Animal Sciences
Vol.07 No.03(2017), Article ID:77443,13 pages
10.4236/ojas.2017.73019
Enzyme-Linked Immunosorbent Assays Using the Recombinant gp51 and p24 of Bovine Leukemia Virus for Immunodetection of the Disease
Alejandra Larsen1, Santiago Corva1, Javier Panei1,2, Christoph Geisler3, Eduardo Mortola1
1Faculty of Veterinary Science, National University of La Plata, La Plata, Argentina
2CONICET (CCT-La Plata), La Plata, Argentina
3Department of Immunology, Faculty of Veterinary Sciences, National University of La Plata, La Plata, Argentina d Department of Molecular Biology, University of Wyoming, Laramie, WY, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: March 7, 2017; Accepted: July 3, 2017; Published: July 6, 2017
ABSTRACT
Enzyme-linked immunosorbent assay (ELISA) is often used to test bovine leukemia virus (BLV) infection. However, commercially available kits test in South America detect only antibodies against the gp51 protein. With the aim to improve the sensitivity of the test, we developed here a two-step indirect dual ELISA test that included both proteins p24 and gp51, expressed and produced in E. coli and baculovirus expression system respectively. Two hundred ten BLV sera, stated as double positive or double negative by the combination of commercial agar gel immunodiffusion (AGID) assay and a gp51- ELISA test, were tested with our in house dual rp24/rgp51 ELISA. Firstly, we checked the purified, optimized and standardized proteins as antigen by the checkerboard technique, and set up our in house ELISA test. The concordance correlation coefficient (CCC) and coefficient of variation (CV) intraplate repeatability levels were within the values established by the international standards. The statistical analysis demonstrated the value of sera correctly ranked highest (93.48%), and for 0.3 cutoff, the sensitivity was 95.65% and the specificity 91.30%. In conclusion, the rp24/rgp51 ELISA developed and standardized here demonstrated to have good analytical characteristics to be considered for screening of BLV.
Keywords:
Bovine Leukemia Virus, Indirect Dual ELISA, Recombinant Antigens

1. Introduction
The bovine leukemia virus (BLV) is the causative agent of Enzootic Bovine Leukosis (EBL), a lymphoproliferative type disease that affects B cells, and the provirus is integrated into the genome, transforming infected animals in Lifetime carriers [1] . Given the multiple transmission routes, both natural and iatrogenic and the minimum volumes of peripheral blood required to transmit the virus, this disease has a high potential for disease transmission [2] [3] .
The economic impact of this disease lies not only in direct losses for confiscation at the slaughterhouse, but also the associated carrier status of the animal, with an important decrease in animal productivity [4] ; that also include the costs of veterinary services, diagnosis and treatment of the disease, as well as the costs of early animal replacement [5] .
The Bovine leukemia virus (BLV) is an exogenous B-lymphotropic retrovirus that naturally provokes B-lymphoproliferative disorders in cattle and can be used to induce a related B-cell-associated pathology in experimentally infected sheep [6] . Structural proteins p24 and gp51 are potent immunogens and the antibody production can be detected in most of the animals infected with VLB. In experimentally infected animals anti-gp51 antibodies seem to be the first to appear with titres consistently higher than anti-p24 antibodies [7] [8] [9] . Other authors reports that high sensitivity tests are able to detect anti-p24 antibodies before the appearance of anti-gp51 antibodies; and the anti-p24 antibodies could be an equal or greater degree than anti-gp51 [3] [10] . The immunoglobulin isotype in the immune response varies according to the stages of LEB disease, in asymptomatic animals IgM and IgG1 anti p24 and gp51were detected, whereas the detection of IgA occurs in animals with lymphoma [11] .
The most frequently used serological tests to detect VLB infection are agar gel immunodiffusion (AGID) and enzyme-linked immunosorbent assay (ELISA). The AGID, that mainly detects anti-gp51 antibodies [9] , is a test with high specificity, low cost, quick and easy to perform, but with some main disadvantages like low sensitivity, time consuming for reading and subjective interpretation of test results and is unable to be performed with milk samples [12] . At the present, the detection of specific antibodies based on the recognition of crude antigens by AGID or ELISA requires mass production of the virus from persistently infected FLK cell lines cultures. The use of Escherichia coli or baculovirus expression systems to produce recombinant antigen(s) would be greatly beneficial in improving standardization of the tests and reducing their production cost.
Enzyme-linked immunosorbent assay (ELISA) tests using VLB antigens have already been reported [12] [13] , but compared to the current serological tests, none of these recombinant antigens has allowed detection of all serologically positive individuals. The aim of this study was to develop an indirect dual ELISA test that included both recombinant proteins p24 and gp51 directly absorb to the 96-well plate, to be used in Argentina and other countries of South America. The use and evaluation of the two recombinant antigens could be a strategy to improve the sensitivity of recombinant antigen-based tests for the serodiagnosis of LEB.
2. Materials and Methods
2.1. Sera Samples
A total of 283 serum samples, collected from naturally infected adult cows from different herds, were obtained from the serum bank of the Virology Laboratory of the Veterinary Faculty of University of La Plata, Argentina. The sera samples were tested for the presence of BLV specific antibodies by AGID and a commercial indirect ELISA test. International reference serum sample E4 (Veterinary Laboratories Agency. New Haw, Adelestope Surrey. UK) was used as control.
2.2. Recombinant Antigens Production
2.2.1. Expression and Purification of Recombinant p24 Antigen in Bacterial Cell
The complete coding region for the p24 gene of the BLV was amplified by PCR using the FLK (fetal lamb kidney) BLV infected cell line as a template. The primers used carried a Bam HI restriction sites for the forward primer (5’-GGC GGA TCC ATG CCA ATC ATA TCT GAA-3’) and a Hind III site for reverse primer (5’-GGC AAG CTT TAG AGA AGT GCA GGC TGT-3’). The primers were designed based on the genomic sequence deposited in GenBank under accession number K02120.1. PCR was carried out by denaturing at 94˚C for 2 minutes, 30 cycles of: annealing at 50˚C for 45 seconds, extension at 72˚C for 2 minutes, followed by a final extension period at 72˚C for 10 minutes. The PCR product was digested with both BamHI and HindIII, cloned into the pQE-30 expression vector (QIAGEN)) and the transformation of competent M15 [pREP4] E. coli cells (QIAGEN) were performed according to the manufacturer’s instructions. To determine the presence of the recombinant plasmid, a PCR was performed and the product was used for confirm the insertion of the gene in frame. The absence of mutations in the p24 gene was confirmed by plasmids sequencing using M13 forward and reverse primers. The sequencing results were analyzed by comparing with databases and using the basic local alignment search tool (BLAST) software.
The selected E. coli p24/clones were inoculated into LB medium contained 0.1 mM of IPTG (isopropyl-bD-thiogalactopyranoside) for induction; the medium was supplemented with ampicillin (100 mg/ml) and kanamycin (25 µg/ml). The purification of the recombinant protein was performed in batch, the buffer used (50 mM NaH2PO4 and 500 mM NaCl), was the same all along the protocol. The cells were pelleted and resuspended in lysis buffer pH7, lysozyme (1 mg/ml) was added and incubate at 37˚C for 30 min. The lysis was completed by few rounds of sonication. The presence of the protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a 12% polyacrylamide gel and the antigenicity of the protein by Western blot analysis according to the standard proceeding, using E4 control sera (Veterinary Laboratories Agency. New Haw, Adelestope Surrey. UK), a commercial BLV p24 monoclonal antibody (VMRD Inc., WA USA) and a pool of negative control serums.
The recombinant protein were purified from the supernatant lysate using Ni-NTA (QIAGEN, USA) and eluted off the resin by buffer pH4.The concentration of the recombinant protein was determined by a quantitative Coomassie blue-stained gel using bovine serum albumin as the standard.
2.2.2. Expression and Purification of Recombinant gp51 Antigen in Insect Cell
The complete coding region for the gp51 gene of the BLV was amplified by PCR using the FLK (fetal lamb kidney) BLV infected cell line as a template and the following set of primers: forward 5’-CAC CAT GCC TAA AGA ACG A-3’, and reverse: 5’-CCG GGT AGG AGG GGC GGA-3’. The primers were designed based on the genomic sequence deposited in GenBank under accession number: K02120.1. PCR was carried out by denaturing at 94˚C for 2 minutes, 30 cycles of: annealing at 55˚C for 45 seconds, extension at 72˚C for 2 minutes, followed by a final extension period at 72˚C for 10 minutes. A CACC sequence (underlined) was introduced in the forward primer for directional cloning into the entry vector (pENTR/D-TOPO―BaculoDirect TM GST Gateway, Invitrogen. USA), following the manufacturer’s instructions.
The rgp51/pENTR/D-TOPO cloning vector was amplified in TOP10 E. coli cells and the absence of mutations in the gp51 gene was confirmed by plasmids sequencing using M13 forward and reverse primers. The sequencing results were analyzed by comparing with databases and using the basic local alignment search tool (BLAST) software. Construction of the Recombinant baculovirus was performed following the manufacturer’s instructions of the BaculoDirect™ GST Gateway®. System (Invitrogen, UK). Briefly, to obtain the recombinant baculovirus, the LR reaction was performed incubating at room temperature the purified entry vector rgp51/pENTR/D-TOPO with baculovirus linear DNA. The recombinant baculovirus directly transfected Sf9 cells in presence of Cellfectin II Reagent® (Invitrogen, UK) and Ganciclovir for the recombinant baculovirus selection. Recombinant viruses were purified by plaque assay and screened for the expression of recombinant gp51 by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and the specificity was analyzed by Western blot analysis according to the standard proceeding, using E4 control sera (Veterinary Laboratories Agency. New Haw, Adelestope Surrey. UK), a commercial BLV gp51 monoclonal antibody (VMRD Inc., WA USA) and a pool of negative control serums. For the production of recombinant gp51, approximately 2 × 106 Sf9 cells were infected with the recombinant virus at a multiplicity of infection of 5, and then cultured at 27˚C for 3 days. After culture centrifugation, the cells were harvest and resuspended in lysis buffer pH7 with 0.3% SDS (Sodium Dodecyl Sulfate), and 1% 2B mercaptoetanol. The complete celllysate was obtained by syringe passage consisting of 10 passes with 25-gauge needle followed by 10 passages through 27-gauge needle. The purification of the recombinant protein from the supernatant lysate was performed as explained above using Ni-NTA (QIAGEN, USA).
2.3. AGID and Commercial gp51-ELISA Tests
The AGID test, produced by Universidad de LaPlata, Argentina (UNLP-AGID) and a commercial indirect ELISA test kit (VMRD Inc., USA) that detect antibodies against BLV gp51 protein, were used according to the manufacturer’s recommendations. Both tests were used as standard of comparison against our in house ELISA to detect specific BLV antibodies.
2.4. In House rp24/rgp51 ELISA Procedure
Two checkerboard tests (CBT) was performed, one for each individual recombinant protein (rp24 and rgp51) and the third with a mix of the two proteins. Foreach individual recombinant protein CBT, an appropriate ranges of serum and conjugate dilutions were identified by preliminary testing to determine the optimal dilution of the conjugate (1/2500) and the serum (1/25, 1/50 and 1/100), using the following range of proteins concentration (10 to 0.25 ug/ml). The best binding ratio was chosen to select the best concentration of the reagents for our test. The third CBT was performed with the same serum dilutions as before and a mix of the two proteins at different concentrations. Mix 1 (250 ng per well of rp24 and 50 ng per well of rgp51), and mix 2 (250 ng per well of rp24 and 25 ng per well of rgp51).
Once the optimal working concentrations for all reagents were determined by CBT, we proceeded to perform our rp24/rgp51 ELISA. NuncPolysorp F plates were coated overnight at 4˚C with 250 ng/well of rp24 and 25 ng/well of rgp51 contained in 100 ul of 0.05 mol/l carbonate buffer (pH 9.6). The antigen-coated plate was washed three times with PBST (0.01 mol/l phosphate-buffered saline, pH 7.4, 0.05% Tween 20) and blocked with PBST containing 1 % (W/V) skim milk at 37˚C for 1 h. After washing, 100 ul of the diluted sera panel was added, and then incubated at 37˚C for 1 h. The assay was performed using 3 different sera dilutions: 1/100, 1/50 and 1/25. After washing, 100 ul of goat anti-bovine HRP-IgG antibodies (1:2500) was added and incubated at 37˚C for 1 h. The plates were washed three times and then incubated with 100 ul of 30 mg of 2,2 0-azino-bis-(3-benzthiazoline-6-sulfonic acid) (ABTS) (KPL, Inc.) substrate in 100mM phosphate-citrate buffer pH 4.0 and 3 ul of 30% H2O, for 40 min at room temperature. The optical density (OD) of each well was read at 405 nm using a microplate reader (Bio-Rad). Duplicate positive and negative control sera were included in each plate.
The checkerboard results are assessed by determine the Binding Ratio (BR) [14] .
2.5. Statistical Analysis
The repeatability within and between plates was analyzed by the reproducibility index called the concordance correlation coefficient (CCC) and coefficient of variation (CV) [14] [15] [16] . The following formula was used to normalize de ELISA results: ODN = OD-Nt/Pst-Nt, where OD is the absorbance value of the sample, Pst and Nt are the mean absorbance values of the positive and negative controls on each plate, respectively [15] .
Calculations to determine test sensitivity and specificity were carried out by Stata SE 11 program (Stata Corporation, College Station, Texas, USA) and TG- ROC program (Two Graphic Receiver Operating Characteristic). For cutoff calculation the correct classification parameter was used [17] .
3. Results
3.1. Expression, Purification and Characterization of rp24 in E. coli
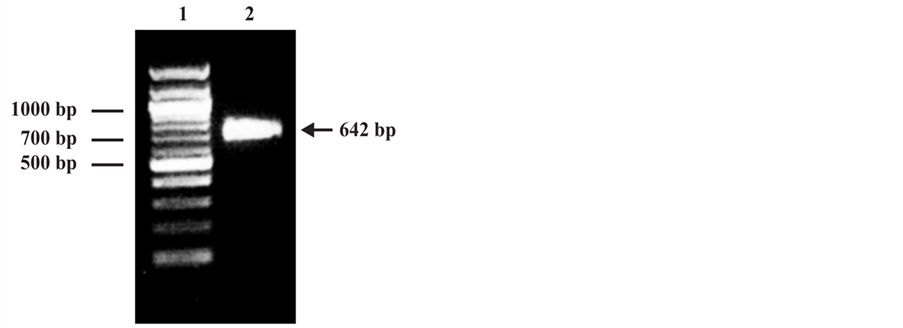
The PCR amplification assay generated a unique fragment with the expected molecular level (642 bp) and visible on agarose gel under ultraviolet light (Figure 1(a)). After cloning of the PCR product to the pQE-30 expression vectorthe present of the gene was confirmed by PCR and the purified selected plasmid, contained the p24 gene in frame, was sequenced. The sequence analyzed by comparing with databases from BLV reference strains showed a 99% of nucleotide identity with BLV cell-line FLK-BLV subclone pBLV913 (GenBankID: EF600696.1) and 98% of nucleotide identity con BLV (GenBankID: K02120.1) [18] .
The analysis by SDS PAGE of the Denatured lysate of induced E. coli M15 (pREP4) showed by Coomassie blue staining the presence of the p24 recombinant protein with the predicted size (24 kDa) corresponding to the molecular weight to the fusion protein contained N-terminally His-tag (Figure 1(b)). The authenticity of the recombinant protein was confirmed by Western blot analysis using the E4 control positive serum (Veterinary Laboratories Agency. New Haw, Adelestope Surrey. UK), a commercial monoclonal anti-p24 antibody (VMRD Inc., WA USA) and a pool of negative control serum from BLV non-infected cow (Figure 2).
The expressed rp24 protein was subsequently purified using Ni-NTA affinity
 (a)
(a)
 (b)
(b)
Figure 1. (a) Agarose gel electrophoresis of PCR product. 1: DNA ladder; 2: Amplified 642 bp PCR product. (b) SDS-PAGE analysis of recombinant p24 protein expressed in E. coli. 1: Lysate from cells expressing the rp24 protein; 2: Prestained molecular weight marker; 3: Lysate from non-expressing cells.

Figure 2. Western Blot analysis of recombinant p24 protein expressed in E. coli. 1: Prestained molecular weight marker; 2: Monoclonal anti- p24 antibody; 3: Pool of negative control serum from BLV non-infected cow; 4: Polyclonal E4 control positive sera.
chromatography by pH elution methods. For yield optimization, several induction parameters were tested, and the highest yield, 5.6 mg per liter culture, was achieved following induction with 0.6 mM IPTG (Isopropyl β-D-1-thiogalacto- pyranoside) for 18 h at 25˚C.
3.2. Expression, Purification and Characterization of rgp51 in Baculovirus
The PCR amplification assay generated a unique fragment with the expected molecular level (894 bp) and visible on agarose gel under ultraviolet light (Figure 3(a)). A consensus Gateway entry clone was used to produce a baculovirus (Acgp51) encoding a C-terminally 6X His-tagged form of the full-length gp51 protein under the transcriptional control of the polyhedrin promoter. After confirming fragment insertion and orientation by PCR, the purified selected plasmid was automated sequenced and the sequence analyzed by comparing with databases from BLV reference strains: 99% of nucleotide identity with BLV cell-line FLK-BLV subclone pBLV913 (GenBankID: EF600696.1) and 99% of nucleotide identity con BLV (GenBankID: K02120.1) [18] .
Acgp51-infected Sf9 cell extracts analyzed by SDS-PAGE showed the presence of a very strong protein band of approximately 34 kDa, which was absent in mock-infected cell extracts (Figure 3(b)). Recombinant gp51 protein was purified by Ni2+ affinity purification procedure by pH elution methods. The concentration of the protein was determined with a commercial kit (Micro BC Assay protein Quantitation Kit, Uptima-Interchim). A total of 0.2 mg of purified recombinant protein was obtained from 5 × 107 Sf9 cells.
The identity of this protein as gp51 was confirmed by Western blot analysis using the E4 control positive serum (Veterinary Laboratories Agency. New Haw, Adelestope Surrey. UK), a commercial monoclonal anti-gp51 antibody (VMRD Inc., WA USA) and a pool of negative control serum from BLV non-infected cow.
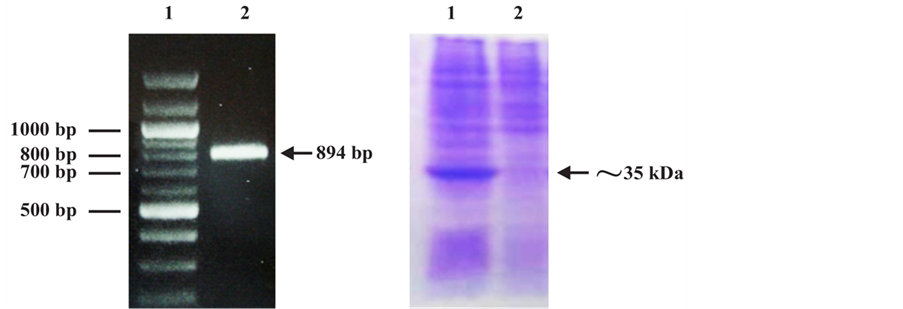
Figure 3. (a) Agarose gel electrophoresis of PCR product.1: DNA ladder; 2: Amplified 894 bp PCR product. (b) SDS-PAGE analysis of recombinant gp51 protein expressed in Sf9insect cells. 1: Lysate from Sf9 insect cells expressing the rgp51 protein; 2: Lysate from non-expressing Sf9 insect cells.
The Figure 4 shows that the antibodies react with a mayor band with molecular mass of approximately 34 kDa recombinant protein and several protein fractions from 34 up to 50 kDa. The molecular mass of recombinant gp51 protein was smaller than that of the native gp51.
3.3. Serum Control Panel Determination by AGID and ELISA Commercial Kit
Of a total of 283 serum samples analyzed by AGID and commercial ELISA kits, were stated 101 as double positive and 109 as double negative. These serum were selected for further analysis, the 73 serum samples that reacted with discordant results between both assays were not included for the in house ELISA test.
3.4. Characteristics Estimation of the in House ELISA Test
The optimal concentration of recombinant proteins used as antigen for the in house ELISA determinate by CBT, were 250 ng per well for rp24 and 25 ng per well for rgp51. Three sera dilution were tested (1/25, 1/50 and 1/100) to determine the optimal performance of the assay, and the 1/25 serum dilution were used to perform the assay according to the best banding ratio value. The software package Stata version 11 calculated the correct classified serum percentage (CC%) and the likelihood ratio for positive sera (LR+), that allow to disclosed the diagnostic sensibility (DSe) and specificity (DSp) of our in house ELISA test showed in the Table 1.
4. Discussion
In the epidemiological disease control of EBL it is necessary to have tests that increasingly sensitive, specific and be economically feasible to performed in large populations of animals. During the immune response against BLV the synthesis and persistence of specific anti-p24 and anti-gp51 antibodies is lifelong [9] [19] [20] , this features make these two highly immunogenic proteins as candidate to be included in any diagnostic test for EBL.

Figure 4. Western Blot analysis of recombinant gp51 protein expressed in Sf9 insect cells. 1: Polyclonal E4 control positive sera; 2: Pool of negative control serum from BLV non-infected cow; 3: Prestained molecular weight marker; 4: Monoclonal anti-gp51 antibody.

Table 1. Dual rp24/rgp51 ELISA assays statistical analysis by TG-ROC. Coff: cutoff; DSe: diagnostic sensitivity; DSp: diagnostic specificity; CC: Correct Classification. LR+: likelihood ratio for positive sera.
Firstly, in this study we successfully produced a recombinant p24 protein with a proved antigenicity against the control sera, the bacterial expression system achieved a mass production of approximately 5.6 mg per liter cultures. However, the production of the recombinant gp51 protein was obtaining from the baculovirus expression system. Although the presence and antigenicity of the recombinant gp51 protein in Sf9 insect cells was evident by Western blot analysis, the recombinant protein recognized by the monoclonal anti-gp51 antibody, showed the presence of several protein fractions with molecular weights ranging from 34 kDa to about 50 kDa. All of these proteins or protein fractions that migrated faster than the native gp51 molecules, could represent partially glycosylated recombinant protein. However, the protein were anyway recognized by the positive control serum and the antigenicity maintaining its character despite incomplete glycosylation. Similar finding were reported in previous works by Giusseppe et al. [21] using the baculovirus system. From this data, we can assume that the heavier fraction corresponded to the band located at the height control molecular weight of 35 kDa, suggesting that part of the recombinant protein could be identified as partially glycosylated orunglycosylated product. The antigenic identity of the recombinant gp51 protein was confirmed also by the use of polyclonal control positive sera in western blot analysis. Following production and purification of recombinant gp51 protein in the baculovirus system, a concentration of 0.2 mg was obtained from 5 × 107 Sf9 cells.
To develop our in house ELISA test, firstly we checked the purified, optimized and standardized proteins asantigen by the checkerboard technique, in order to determine the concentration of the antigen and the sera dilution to be use in the test. Two independent ELISA tests for rp24 and rgp51 and also the dual ELISA rp24/rgp51 integrated proteins were tested. We tried different brands of 96-well plates and found that the 96-well plates from Nunc-Immuno Plates Polysorb (InterMed, Co. Denmark) were the more appropriate support to perform our in house ELISA test, because were where the recombinant antigens were best adsorbed to the plate. Also, the antigenicity of the joined together recombinant proteins were kept unalterable during the development of the indirect ELISA test.
From the analysis of the checkerboard results, we can concluded that a high BR at a concentration within the parameters suggested by R. Crowther [14] , were obtained using a concentration of 250 ng per well of rp24 and 25 ng per well of rgp51. These antigen concentrations were maintained throughout all the tests. To perform the calculations and analysis of the results, the absorbance values were standardized as percentage of seropositive values.
According to the results presented here (CCC and CV analysis) intraplate repeatability levels were within the values stablished by international standards [22] . Through the experiment, several dual rp24/rgp51 ELISA tests were performed using 3 different sera dilutions (1/100, 1/50 and 1/25), in behalf of that it was possible to improve the performance of the test, achieving significant advances in DSe and DSp attached to the higher percentage of correct classification (predicted probabilities), together with a likelihood ratio of 11.
In order to validate the results of our in house ELISA, we determine the number of samples to be processed according to the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals Table No. IV.2 [22] , where the theoretical number of samples from animals of known infection status required for establishing DSe and DSp estimates depending on likely value of DSe or DSp and desired error margin and confidence.
Given the sensitivity value of 95.65% obtained for the test at 1/25 dilution, we determined the number of samples to be processed with 5% of estimated error and a confidence interval of 90%. In our study, a total of 210 double positive or double negative sera were analyzed. The statistical analysis shows the value of sera correctly ranked highest (93.48%) and for 0.3 cutoff, DSe was 95.65% and DSp 91.30%.
The World Organization for Animal Health (OIE) determines that the tests for the diagnosis of LEB are the Indirect ELISA and AGID. In our country the health authority accepts these two official tests, but so far there is no domestic production of commercial ELISA. The aim of developing a diagnostic test that includes together the two recombinant proteins (gp51 and p24), is to improve the DSe and DSp when the two proteins go together directly absorb to the 96- well-plate.
Considering the prevalence of the disease and its consequences in the reduction of the productivity of livestock and indirect economic losses from the restriction in international market [4] [23] , it is required to developed diagnosis tools that increase sensitive, specific and also be economically feasible to use in large livestock farms.
5. Conclusion
In conclusion, we developed here a dual rp24/rgp51 ELISA test with two very promising recombinant antigens. This assay is a two-step ELISA which involves two binding process of primary antibody and labeled secondary antibody, without using monoclonal antibodies precoating the plate. Despite a larger number of samples should be analyzed to validate the preliminary data obtained in this study; our ELISA assay has proven to be sensitive and specific, to have a good reproducibility, to be easy to perform, and to be suitable for screening animals for BLV infection. This test could be taken into account as a valid alternative to the AGID, not only in our country but also in others countries of South America.
Acknowledgements
We thank Prof. Dr. Donald L. Jarvis at the University of Wyoming for kindly providing us with laboratory facilities and research reagents.
Cite this paper
Larsen, A. Corva, S., Panei, J., Geisler, C. and Mortola, E. (2017) Enzyme-Linked Immunosorbent Assays Using the Recombinant gp51 and p24 of Bovine Leukemia Virus for Immunodetection of the Disease. Open Journal of Animal Sciences, 7, 241-253. https://doi.org/10.4236/ojas.2017.73019
References
- 1. Lucas, M.H. (1992) Viral Diseases. Enzootic Bovine Leukosis. In: Andrews, A.H., Blowey, R.W., Boyd, H. and Eddy, R.G., Eds., Bovine Medicine: Diseases and Husbandry, 2nd Edition. Wiley-Blackwell, Hoboken, 530-537.
- 2. Robert, D.H., Lucas, M.H. and Wibberley, G. (1986) To Compare the Incubation Period Following Intratracheal and Subcutaneous Inoculation of Bovine Leucosis Virus Infected Lymphocytes and to Study Their Clearance from the Circulation. Veterinary Immunology and Immunopathology, 11, 351-358.
https://doi.org/10.1016/0165-2427(86)90037-1 - 3. Gonzalez, E.T., Oliva, G.A., Valera, A.R., Bonzo, E., Licursi, M. and Etcheverrigaray, M.E. (2001) Leucosis Enzoótica Bovina: Evaluación De Técnicas Diagnósticas (ID, ELISA-1, WB, PCR) En Bovinos Inoculados Experimentalmente. Analecta Veterinaria, 21, 12-20.
- 4. Brenner, J., Van-Haam, M., Savir, D. and Trainin, Z. (1989) The Implication of BLV Infection in the Productivity, Reproductive Capacity and Survival Rate of a Dairy Cow. Veterinary Immunology and Immunopathology, 22, 299-305.
https://doi.org/10.1016/0165-2427(89)90017-2 - 5. Monti, G.E., Frankena, K. and De Jong, M.C.M. (2007) Evaluation of Natural Transmission of Bovine Leukaemia Virus within Dairy Herds of Argentina. Epidemiology and Infection, 135, 228-237.
https://doi.org/10.1017/S0950268806006637 - 6. Weiss, R.A. (2006) The Discovery of Endogenous Retroviruses. Retrovirology, 3, 67.
https://doi.org/10.1186/1742-4690-3-67 - 7. Troiano, L.D.C., Thomaz-Soccol, V., Agottani, J.V.B., Brodzinski, J., Penha, T.R., and Ozaki, S.C. (2013) Production, Characterization, and Use of Monoclonal Antibodies Against gp51 Protein to Diagnose Bovine Leukemia Virus Infection. BioResearch Open Access, 2, 55-60.
https://doi.org/10.1089/biores.2012.0295 - 8. González, E.T., Licursi, M., Oliva, G.A. and Etcheverrigaray, M.E. (1999) Diagnóstico De La Leucosis Enzoótica Bovina (LEB) En Animales Naturalmente Infectados. XIV Congresso Estadual De Medicina Veterinaria, III Congreso De Medicina Veterinaria Do Cone Sul. Gramado, Brasil. Proceeding, 192.
- 9. Monti, G.E., Frankena, K., Engel, B., Buist, W., Tarabla, H.D. and de Jong, M.C. (2005) Evaluation of a New Antibody-Based Enzyme-Linked Immunosorbent Assay for the Detection of Bovine Leukemia Virus Infection in Dairy Cattle. Journal of Veterinary Diagnostic Investigation, 17, 451-457.
https://doi.org/10.1177/104063870501700507 - 10. Kittelberger, R., Reichel, M.P., Meynell, R.M, Tham, K.M. and Molloy, J.B. (1999) Detection of Antibodies against the Core Protein p24 of the Bovine Leukaemia Virus in Cattle for Confirmatory Serological Testing. Journal of Virological Methods, 77, 109-114.
https://doi.org/10.1016/S0166-0934(98)00143-8 - 11. Heeney, J.L., Valli, V.E. and Montesanti J. (1988) Alterations in Humoral Immune Response to Bovine Leukaemia Virus Antigens in Cattle with Lymphoma. Journal of General Virology, 69, 659-666.
https://doi.org/10.1099/0022-1317-69-3-659 - 12. Simard, C., Richardson, S., Dixon, P., Bélanger, C. and Maxwell, P. (2000) Enzyme-Linked Immunosorbent Assay for the Diagnosis of Bovine Leukosis: Comparison with the Agar Gel Immunodiffusion Test Approved by the Canadian Food Inspection Agency. Canadian Journal of Veterinary Research, 64, 101-106.
- 13. Mammerickx, M., Potetelle, D., Bruck, C. and Burny, A. (1984) Diagnosis of Enzootic Bovine Leukosis with an Immunossay Test (Elisa) Utilizing a Monoclonal Antibody Ann. MedVet Medical, 123, 55-63.
- 14. Crowther, J.R. (2009) Tritation of Reagents. In: Crowther, J.R., Ed., The ELISA Guidebook (Methods in Molecular Biology), 2nd Edition. Humana Press, New York City, 79-109.
- 15. Sanchez, J., Dohoo, I.R., Markham, F., Leslie, K. and Conboy, G. (2002) Evaluation of the Repeatability of a Crude Adult Indirect Ostertagia Ostertagi ELISA and Methods of Expressing Test Results. Veterinary Parasitology, 109, 75-90.
https://doi.org/10.1016/S0304-4017(02)00194-2 - 16. OIE. (2008) Enzootic Bovine Leukosis. In: World Organisation for Animal Health, Ed., Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, World Organisation for Animal Health, Paris, Volume 2, Chapter 2.4.11, 729-738.
- 17. Xu, H., Lohr, J. and Greiner, M. (1997) The Selection of ELISA Cut-Off Points for Testing Antibody to Newcastle Disease by Two-Graph Receiver Operating Characteristic TG-ROC Analysis. Journal of Immunological Methods, 208, 61-64.
https://doi.org/10.1016/S0022-1759(97)00128-2 - 18. Sagata, N., Yasunaga, T., Tsuzuku-Kawamura, J., Ohishi, K., Ogawa, Y. and Ikawa, Y. (1985) Complete Nucleotide Sequence of the Genome of Bovine Leukemia Virus: Its Evolutionary Relationship to Other Retroviruses. Proceedings of the National Academy of Sciences of the United States of America, 82, 677-681.
https://doi.org/10.1073/pnas.82.3.677 - 19. González, E.T., Oliva, G.A., Ando, Y. and Etcheverrigaray, M.E. (1988) Bovine Leukemia Virus. Antigen Production and Serologic Detection. 2nd International Conference on the Impact of Viral Diseases on the Development of Latin American Countries and the Caribbean Region, Mar del Plata, Argentina.
- 20. Van der Maaten, M. and Miller, J.M. (1990) Bovine Leukosis Virus. In: Dinter, Z. and Morein, B., Eds., Virus Infections of Ruminants, Elsevier Science Publishers, Amsterdam, 419-429.
https://doi.org/10.1016/b978-0-444-87312-5.50058-8 - 21. De Giuseppe, A., Feliziani, F., Rutili, D. and De Mia, G.M. (2004) Expression of the Bovine Leukemia Virus Envelope Glycoprotein (gp51) by Recombinant Baculovirus and Its Use in an Enzyme-Linked Immunosorbent Assay. Clinical and Diagnostic Laboratory Immunology, 11, 147-151.
https://doi.org/10.1128/cdli.11.1.147-151.2004 - 22. OIE. (2013) Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. In: OIE, Ed., Principles and Methods of Validation of Diagnostic Assays for Infectious Diseases, Chapter 1.1.6. (NB: Version adopted in May 2013).
- 23. Pelzer, K.D. (1997) Economics of Bovine Leukemia Virus Infection. The Veterinary Clinics of North America. Food Animal Practice, 13, 129-141.
https://doi.org/10.1016/S0749-0720(15)30368-6