Open Journal of Genetics
Vol.06 No.04(2016), Article ID:71545,13 pages
10.4236/ojgen.2016.64007
An Adaptive Weighted Sum Test for Family-Based Multi-Marker Association Studies
Renfang Jiang, Jianping Dong, Yilin Dai
Department of Mathematical Sciences, Michigan Technological University, Houghton, USA
![]()
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 22, 2016; Accepted: October 24, 2016; Published: October 27, 2016
ABSTRACT
Backgrounds: Although many disease-associated common variants have been discovered through genome-wide association studies, much of the genetic effects of complex diseases have not been explained. Population-based association studies are vulnerable to population stratification. A possible solution is to use family-based tests. However, if tests only estimate the genetic effect from the within-family variation to avoid population stratification, they may ignore the useful genetic information from between-family variation and lose power. Methods: We have developed an adaptive weighted sum test for family-based association studies. The new test uses data driven weights to combine two test statistics, and the weights measure the strength of population stratification. When population stratification is strong, the proposed test will automatically put more weight on one statistic derived from within-family variation to maintain robustness against spurious positives. On the other hand, when the effect of population stratification is relatively weak, the proposed test will automatically put more weight on the other statistic derived from both within-family and between-family variation to make use of both sources of genetic variation; and at the same time, the degrees of freedom of the test will be reduced and power of the test will be increased. Results: In our study, the proposed method achieves a higher power in most scenarios of linkage disequilibrium structure as well as Hap Map data from different genes under different population structures while still keeping its robustness against population stratification.
Keywords:
Family Data, genetic Association Study, population Stratification

1. Introduction
In past decades, many disease-associated common variants have been discovered through genome-wide association studies (GWASs). However, the majority of the genetic effects of complex diseases still cannot be explained. Recent advances in next-generation sequencing technologies provide new opportunities to study the genetic effects of low- frequency variants and rare variants. Many of those complex-trait rare-variant association studies are population based [1] . Since rare variations can differ greatly among populations, population-based rare variant association studies are vulnerable to population stratification. Several rare-variant transmission disequilibrium tests have been proposed [2] [3] . Traditionally, family-based association studies test one SNP at a time. Multi-marker tests usually work better to detect an underlying genetic variance over a genomic region than single marker tests, especially in the detection of complex diseases, because multi-marker tests consider the joint information over the whole region. Many multi-marker family association tests have been proposed, some are based on gene- ralized estimating equations (GEEs) [4] , and some use linear combinations of single marker contributions [3] . After a genome-wide association study, people often use genotype imputation for further studies. A recently developed program GIGI is efficient to impute genotypes in a large pedigree [5] , and it is used for rare-variant family association studies [6] . One distinct advantage of family-based association tests (FBAT) is their robustness against population admixture and stratification. However, if tests only estimate the genetic effect from the within-family variation to avoid population stratification, they may ignore the useful genetic information from between-family variation and lose power. Imputed allele dosages are used in FBATdosage [7] . To correct the bias introduced by genotype uncertainty, FBAT-LRT is proposed [8] . In this article, we introduce an adaptive weighted sum association test to capture more important information from multiple loci in family-based studies by considering the genetic effect from both within-family and between-family variation while maintaining robustness to population stratification.
The test is proposed for family-based association studies of quantitative trait in either a candidate region study or a genome-wide scan. The data-driven weights are based on a measure of population stratification. Since population stratification and linkage disequilibrium (LD) cause a bias for the estimate, a permutation procedure is employed to find the p-value. Extensive simulation studies are carried out under various LD structures as well as Hap Map data from different genes under different population structures. In these simulation studies, we examine the Type I error rate and compare the power of the proposed method with other FBAT tests. Simulation results show that the proposed method has a correct Type I error rate and consistently achieves a higher or similar power in all scenarios. In summary, we believe the adaptive weighted sum based FBAT is a potentially powerful method for family-based genetic study of multiple markers and it can also be used as an alternative tool for the detection of underlying causative genetics variances.
2. Method
In family-based association studies, FBAT, a general unified approach, has been proposed to permit any type of genetic models, a general family design, different phenotypes and multiple markers [9] . Family-based tests are generally robust to population stratification and those tests can avoid any population bias in other standard designs. Recently, the multi-marker test FBATMM [10] , which is similar to the Hotelling 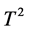 test, has been proposed for family-based studies. Another multi-marker test FBATLC [11] linearly combines single-marker test statistics using data-driven weights derived by conditional mean model [12] . The weights are least square estimates of genetic effects. The data-driven weights are regarded as fixed for FBAT. These two methods have been implemented in the program FBAT, which has been widely used in family-based association studies. The data-driven weights in FBATLC are the estimates of genetic effect considering between-family variation. It is a biased estimator and is sensitive to population structure. We investigate the data-driven weights used in FBATLC and provide a new methodology to analyze the multiple correlated markers for family-based association studies.
test, has been proposed for family-based studies. Another multi-marker test FBATLC [11] linearly combines single-marker test statistics using data-driven weights derived by conditional mean model [12] . The weights are least square estimates of genetic effects. The data-driven weights are regarded as fixed for FBAT. These two methods have been implemented in the program FBAT, which has been widely used in family-based association studies. The data-driven weights in FBATLC are the estimates of genetic effect considering between-family variation. It is a biased estimator and is sensitive to population structure. We investigate the data-driven weights used in FBATLC and provide a new methodology to analyze the multiple correlated markers for family-based association studies.
We use FBATWS to denote the new test. It is based on weighted sum of two association tests. One of which estimates the genetic effect from both within-family and between-family variation and the other is from within-family variation only. The weights are computed automatically based on a measure of the population stratification str- ength in family data. If the strength of the population stratification is strong, including between-family variation will produce false positives. At this time we need to decrease the weight of the test estimating the genetic effect from both within-family and between-family variation, and increase the weight of the other test to reduce false positive rates. If the strength of the population stratification is weak, it will not produce much false positive. Including between-family variation will increase power of the test, and at the same time it will not produce much false positive. That is why we want to increase the weight of the test estimating the genetic effect from both within-family and between-family variation. The proposed method can capture more important information from multiple loci in the family data while maintaining robustness to population stratification. Since population stratification and linkage disequilibrium cause a bias for the estimate, a permutation procedure is employed conditional on the traits, parental genotypes, and haplotypes.
The general idea of FBAT [9] is to regard the offspring genotype as random conditional on the traits and parental genotypes. The test statistic is computed from the distribution of offspring genotype under the null hypothesis. Let 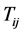 denote the coded trait for the jth offspring in the ith family and
denote the coded trait for the jth offspring in the ith family and 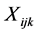 denote the coded genotype score for the kth marker of the jth offspring in the ith family, where
denote the coded genotype score for the kth marker of the jth offspring in the ith family, where 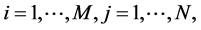 and
and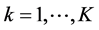 .
.
Following the standardized FBAT [9] , let:
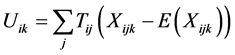 (1)
(1)
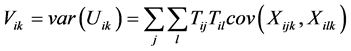 (2)
(2)
With a large number of families, FBAT statistic for the kth marker:
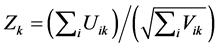 (3)
(3)
is approximately N(0,1).
Another approach to the multi-marker family-based association testing is to linearly combine single-marker test statistics using data-driven weights (FBATLC) [11] . Conditional on the traits and parental genotypes, the weights can be derived by the conditional mean model of trait T for the kth marker as follows:
![]() (4)
(4)
where ![]() for offspring in the informative families and
for offspring in the informative families and ![]()
for the others (include offspring in the non-informative families and all parents).
Let ![]() where
where ![]() is the standardized least square esti- mator of
is the standardized least square esti- mator of![]() . Then the multi-marker FBATLC test statistic:
. Then the multi-marker FBATLC test statistic:
![]() (5)
(5)
is approximately N(0,1), where ![]() is the vector of single FBAT test statistics and
is the vector of single FBAT test statistics and ![]() can be derived from the conditional pairwise haplotype distribution in offspring or from the empirical estimator of the covariance matrix [10] .
can be derived from the conditional pairwise haplotype distribution in offspring or from the empirical estimator of the covariance matrix [10] .
Although the data-driven weights are independent of Z under ![]() because the FBAT test is computed conditional on traits and on parental genotypes, the power of FBATLC will be highly dependent on the estimate of the optimal weights. In the conditional mean model, the weights are estimates of genetic effects using population data, which can be regarded as estimates of the genetic effects using between-family variation. It has been shown that this estimator is biased unless there is no population stratification. Intuitively, the more accurate the estimate is, the closer the weights to the optimal weights, and the more power the test can gain. However it will lose power if the effect of population stratification is significant. Thus, we proposed a new multi-marker test FBATWS using adaptive weights to combine two test statistics based on the estimate of the existing population stratification.
because the FBAT test is computed conditional on traits and on parental genotypes, the power of FBATLC will be highly dependent on the estimate of the optimal weights. In the conditional mean model, the weights are estimates of genetic effects using population data, which can be regarded as estimates of the genetic effects using between-family variation. It has been shown that this estimator is biased unless there is no population stratification. Intuitively, the more accurate the estimate is, the closer the weights to the optimal weights, and the more power the test can gain. However it will lose power if the effect of population stratification is significant. Thus, we proposed a new multi-marker test FBATWS using adaptive weights to combine two test statistics based on the estimate of the existing population stratification.
The strength of population stratification will be measured by
 (6)
(6)
where  for
for  Then the test statistic can be written as:
Then the test statistic can be written as:
 (7)
(7)
Under the null hypothesis: no genetic effect and no population stratification,  and
and  are independent standard normal random variables. Therefore,
are independent standard normal random variables. Therefore,  is a folded normal random variable with
is a folded normal random variable with  and
and . It is clear that the strength of population stratification increases as
. It is clear that the strength of population stratification increases as  increases. When population stratification is strong, FBATWS will automatically put more weight on the second term to maintain robustness against spurious positives. On the other hand, when the effect of population stratification is relatively weak, FBATWS will automatically put more weight on the first term to make use of both sources of genetic variation: between-family and within-family. In latter case, the degrees of freedom of the test will be reduced, and power of the test will be increased. Because LD structure will be maintained in the permutation procedure, in order to improve the computational efficiency, FBATWS does not consider LD structures.
increases. When population stratification is strong, FBATWS will automatically put more weight on the second term to maintain robustness against spurious positives. On the other hand, when the effect of population stratification is relatively weak, FBATWS will automatically put more weight on the first term to make use of both sources of genetic variation: between-family and within-family. In latter case, the degrees of freedom of the test will be reduced, and power of the test will be increased. Because LD structure will be maintained in the permutation procedure, in order to improve the computational efficiency, FBATWS does not consider LD structures.
The second term  can be written as:
can be written as:
 (8)
(8)
 is a vector and
is a vector and  is an empirical estimator of the
is an empirical estimator of the
covariance matrix . The entry of
. The entry of  at the
at the  th row and the
th row and the  th column is
th column is
 (9)
(9)
 is the coded genotype score for the kth marker, of the jth offspring in the ith family.
is the coded genotype score for the kth marker, of the jth offspring in the ith family.  is the coded trait for the jth offspring in the ith family. Therefore, the second term
is the coded trait for the jth offspring in the ith family. Therefore, the second term  is one of the asymptotic tests in [13] , which has been proposed recently to gain more power under strong LD structures. When the parental haplotypes are known, a permutation procedure will be employed to compute the p-value of FBATWS. For each child with fixed trait in any family, each parental haplotype is transmitted to the child with equal probability, so that, for any given parental hypostyles, there are four different permutations of the data. When the parental haplotypes are unknown, inferring haplotype is needed. There are several methods to infer haplotypes. For example, Thunder [14] , Beagle [15] , Impute 2 [16] , and SNPtools [17] . Haplotype can also be inferred by using sequencing reads [18] .
is one of the asymptotic tests in [13] , which has been proposed recently to gain more power under strong LD structures. When the parental haplotypes are known, a permutation procedure will be employed to compute the p-value of FBATWS. For each child with fixed trait in any family, each parental haplotype is transmitted to the child with equal probability, so that, for any given parental hypostyles, there are four different permutations of the data. When the parental haplotypes are unknown, inferring haplotype is needed. There are several methods to infer haplotypes. For example, Thunder [14] , Beagle [15] , Impute 2 [16] , and SNPtools [17] . Haplotype can also be inferred by using sequencing reads [18] .
3. Simulation Results
In the simulation study, we apply the proposed test FBATWS on two sets of data. One is simulated with six scenarios of LD structure. The other is downloaded haplotype data from 170 unrelated samples of JPT + CHB (Japanese in Tokyo, Japan + Han Chinese in Beijing, China) in the HapMap3 Phased Haplotypes. We compare the power of the proposed test FBATWS with the following three FBAT tests: 1) the single-marker test with Bonferroni multiple testing adjustment FBATB the Bonferroni adjusted p-value  where
where  is the minimal p-value among the single-marker tests 2) the multi-marker test FBATMM [10] , which is similar to the Hotelling
is the minimal p-value among the single-marker tests 2) the multi-marker test FBATMM [10] , which is similar to the Hotelling  test, 3) the multi-marker test FBATLC [11] that linearly combines the single-marker test statistics using data-driven weights.
test, 3) the multi-marker test FBATLC [11] that linearly combines the single-marker test statistics using data-driven weights.
One goal of the simulation study is to examine whether the proposed multi-marker test is robust to the underlying LD structure. We consider six different LD structures and assume additive genetic effect. A target region with eight observed SNPs and an unobserved causative SNP in the middle is simulated. For each nuclear family, both parental haplotypes for nine correlated SNP markers are simulated on the basis of a multivariate normal distribution with LD structure  where
where  Each allele on the haplotype is generated with the cut-off of the minor allele frequency which is obtained from a uniform distribution between 0.1 and 0.3. The haplotypes of off spring are obtained by the simulated Mendelian transmission without recombination based on the parental haplotypes. The genotypes for each individual are generated by the sum of two haplotypes. The six scenarios of LD pattern are defined by the following pairwise
Each allele on the haplotype is generated with the cut-off of the minor allele frequency which is obtained from a uniform distribution between 0.1 and 0.3. The haplotypes of off spring are obtained by the simulated Mendelian transmission without recombination based on the parental haplotypes. The genotypes for each individual are generated by the sum of two haplotypes. The six scenarios of LD pattern are defined by the following pairwise  if
if  0, otherwise. The formula of
0, otherwise. The formula of  is shown in Table 1. For all scenarios, the correlation between the causal SNP and the observed SNPs is
is shown in Table 1. For all scenarios, the correlation between the causal SNP and the observed SNPs is  where d is the index of causal SNP and t has the equal possibility to be +1 or −1. The results are shown in Figure 1.
where d is the index of causal SNP and t has the equal possibility to be +1 or −1. The results are shown in Figure 1.
The quantitative phenotype of each individual is determined by:
 (8)
(8)
where  is the overall mean for one family following a normal distribution
is the overall mean for one family following a normal distribution ,
,  is the trait correlation within one family, G is the genetic effect term and
is the trait correlation within one family, G is the genetic effect term and  is a independent error term following a normal distribution
is a independent error term following a normal distribution , where
, where  so that the total variance of the trait is 1.We consider all the samples come from one population and set
so that the total variance of the trait is 1.We consider all the samples come from one population and set  to be 0 in this simulation study. The Heritability
to be 0 in this simulation study. The Heritability  for this model will be given from 0 to 0.09, thus the variance of the genetic effect can be obtained by
for this model will be given from 0 to 0.09, thus the variance of the genetic effect can be obtained by . The genetic effect G is determined by the genotype score
. The genetic effect G is determined by the genotype score

Table 1. Six scenarios of LD pattern (t has a equal possibility to be +1 or −1).

Figure 1. LD structures for simulation.
 of the unobserved causal SNP:
of the unobserved causal SNP:  where a is genetic effect value which is determined by
where a is genetic effect value which is determined by  (p is the minor allele frequency at the causal SNP) for the additive model [11] . 500 trios with 1000 simulation replicates are considered and the significance level is set at 0.05.
(p is the minor allele frequency at the causal SNP) for the additive model [11] . 500 trios with 1000 simulation replicates are considered and the significance level is set at 0.05.
Next, our simulation study will be based on real LD structure. We download haplotype data from 170 unrelated samples of JPT + CHB (Japanese in Tokyo, Japan + Han Chinese in Beijing, China) in the HapMap3 Phased Haplotypes. We consider three genes CHI3L2 (in the region of 15.78 kb), CTLA4 (in the region of 10 kb) and IL21R (in the region of 47.69 kb), which have also been analyzed in other simulation studies [19] [20] [21] [22] . Their LD pattern can be visualized on the HapMap site. We perform the simulation study using SNPs with minor allele frequency (MAF) >0.01, and we remove the redundant SNPs that are perfectly correlated with other SNPs. We have 12 SNPs left for CHI3L2, seven SNPS for CTLA4 and 10 SNPs for IL21R. We calculate haplotype frequencies from the samples of each gene and generate the parents of each family based on the known haplotype frequencies. The disease marker is randomly chosen as unobserved SNP. Other SNPs are observed as haplotype data and the quantitative phenotypes of offspring in each family are generated from a quantitative phenotype model. Two scenarios (500 trios under one population and two populations) are considered in the simulation study with 1000 simulation replicates and a significance level of 0.05. To generate quantitative phenotypes for samples from one population, let  for samples from two distinct populations, let
for samples from two distinct populations, let  be 0.5 or −0.5.
be 0.5 or −0.5.
Type I error rate for the case of six mimicked LD structures is shown in Table 2. All tests have a correct Type I error rate. It is expected that the proposed method will have a correct Type I error rates due to the permutation procedure. The result of power comparison is shown in Figure 2.
Four FBAT tests are considered for power comparisons with six different LD structures. The unobserved casual SNP has an equal chance to be positively or negatively correlated to those observed SNPs in all scenarios. In Figure 2, FBATB (B), (MM), FBATLC (LC), and FBATWS (WS) are indicated by the blue dot-dashed line, the green dotted line, the red dash line, and the black solid line, respectively. In the first simulation study, the goal is to compare the performance of the proposed method with other FBAT methods. We fix the window size for each scenario and assume the sample come from the same population. An examination of the results show that FBATWS has a consistently higher power in all cases, followed by FBATLC, FBATMM and FBATB FBATB is considered as the most conservative test in this study, because the independent assumption is violated. The power of FBATMM is improved since it considers the variance-covariance matrix. On the other hand, it also suffers from the relatively high degrees of freedom, especially when the region under consideration is large. The power of FBATLC is improved since it has only one degree of freedom, it uses the optimal weights to combine single-marker tests, and it overcomes the degrees of freedom problem raised by FBATMM. In a genetic region with strong LD, we do not have any clue of how the underlying casual marker is related to the observed SNPs. The optimal weights in FBATLC are biased estimates of genetic effects [23] . Therefore, using incorrect estimation of genetic effect as weights in FBATLC will lose some power. The power of FBATWS is improved since it not only considers the optimal weights to combine single-marker tests like FBATLC, but also automatically adjusts the weights based on the estimate of the genetic effect from between-family variants and within-family variants.
Type I error rates for the simulated HapMap data on CHI3L2, IL21R, and CTLA4 are given in Table 3. Type I error rate of all tests are well controlled under 0.05 level of
Table 2. Type I error rates for four FBAT tests using simulated data.
B, MM, LC, WS indicates FBATB, FBATMM, FBATLC, FBATWS, respectively. L1, L2, L3, L4, L5, L6, indicate six scenarios of LD structure given in Table 1.

Figure 2. Power comparisons using simulated data.
significance. We also found that FBATB has a lower type 1 error rate than other tests, because the strong LD structure existed in all three regions. The results of power comparison in one population and two populations are shown in Figure 3 and Figure 4. The underlying casual marker is randomly selected each time, which make the LD structures relatively complicated in these scenarios.
Table 3. Type I error rates of four FBAT tests using HapMap data, * denotes the cases in mixed populations of two.
B, MM, LC, WS indicates FBATB, FBATMM, FBATLC, FBATWS, respectively.

Figure 3. Power comparisons using Hapmap data.

Figure 4. Power comparisons using Hapmap data.
Four FBAT tests are considered for power comparisons under different LD structures of three genes CHI3L2 (in the region of 15.78 kb), CTLA4 (in the region of 10 kb) and IL21R (in the region of 47.69 kb). The unobserved casual SNP is randomly selected in all scenarios. In Figure 3 and Figure 4, FBATB (B), FBATMM (MM), FBATLC (LC), and FBATWS (WS) are denoted by the blue dot-dashed line, the green dotted line, the red dash line, and the black solid line, respectively.
We consider all samples from one population first. The power of FBATWS is relatively high in most scenarios. For gene CHI3L2, where SNPs are dense and highly correlated with each other, FBATWS is the most powerful test, followed by FBATWS, FBATMM and FBATB when the heritability is relatively low. As heritability increasing, the power of FBATMM is the highest, and FBATWS is the second among all tests. This implies FBATWS is more sensitive to the genetic effect with low heritability. FBATMM is adept to deal with genetic region with strong LD and high heritability. For the gene CTLA4, where the number of markers is relatively small and LD pattern is relatively weak, FBATWS is again the most powerful test, followed by FBATLC, FBATB and FBATMM. For the gene IL21R, where SNPs are loose and LD pattern is relatively weak, FBATWS is the most powerful test, followed by FBATB, FBATLC, and FBATMM. For genetic region with weak LD like CTLA4 and IL21R, FBATMM lose its potential power due to the issue of degrees of freedom. In all scenarios of two populations, the results are similar that FBATWS is the most powerful test except for simulated data based on gene CTLA4 with high heritability. In practice, most undiscovered genetic variants have low heritability. The power of tests depends on the LD patter. In general, FBATWS automatically adjusted the weights to combine the estimates of genetic effect from various source of genetic variants, therefore is a powerful test for family-based association studies. It is robust to population stratification and the underlying LD structure. Our simulated results demonstrate that V is a potentially powerful test among multi-marker tests.
4. Concluding Remarks
We propose a novel multi-marker family-based association test for multi-marker testing using data-driven weights to automatically combine statistics, which are based on different sources of genetic variation. One of the statistics comes from the estimation of the genetic effects from both within-family and between-family variations, which is more like a population-based statistic. The other is from estimation of within-family variation, which is a family-based statistic. The data driven weights are computed automatically, and they measure the strength of the population stratification existed in the family data. The advantage of family-based studies is its ability to avoid spurious positives caused by population stratification. For the FBAT test, we regard the offspring genotypes as a random variable given trait and parental genotypes or haplotypes. On the other hand, FBAT tests do not consider the genetic information from between- family variation, since those can raise the issue of population stratification. By using adaptive weighted sum to combine this information efficiently into the test statistics can improve the power of the test.
The proposed method tries to use the most information of genetic variance for family based association studies. Data driven weights are employed to make our test robust to population stratification and linkage disequilibrium between multiple markers. Since population stratification and linkage disequilibrium cause the bias of the estimation, a permutation procedure is employed and descried for this situation. The new test is a potentially powerful method for family-based genetic study of multiple markers by considering genetic variance in different aspects and can also provide an alternative tool for the detection of underlying causal genetics variances. In our simulation studies using mimicked LD patterns and three genes from HapMap data, the results show that the proposed test achieves a higher power in most scenarios than the single-marker test with Bonferroni correction, the multi-marker test similar to the Hotelling  test, and the multi-marker test that linearly combines the single marker tests using data-driven weights. Although the proposed test can achieve a higher power in some complex situations, it is not optimal in all situations. For example among some SNPs or tag SNPs, if there is a super SNP strongly or perfectly associated with the disease or causal locus, then the single-marker test with Bonferroni correction should have a higher power than other multi-marker tests.
test, and the multi-marker test that linearly combines the single marker tests using data-driven weights. Although the proposed test can achieve a higher power in some complex situations, it is not optimal in all situations. For example among some SNPs or tag SNPs, if there is a super SNP strongly or perfectly associated with the disease or causal locus, then the single-marker test with Bonferroni correction should have a higher power than other multi-marker tests.
Cite this paper
Jiang, R.F., Dong, J.P. and Dai, Y.L. (2016) An Adaptive Weigh- ted Sum Test for Family-Based Multi-Mar- ker Association Studies. Open Journal of Genetics, 6, 61-73. http://dx.doi.org/10.4236/ojgen.2016.64007
References
- 1. Lee, S., Abecasis, G.R., Boehnke, M. and Lin, X. (2014) Rare-Variant Association Study Designs and Statistical Tests. American Journal of Human Genetics, 95, 5-23.
http://dx.doi.org/10.1016/j.ajhg.2014.06.009 - 2. He, Z., O’Roak, B., Smith, J.D., Wang, G., Hooker, S., Santos-Cortez, R.L.P., Li, B., Kan, M., Krumm, N., Nickerson, D.A., Shendure, J., Eichler, E.E. and Leal, S.M. (2014) Rare-Variant Extensions of the Transmission Disequilibrium Test: Application to Autism Exome Sequence Data. American Journal of Human Genetics, 94, 33-46.
http://dx.doi.org/10.1016/j.ajhg.2013.11.021 - 3. Jiang, Y., Satten, G.A., Han, Y., Epstein, M.P., Heinzen, E.L., Goldstein, D.B. and Allen, A.S. (2014) Utilizing Population Controls in Rare-Variant Case-Parent Association Tests. American Journal of Human Genetics, 94, 845-853. http://dx.doi.org/10.1016/j.ajhg.2014.04.014
- 4. Wang, X., Lee, S., Zhu, X., Redline, S. and Lin, X. (2013) GEE-Based SNP Set Association Test for Continuous and Discrete Traits in Family-Based Association Studies. Genetic Epidemiology, 37, 778-786.
http://dx.doi.org/10.1002/gepi.21763 - 5. Cheung, C.Y., Thompson, E.A. and Wijsman, E.M. (2013) GIGI: An Approach to Effective Imputation of Dense Genotypes on Large Pedigrees. American Journal of Human Genetics, 92, 504-516.
http://dx.doi.org/10.1016/j.ajhg.2013.02.011 - 6. Saad, M. and Wijsman, E. (2013) Power of Family-Based Association Designs to Detect Rare Variants in Large Pedigrees Using Imputed Genotypes. Genetic Epidemiology, 38, 1-9.
http://dx.doi.org/10.1002/gepi.21776 - 7. Cobat, A., Abel, L., Alcais, A. and Schurr, E. (2014) A General Efficient and Flexible Approach for Genome-Wide Association Analyses of Imputed Genotypes in Family-Based Designs. Genetic Epidemiology, 38, 560-571.
http://dx.doi.org/10.1002/gepi.21842 - 8. Yu, Z. (2012) Family-Based Association Tests Using Genotype Data with Uncertainty. Biostatiistics, 13, 228-240.
http://dx.doi.org/10.1093/biostatistics/kxr045 - 9. Laird, N.M., Horvath, S. and Xu, X. (2000) Implementing a Unified Approach to Family-Based Tests of Association. Genetic Epidemiology, 19, S36-S42.
http://dx.doi.org/10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M - 10. Rakovski, C.S., Xu, X., Lazarus, R., Blacker, D. and Laird, N.M. (2007) A New Multimarker Test for Family-Based Association Studies. Genetic Epidemiology, 31, 9-17.
http://dx.doi.org/10.1002/gepi.20186 - 11. Xu, X., Rakovski, C., Xu, X.P. and Laird, N. (2006) An Efficient Family-Based Association Test Using Multiple Markers. Genetic Epidemiology, 30, 620-626.
http://dx.doi.org/10.1002/gepi.20174 - 12. Lange, C., De Meo, D., Silverman, E.K., Weiss, S.T. and Laird, N.M. (2003) Using the Noninformative Families in Family-Based Association Tests: A Powerful New Testing Strategy. American Journal of Human Genetics, 73, 801-811. http://dx.doi.org/10.1086/378591
- 13. Pan, W. (2009) Asymptotic Tests of Association with Multiple SNPs in Linkage Disequilibrium. Genetic Epidemiology, 33, 497-507.
http://dx.doi.org/10.1002/gepi.20402 - 14. Li, Y., Willer, C.J., Ding, J., Scheet, P. and Abecasis, G.R. (2010) MaCH: Using Sequence and Genotype Data to Estimate Haplotypes and Unobserved Genotypes. Genetic Epidemiology, 34, 816-834.
http://dx.doi.org/10.1002/gepi.20533 - 15. Browning, B.I. and Browning, S.R. (2009) A Unified Approach to Genotype Imputation and Haplotype-Phase Inference for Large Data Sets of Trios and Unrelated Individuals. American Journal of Human Genetics, 84, 210-223. http://dx.doi.org/10.1016/j.ajhg.2009.01.005
- 16. Montgomery, S.B., Goode, D.L., Kvikstad, E., Albers, C.A., Zhang, Z.D., Mu, X.J., Ananda, G., Howie, B., Karczewski, K.J., Smith, K.S., et al. (2013) 1000 Genomes Project Consortium. The Origin, Evolution, and Functional Impact of Short Insertion-Deletion Variants Identified in 179 Human Genomes. Genome Research, 23, 749-761.
http://dx.doi.org/10.1101/gr.148718.112 - 17. Wang, Y., Lu, J., Yu, J., Gibbs, R.A. and Yu, F. (2013) An Integrative Variant Analysis Pipeline for Accurate Geno-type/Haplotype Inference in Population NGS Data. Genome Research, 23, 833-842.
http://dx.doi.org/10.1101/gr.146084.112 - 18. Delaneau, O., Howie, B., Cox, A.J., Zagury, J.F. and Marchini, J. (2013) Haplotype Estimation Using Sequencing Reads. Genetic Epidemiology, 93, 687-696.
http://dx.doi.org/10.1016/j.ajhg.2013.09.002 - 19. Chapman, J. and Whittaker, J. (2008) Analysis of Multiple SNPs in a Candidate Gene or Region. Genetic Epidemiology, 32, 560-566.
http://dx.doi.org/10.1002/gepi.20330 - 20. Jiang, R.F., Dong, J.P. and Dai, Y.L. (2009) Improving Power in Genetic-Association Studies via Wavelet Transformation. BMC Genetics, 10, 53.
http://dx.doi.org/10.1186/1471-2156-10-53 - 21. Wang, K. and Abbott, D. (2008) A Principal Components Regression Approach to Multilocus Genetic Association Studies. Genetic Epidemiology, 32, 108-118.
http://dx.doi.org/10.1002/gepi.20266 - 22. Wang, T. and Elston, R.C. (2007) Improved Power by Use of a Weighted Score Test for Linkage Disequilibrium Mapping. American Journal of Human Genetics, 80, 353-360.
http://dx.doi.org/10.1086/511312 - 23. Abecasis, G.R., Cardon, L.R. and Cookson, W.O.C. (2000) A General Test of Association for Quantitative Traits in Nuclear Families. American Journal of Human Genetics, 42, 279-292.
http://dx.doi.org/10.1086/302698
Abbreviations
LD: Linkage disequilibrium,
GWASs: Genome-wide association studies,
FBAT: Family-based association test,
GEE: Generalized estimating equation,
FBAT dosage: Imputing allele dosages in FBAT,
FBATMM: Multi-marker family-based association test,
FBATLC: Linearly combined single-marker test statistics,
FBATWS: Proposed test in this article,
FBATB: Single-marker test with Bonferroni multiple testing adjustment,
SNP: Single-nucleotide polymorphism.

Submit or recommend next manuscript to SCIRP and we will provide best service for you:
Accepting pre-submission inquiries through Email, Facebook, LinkedIn, Twitter, etc.
A wide selection of journals (inclusive of 9 subjects, more than 200 journals)
Providing 24-hour high-quality service
User-friendly online submission system
Fair and swift peer-review system
Efficient typesetting and proofreading procedure
Display of the result of downloads and visits, as well as the number of cited articles
Maximum dissemination of your research work
Submit your manuscript at: http://papersubmission.scirp.org/
Or contact ojgen@scirp.org