Open Journal of Genetics
Vol.07 No.01(2017), Article ID:73977,8 pages
10.4236/ojgen.2017.71001
Multiple z-Score Based Method for Noninvasive Prenatal Test Using Cell-Free DNA in Maternal Plasma
Hyuk Jung Kwon, Amit Goyal, Heesu Im, Kichan Lee, Seon Young Yun, Yoon Hee Kim, Sungjong Lee, Mi-Gyeong Lee, Hyuna Lee, Reena Garg, Boram Park, Soyoung Choi, Joungsu Joo, Jin-Sik Bae, Min-Jeong Kim, Min Seob Lee, Sunghoon Lee*
EONE-DIAGNOMICS Genome Center Co. Ltd., Inchon, Korea

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 1, 2017; Accepted: February 4, 2017; Published: February 7, 2017
ABSTRACT
Objective: To improve the detecting accuracy of chromosomal aneuploidy of fetus by non-invasive prenatal testing (NIPT) using next generation sequencing data of pregnant women’s cell-free DNA. Methods: We proposed the multi-Z method which uses 21 z-scores for each autosomal chromosome to detect aneuploidy of the chromosome, while the conventional NIPT method uses only one z-score. To do this, mapped read numbers of a certain chromosome were normalized by those of the other 21 chromosomes. Average and standard deviation (SD), which are used for calculating z-score of each sample, were obtained with normalized values between all autosomal chromosomes of control samples. In this way, multiple z-scores can be calculated for 21 autosomal chromosomes except oneself. Results: Multi-Z method showed 100% sensitivity and specificity for 187 samples sequenced to 3 M reads while the conventional NIPT method showed 95.1% specificity. Similarly, for 216 samples sequenced to 1 M reads, Multi-Z method showed 100% sensitivity and 95.6% specificity and the conventional NIPT method showed a result of 75.1% specificity. Conclusion: Multi-Z method showed higher accuracy and robust results than the conventional method even at low coverage reads.
Keywords:
Cell-Free DNA, z-Score, Multiple Thresholds, Coefficient of Variance, Noninvasive Prenatal Testing, NIPT

1. Introduction
The most common chromosomal aneuploidy for a new born infant is Trisomy 21. The overall occurrence of trisomy 21 is around 0.001%, but the risk increases up to 0.02% for women above 45 years old [1] [2] [3] . Traditional methods of prenatal screening for fetal aneuploidy have a high miscarriage risk since it involves invasive sampling. Recent technology advancement in Next Generation Sequencing (NGS) and Bioinformatics led to a novel Non-Invasive Prenatal Test (NIPT) method to analyze fetus aneuploidy using cell-free DNA (cfDNA) in the plasma of pregnant women. This NIPT has been shown to be both highly sensitive and highly specific across numerous studies [4] . The whole chromosome analysis uses massive parallel sequencing data and applies statistical normalization of each chromosome read count. Sequence reads are mapped to the human reference genome and quantified according to their genomic locus. After normalizing the read count, one z-score per chromosome was calculated to determine fetal aneuploidy [5] . Most of published NIPT studies rely on the z-score which represents the quantitative variations of the chromosome of interest and they show the results as positive or negative by checking if the z-score exceeds the predefined threshold [6] .
Even though these methods become highly accurate, they still have a 0.1% possibility of reporting false positives and need enough read count to score the high sensitivity [7] [8] [9] .
While many NIPT methods have been developed and introduced, studies are under way to increase accuracy. Sunshin Kim et al. [10] , for example, introduced a new algorithm based on selecting reference samples adaptively using CV according to the shared ranges of GC content and DNA reads fraction. They showed reliable results within GC (0.424 ± 0.001) for 7.4 ± 2.1 million raw reads, but the insufficient, yet large sample size for selecting reference samples is a concerning issue.
In order to save sequencing cost and time, some research is being conducted with less reads, for example, Lau, T.K. et al. [11] , reported the clinical performance of NIPT based on low-coverage whole-genome sequencing as 0.1× on average with approximately 300 bp which produces the minimal amount of unique sequencing reads less than 3.5 million.
In this study, we devised a new algorithm which uses multiple z-scores to determine the fetal aneuploidy. Multi-Z algorithm shows 100% sensitivity and specificity for 3 M-reads samples, and 100% sensitivity and 95.6% specificity for 1 M-reads samples.
2. Method
2.1. cfDNA Sequencing
About 10 mL of blood was collected from 216 pregnancies into a cfDNA Vangenes Cell Free DNA Tubes (Vangenes, US) and centrifuged at 1900 g for 15 min at RT. The plasma was transferred into 1.5 mL tubes and then centrifuged at 16,000 g for 15 min at RT. The separated plasma was transferred to 5 mL Cryogenic Tube and stored at −40˚C. cfDNA was isolated from 2 mL plasma by using the QIAsymphony DSPvirus/Pathogen midi kit (Qiagen, Germany) according to the manufacturer’s instructions. Ion Proton sequencing libraries were prepared by using cfDNA samples (<100 ng) according to the manufacturer’s instructions (Life Technology, US). Ion PI™ Chip kit v3 was used to yield an average 7 million and 1 million sequencing reads for nucleotide.
2.2. Data Analysis
The DNA fragments were mapped to the human reference genome sequence (hg19) by using BWA 0.7.10 [12] and duplicated DNA reads were removed by using Picard 1.81 [13] . Finally, uniquely aligned counts for each chromosome were calculated by Samtools 0.1.18 [14] .
We generated artificial samples consist of 1 M and 3 M sequence reads by randomly selecting 1 million and 3 million reads from the samples sequenced up to 7 M reads, respectively. In this study, we calculated the multiple z-score for each chromosome of interest in two steps. First dividing the read count of the chromosome of interest to the all remaining autosomal chromosome one by one to get normalized read count as shown in Figure 1, and then, z-score calculation using Equation (1), by utilizing the average and SD obtained from sample dataset which are composed of 200 normal samples. For example, if chr13 is our chromosome of interest, read count of chromosome 13 is divided by chr1, chr2, chr3, etc. except the chr13. So, it will yield 21 normalized read count for the chromosome 13. Now using the average and SD for each normalized read count in sample dataset, we developed the multiple z-scores for the chromosome 13, i.e. Zscore13,1, Zscore13,2, Zscore13,3, etc. Similarly, multiple z-scores were developed for each of the autosomal chromosome.
We also calculated the CV through the calculated SD in sample dataset and it is used to apply the order of z-score thresholds gradually lowest to highest, e.g. chr7 is the lowest and chr12, chr14, chr9, chr11, followed in ascending order.
Multiple z-scores are calculated for normal samples using the reference mean and SD as follows:

Figure 1. Normalization between chromosomes. Normalized value between two chromosomes is calculated by dividing the value of interested chromosome by that of each chromosome.
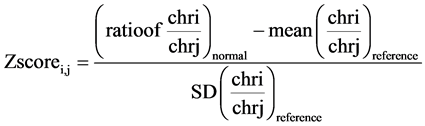 (1)
(1)
The equation is a multiple z-scores calculation. In this equation, i is for chromosome of interest and j is for other chromosomes to normalize.
Now, Multi-Z algorithm classifies negative versus positive case for test samples by applying the smallest z-score of a certain chromosome. Since we used 70 samples as thresholds in this paper, there are 70 z-scores for a chromosome of interest and the smallest z-score among them is chosen as an applicable threshold. Repeatedly, Multi-Z algorithm applies the next threshold of another chromosome according to the ascending order of CV.
3. Results
The algorithm was tested on a cohort of 216 pregnant women including seven T21 and we found the Multi-Z algorithm produced reliable results with higher sensitivity and specificity. Up to 70 control samples which had undergone the amniocentesis were used for determining thresholds. We generated 70 randomly selected samples from seven trisomy samples to make robust thresholds by increasing number of control samples.
As for 3 M-reads samples, the amount of unique sequencing reads is around 2 million and the average length of sequenced read is around 160 bp that can be calculated approximately 0.1× low coverage depth. The uniquely mapped read for 1 M samples is 0.7 M and the coverage depth is 0.035× as same calculation as 3 M-reads.
In this paper, we used the term 3 M-reads, 1 M-reads as the amount of sequenced reads
In Figure 2, the results of Multi-Z for 187 normal samples randomly generated at 3 M read length (black dots) with 70 control samples (white dots) show 100% specificity at only seven thresholds as listed in Table 1. The red-dashed line means the least z-score of interested chromosome and this line classifies the normal samples as trisomy or normal while repeating the chromosomes in interest. The number of normal samples is decreased as we repeat applying the algorithm and all the normal samples, finally, are categorized as normal after applying chromosome 6 which is 7th threshold.
In Figure 3, we added 29 samples which were experimentally sequenced at 1 million reads, represented by red dots. Seven of these samples among 216 samples were judged to be trisomy by using eight thresholds without any false positives, while nine false positive samples were found in 209 normal samples resulting in 95.6% specificity as listed Table 1.
In Figure 4, we found the conventional NIPT detection algorithm shows lower specificity compared to the multi-dimensional detecting algorithm. In this figure, we can find the only one threshold as red-dashed line which is the smallest z-score of 70 control samples. There are 9 falsely detected samples as trisomy

Table 1. Accuracy comparison between the conventional NIPT and Multi-Z.

Figure 2. 3 M-reads by Multi-Z. Every normal sample (close black dots) is classified as normal by using seven thresholds of control sample (open black dots). Red dashed line in each figure represents the smallest z-score for applied threshold of control samples. Samples remained above red dashed line are considered as trisomy.
for 3 M-reads samples which yield 95.1% Specificity as listed in Table 1. As for 1 M-reads samples, we found 52 false positive samples and it shows the worse specificity as 75.1% in Table 1. This result shows the conventional NIPT method is not applicable for low reads samples.
4. Discussion
We compared the sensitivity and the specificity between the conventional NIPT

Figure 3. 1 M-reads by Multi-Z. Seven of 1M sequencing samples (red dots) are judged as trisomy, and 9 normal samples (close black dots) are defined as trisomy (False Positive) by using 19 z-scores. Red dashed line in each figure represents the smallest z-score for applied threshold of control samples.
and Multi-Z algorithm for 1 M-reads and 3 M-reads. The results showed that Multi-Z algorithm can be used for low coverage cfDNA NIPT samples with higher specificity. When applying single Z-score threshold, it is often difficult to distinguish between the anueploid and the euploid, especially when the z-score is close to borderline, besides it may cause false positives call for some samples. We observed that the use of Multi Z-score approach for such ambiguous samples, using the correlation between specific chromosomes, showed a higher z-score and the same tendency was observed for all our test samples. Therefore, we concluded that more thresholds can be used through correlation with other chromosomes, and confirmed that it reduced the possibility of false positives. Also, we found the proper order of applying thresholds according to the CV.
Since the Multi-Z can be applied to low reads samples, it has potential competitive advantages such as reduction of experiment cost and the rapid analysis in the emerging NIPT market. A major concern in this study was to determine the precise number of thresholds to call the aneuploidy, i.e. how many z-score thresholds would be effective to detect Trisomy 21 and how to decide borderline range with selected thresholds. If the number of applied thresholds are increased, it may cause false negative and vice versa. Although, this method can

Figure 4. Accuracy of previous NIPT method at low read sequencing data. Nine and 52 normal samples were detected falsely as aneuploidy in (a) 3 M-reads data and (b) 1 M-reads data, respectively. Open black circles in (a) and (b) represent the trisomy sample’s z-scores, and the red dashed line represents the chosen threshold among all z-scores. Closed black circle represents normal samples’ z-score. red circles in (b) represent the samples sequenced at 1 million, and open red circles represent seven trisomy samples. Red dashed line represents the threshold which is the smallest z-score among control samples and we assume the samples above the red line as trisomy.
detect all the chromosome 21 aneuploidies accurately in our current dataset, a large number of samples would be required to validate our approach further.
5. Conclusion
In conclusion, we confirmed that the Multi-Z method provides an optimal way to reduce false positives of T21 for low coverage samples compared to the conventional NIPT algorithm. It is expected that detecting T13, T18 and sex chromosome aneuploidy can be applied in the same way after the research on T21 is completed.
Cite this paper
Kwon, H.J., Goyal, A., Im, H., Lee, K., Yun, S.Y., Kim, Y.H., Lee, S., Lee, M.-G., Lee, H., Garg, R., Park, B., Choi, S., Joo, J., Bae, J.-S., Kim, M.-J., Lee, M.S. and Lee, S. (2017) Multiple z-Score Based Method for Noninvasive Prenatal Test Using Cell-Free DNA in Maternal Plasma. Open Journal of Genetics, 7, 1-8. https://doi.org/10.4236/ojgen.2017.71001
References
- 1. Savva, G.M., Morris, J.K., Mutton, D.E. and Alberman, E. (2006) Maternal Age-Specific Fetal Loss Rates in Down Syndrome Pregnancies. Prenatal Diagnosis, 26, 499-504.
https://doi.org/10.1002/pd.1443 - 2. Morris, J.K., Mutton, D.E. and Alberman, E. (2002) Revised Estimates of the Maternal Age Specific Live Birth Prevalence of Down’s Syndrome. Journal of Medical Screening, 9, 2-6.
https://doi.org/10.1136/jms.9.1.2 - 3. Haddow, J.E. (1990) Prenatal Screening for Open Neural Tube Defects, Down’s Syndrome, and Other Major Fetal Disorders. Seminars in Perinatology, 14, 488-503.
- 4. Bianchi, D.W., Parker, R.L., Wentworth, J., Madankumar, R., Saffer, C., Das, A.F., Craig, J.A., Chudova, D.I., Devers, P.L., Jones, K.W., Oliver, K., Rava, R.P., Sehnert, A.J. and Group, C.S. (2014) DNA Sequencing versus Standard Prenatal Aneuploidy Screening. New England Journal of Medicine, 370, 799-808.
https://doi.org/10.1056/NEJMoa1311037 - 5. Fan, H.C., Blumenfeld, Y.J., Chitkara, U., Hudgins, L. and Quake, S.R. (2008) Noninvasive Diagnosis of Fetal Aneuploidy by Shotgun Sequencing DNA from Maternal Blood. Proceedings of the National Academy of Sciences of the USA, 105, 16266-16271.
https://doi.org/10.1073/pnas.0808319105 - 6. Jiang, F., Ren, J., Chen, F., Zhou, Y., Xie, J., Dan, S., Su, Y., Xie, J., Yin, B., Su, W., Zhang, H., Wang, W., Chai, X., Lin, L., Guo, H., Li, Q., Li, P., Yuan, Y., Pan, X., Li, Y., Liu, L., Chen, H., Xuan, Z., Chen, S., Zhang, C., Zhang, H., Tian, Z., Zhang, Z., Jiang, H., Zhao, L., Zheng, W., Li, S., Li, Y., Wang, J., Wang, J. and Zhang, X. (2012) Noninvasive Fetal Trisomy (NIFTY) Test: An Advanced Noninvasive Prenatal Diagnosis Methodology for Fetal Autosomal and Sex Chromosomal Aneuploidies. BMC Medical Genomics, 5, 57.
https://doi.org/10.1186/1755-8794-5-57 - 7. Lo, Y.M., Corbetta, N., Chamberlain, P.F., Rai, V., Sargent, I.L., Redman, C.W. and Wainscoat, J.S. (1997) Presence of Fetal DNA in Maternal Plasma and Serum. Lancet, 350, 485-487.
https://doi.org/10.1016/S0140-6736(97)02174-0 - 8. Chiu, R.W., Chan, K.C., Gao, Y., Lau, V.Y., Zheng, W., Leung, T.Y., Foo, C.H., Xie, B., Tsui, N.B., Lun, F.M., Zee, B.C., Lau, T.K., Cantor, C.R. and Lo, Y.M. (2008) Noninvasive Prenatal Diagnosis of Fetal Chromosomal Aneuploidy by Massively Parallel Genomic Sequencing of DNA in Maternal Plasma. Proceedings of the National Academy of Sciences of the USA, 105, 20458-20463.
https://doi.org/10.1073/pnas.0810641105 - 9. Fairbrother, G., Johnson, S., Musci, T.J. and Song, K. (2013) Clinical Experience of Noninvasive Prenatal Testing with Cell-Free DNA for Fetal Trisomies 21, 18, and 13, in a General Screening Population. Prenatal Diagnosis, 33, 580-583.
https://doi.org/10.1002/pd.4092 - 10. Kim, S., Jung, H., Han, S.H., Lee, S., Kwon, J., Kim, M.G., Chu, H., Han, K., Kwak, H., Park, S., Joo, H.J., An, M., Ha, J., Lee, K., Kim, B.C., Zheng, H., Zhu, X., Chen, H. and Bhak, J. (2016) An Adaptive Detection Method for Fetal Chromosomal Aneuploidy Using Cell-Free DNA from 447 Korean Women. BMC Medical Genomics, 9, 61.
https://doi.org/10.1186/s12920-016-0222-5 - 11. Lau, T.K., Cheung, S.W., Lo, P.S., Pursley, A.N., Chan, M.K., Jiang, F., Zhang, H., Wang, W., Jong, L.F., Yuen, O.K., Chan, H.Y., Chan, W.S. and Choy, K.W. (2014) Non-Invasive Prenatal Testing for Fetal Chromosomal Abnormalities by Low-Coverage Whole-Genome Sequencing of Maternal Plasma DNA: Review of 1982 Consecutive Cases in a Single Center. Ultrasound in Obstetrics & Gynecology, 43, 254-264.
https://doi.org/10.1002/uog.13277 - 12. Li, H. and Durbin, R. (2009) Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics, 25, 1754-1760.
https://doi.org/10.1093/bioinformatics/btp324 - 13. Picard Tools. Broad Institute.
http://broadinstitute.github.io/picard/ - 14. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009) The Sequence Alignment/Map Format and SAMtools. Bioinformatics, 25, 2078-2079.
https://doi.org/10.1093/bioinformatics/btp352