Open Journal of Genetics
Vol.2 No.2(2012), Article ID:20383,5 pages DOI:10.4236/ojgen.2012.22011
Mutation screening of muscle development genes in patients with idiopathic clubfoot
![]()
1Department of Orthopaedic Surgery and Rehabilitation, Pediatrics University of Iowa, Iowa City, USA
2Howard Hughes Medical Institute, University of Iowa, Iowa City, USA
Email: jose-morcuende@uiowa.edu
Received 18 March 2012; revised 22 April 2012; accepted 6 May 2012
Keywords: Clubfoot; Genetics; Muscle; SPRY2; RAF1; IQSEC1; LMO7; UCHL3
ABSTRACT
Background: Congenital idiopathic clubfoot is a very common musculoskeletal birth defect, but with no known etiology. Dietz et al. have shown possible linkage in chromosome 3 and 13 in a large, multigenerational family with congenital idiopathic clubfoot. Current evidence suggests that muscle development is impaired in patients with congenital idiopathic clubfoot, therefore we hypothesized that mutations in genes related to muscle development could be associated with this deformity. From the areas identified in the linkage study, candidate genes SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 were selected based upon their presence in skeletal muscle as well as their involvement in muscle development. Methods: The exons and splice sites of the five genes were screened via sequence-based analysis in a group of 24 patients with congenital idiopathic clubfoot. All single nucleotide polymorphisms (SNPs) found were compared to public databases to determine allelic frequency and amino acid modification. Results: While many SNPs were found, none proved to be significantly associated with the phenotype of congenital idiopathic clubfoot. The SNPs found were shown to be common amongst a non-clubfoot population and to follow the allelic frequency of the general population. Conclusions: Based upon these results, SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 are not likely to be the major causes of congenital idiopathic clubfoot. Given the complexity of myogenesis, many other candidate genes remain that could cause defects in the hypaxial musculature that is invariably observed in congenital idiopathic clubfoot. Clinical Relevance: This study further identifies genes which are unlikely to be the direct cause of congenital idiopathic clubfoot. It also helps to eliminate suspected genes found within the given bounds of chromosome 3 and 13.
1. INTRODUCTION
Congenital idiopathic clubfoot is the most common pediatric musculoskeletal deformity affecting 1 - 7 per 1000 newborns. Despite extensive clinical and basic research, its etiology is unknown. However, a genetic component has been suggested. Twin studies have shown a concordance rate of 17% in monozygotic twins, 9% in dizygotic twins of the same sex, and 5% in dizygotic twins of opposite sex [1]. Also, first degree relatives of patient’s with clubfoot have a higher probability of having clubfoot than the general population. For example, if a parent and one child are affected by clubfoot, then there is a 30% chance that subsequent children will also have clubfoot [2]. In addition, several studies have indicated the etiology of clubfoot may be caused by a single, major gene with incomplete dominant inheritance [3-5].
Based upon the growing evidence that a single gene may be responsible for the development of clubfoot, several studies have been reported looking at candidate genes. Genes involved in hind limb development (PITX1, HOXD13, CAND2, WNT7A), biotransformation of tobacco smoke (NAT2), apoptosis (Casp8, Casp10, and CFLAR), and collagen type IX (COL9A1) have all been evaluated and shown to not be related to the development of congenital idiopathic clubfoot [6-11]. Interestingly, Dietz et al. using a genome-wide screening of a fourgeneration family Figure 1 with 13 affected and 41 unaffected members identified linkage regions on chromosome 3 and 13 (LOD score of 2.3) [12]. Therefore, the gene(s) causing idiopathic clubfoot could reside in these areas and a genetic mutation screening of candidate genes may provide insight into its etiology.
Several recent studies have reported evidence of a relationship between clubfoot and muscle development. Ippollito found muscle atrophy in patients with clubfoot and since this atrophy continues throughout life, impairment in muscle growth could be a likely pathological cause [13]. Ippolito et al. also described a “retracting fibrosis” that began early in fetal development; and beta-catenin levels have been shown to be increased in the connective
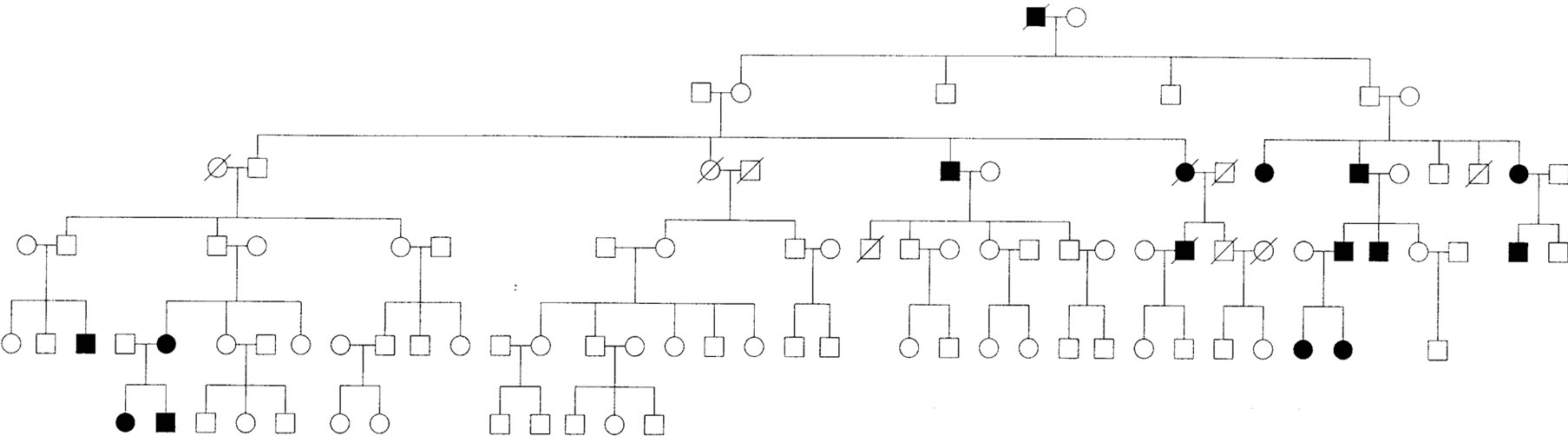
Figure 1. Omaha family pedigree.
tissue of clubfoot in a similar manner as in other fibroproliferative diseases such as palmar fibromatosis [14, 15]. Furthermore, histological and electron microscopy studies have shown that muscle fibers exhibited a loss of organization and directionality, increased variability in the fiber size, increase in Type I fibers, and a decrease in Type II fibers [16-18]. The normal ratio of Type I:Type II fibers is 1:2, while in clubfoot patients the average ratio was 7:1, a 14-times decrease in Type II fiber [18]. Several other studies have found distinct neuromuscular abnormalities in muscle biopsies obtained from patients with congenital idiopathic clubfoot [16-23].
Based on these findings, 5 candidate genes—SPRY2, RAF1, IQSEC1, LMO7, and UCHL3—from the linkage areas by Dietz et al. were selected because their relationship with muscle development. Specifically, SPRY2 has been shown to be a negative regulator of fibroblast growth factor (FGF) signaling, involved in myogenesis, and leads to non-formation of myotubes if mutated [24,25]. RAF1 is involved in a pathway that regulates myoblast proliferation and differentiation. It also enhances the transcripttional activity of MyoD, a controller of muscle cell differentiation [26,27]. IQSEC1 is involved in the fusion of mammalian myoblasts, and when knocked down results in abnormal myotubes [28]. LMO7 interacts with beta-catenins and MyoD, and when deleted in mice, results in the development of dystrophic muscles [29]. Deletion of UCHL3 can cause a mild muscular dystrophy, and when deleted along with LMO7, the severity of muscle degeneration is increased [29,30]. This study was conducted based on the hypothesis that a primary mutation in any one of the five chosen candidate genes (SPRY2, RAF1, IQSEC1, LMO7, and UCHL3) could be associated with congenital idiopathic clubfoot.
2. MATERIALS AND METHODS
2.1. Study Subjects and Ascertainment Strategy
All protocols were approved by the human subjects review board of the University of Iowa Hospitals and Clinics. Informed consent was obtained from all participants.
Twenty-four patients with congenital idiopathic clubfoot without any other congenital abnormalities were identified. Patients with neuromuscular or other recognizable syndromes involving clubfoot were excluded. The Single Nucleotide Polymorphism reference through NCBI was used to identify any previously known SNPs and to compare the results of ethnically-matched allelic frequencies. All exons and splice-sites were screened.
2.2. DNA Extraction, PCR Amplification, and Direct Sequencing
DNA was extracted from cheek swabs using a standard DNA extraction method [31]. Saliva samples were used rather than venipucture due to the pediatric population. The final concentration of the DNA was obtained using spectroscopy and all samples were diluted to 20 ng/µl and stored at 4˚C during the duration of the experiments. The mRNA sequences for SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 were obtained through a nucleotide search on the Ensembl website (http://www.ensembl.org), and the complete cds were used. At the time of sequencing, for SPRY2 (3 exons), the mRNA was 2126 bps long, the accession number is NM_005842. For RAF1 (18 exons), the mRNA was 3291 bps long, the accession number is NM_002880. For IQSEC1 (18 exons), the mRNA was 7229 bps long, the accession number is NM_001134382. For LMO7 (33 exons), the mRNA was 7253 bps long, the accession number is NM_005358. For UCHL3 (11 exons), the mRNA was 899 bps long, the accession number is NM_006002.
2.3. Sequence Analysis
After sequencing the forward strand first, the sequences were viewed using the program Sequencher and if a sequence variant was observed, then the reverse strand was sequenced to verify the result. Insertions, substitutions, deletions, and unknown nucleotides on one strand were checked against the other strand in a BLAST search. When a change was observed in the sequence of both strands, the change was labeled as a polymorphism. If it was shown to be a common variant over a wide, ethnically matched population, and our allelic frequency was similar to the general population, the single nucleotide polymorphism (SNP) was considered unrelated to the development of clubfoot. However, if the SNP was not seen before, then the polymorphism would be considered novel and would then be run in a larger sample population and compared to an equally large control population to verify that the polymorphism was actually novel.
3. RESULTS
Based on sequence-comparison, no previously reported mutations in any of these genes were observed. Many missense and untranslated region variants were found, in addition to SNPs in the intronic areas of SPRY2, RAF1, IQSEC1, LMO7, and UCHL3. However, none of these variants differed compared to previously reported allelic frequencies or they were present in an untranslated region. In SPRY2, one missense Pro-Ser variant was found along with several SNPs in UTRs. In RAF1, one variant was found that was not previously reported, but it was in a UTR. In IQSEC1, only one missense Pro-Ser variant was found. In LMO7, three intronic and three missense variants were found. In UCHL3, three intronic splice site polymorphisms were found. With no new variant SNPs found, no separate control samples were run as the BLAST SNP program served as our control.
4. DISCUSSION
During the development of congenital idiopathic clubfoot, the musculoskeletal components of the lower extremity develop normally during the period of embryogenesis; however, between 12 and 32 weeks, as it has been observed by ultrasonography, the anatomy of the foot begin to deviate and leads to the deformity [32]. While congenital idiopathic clubfoot is usually described as a defect in the foot, the musculature between the knee and the ankle is affected to some degree, and most importantly, the size difference in the affected leg persists for the life of the patient [16,33]. Interestingly, relapses after correction significantly decreases after the child reaches 3 - 4 years of age [34]. This observations lead to the hypothesis that a genetic defect in muscle genes expressed early in life could be the cause of clubfoot.
The genes SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 selected for this study are involved in the embryonic development of muscle. They have been shown to regulate muscle formation through their respective affects on myotube formation, myoblast proliferation, and gene regulation. In addition, two of the genes (LMO7 and UCHL3), when mutated, have been shown to result in a type of muscular dystrophy. This involvement in early muscle development and their location in the linkage regions of chromosome 3 and 13 make them very appealing candidate genes for idiopathic clubfoot.
However, in this study, no mutations were observed that could explain the etiology of clubfoot. Of the many missense, UTR, and intronic variants found in the selected group of genes, none differed compared to the reported allelic frequencies. This showed that the genes SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 were unlikely to be directly involved with the development of congenital idiopathic clubfoot as the single major causative gene.
The limitations of this study were the relatively small sample size, intronic regions besides the splice-sites were not screened, and regulatory sequences for protein-DNA interactions were not investigated. Given that clubfeet of different syndromes appear and are treated similarly, it is possible that there are several distinct etiologies that combine to explain the development of congenital idiopathic clubfeet, in which case a large patient population would need to be screened in order to achieve an adequate representation of all possible pathology. However, based on our hypothesis that a single, major gene mutation is causing congenital idiopathic clubfoot, screening small cohorts is acceptable because the predicted mutation should be in the vast majority of patients screened. Also, our study is limited by only screening candidate genes selected from a linkage region specified by Dietz, et al. This region did not have a LOD score that exceeded 3, which is the standard score for linkage significance. However, we believed the linkage still deserved to be further investigated as it was the first and only genomewide linkage analysis conducted in clubfoot families, and the LOD score > 2 still encouraged investigation. Lastly, we also only screened muscle development genes. While other proposed causes, such as neurologic development, might also contribute to clubfoot development, these were not screened based on the hypothesis that clubfoot may be primarily a muscle disorder.
In summary, screening of the exons and splice sites of SPRY2, RAF1, IQSEC1, LMO7, and UCHL3 demonstrated no mutations associated with the phenotype of congenital idiopathic clubfoot. This result does not exclude the possibility that there exist genetic variants in the studied genes that affect clubfoot in a more complicated manner through protein-protein interactions or enhancer and suppressor sites. Defects in regulatory pathways or proteins that interact with these genes could act on them and lead to clubfoot. Thus, these muscle development genes would be an important, but not direct cause of clubfoot. Given the complexity of myogenesis, many regulatory candidate genes remain that could cause developmental defects in the hypaxial musculature that is invariably observed in clubfoot. Lastly, future research will focus on performing genome-wide analysis of the Omaha pedigree Figure 1 again with new technology that was not available to Dietz, et al. The family shows a strong genetic component that will increase the possibility of finding a potentially causative gene for the development of clubfoot.
5. ACKNOWLEDGEMENTS
The authors would like to thank the Carver College of Medicine Summer Research Program for their financial support. Dr. Val Sheffield is an investigator of the Howard Hughes Medical Institute.
REFERENCES
- Engell, V., Damborg, F., Andersen, M., Kyvik, K.O. and Thomsen, K. (2006) Club foot: A twin study. Journal of Bone & Joint Surgery, British Volume, 88, 374-376. doi:10.1302/0301-620X.88B3.16685
- Beals, R.K. (1978) Club foot in the Maori: A genetic study of 50 kindreds. The New Zealand Medical Journal, 88, 144-146.
- Rebbeck, T.R., Dietz, F.R., Murray, J.C. and Buetow, K.H. (1993) A single-gene explanation for the probability of having idiopathic talipes equinovarus. The American Journal of Human Genetics, 53, 1051-1063.
- Yang, H.Y., Chung, C.S. and Nemechek, R.W. (1987) A genetic analysis of clubfoot in Hawaii. Genetic Epidemiology, 4, 299-306. doi:10.1002/gepi.1370040408
- Chapman, C., Stott, N.S., Port, R.V. and Nicol, R.O. (2000) Genetics of club foot in Maori and Pacific people. Journal of Medical Genetics, 37, 680-683. doi:10.1136/jmg.37.9.680
- Gurnett, C.A., Alaee, F., Kruse, L.M., Desruisseau, D.M., Hecht, J.T., Wise, C.A., et al. (2008) Asymmetric lowerlimb malformations in individuals with homeobox PITX1 gene mutation. The American Journal of Human Genetics, 83, 616-622. doi:10.1016/j.ajhg.2008.10.004
- Wang, L.L., Fu, W.N., Li, L.J., Li, Z.G., Li, L.Y. and Sun, K.L. (2008) HOXD13 may play a role in idiopathic congenital clubfoot by regulating the expression of FHL1. Cytogenetic and Genome Research, 121, 189-195. doi:10.1159/000138884
- Shyy, W., Dietz, F., Dobbs, M.B., Sheffield, V.C. and Morcuende, J.A. (2009) Evaluation of CAND2 and WNT7a as candidate genes for congenital idiopathic clubfoot. Clinical Orthopaedics and Related Research, 467, 1201- 1205. doi:10.1007/s11999-008-0701-x
- Hecht, J.T., Ester, A., Scott, A., Wise, C.A., Iovannisci, D.M., Lammer, E.J., et al. (2007) NAT2 variation and idiopathic talipes equinovarus (clubfoot). American Journal of Medical Genetics A, 143A, 2285-2291. doi:10.1002/ajmg.a.31927
- Ester, A.R., Tyerman, G., Wise, C.A., Blanton, S.H. and Hecht, J.T. (2007) Apoptotic gene analysis in idiopathic Talipes equinovarus (clubfoot). Clinical Orthopaedics and Related Research, 462, 32-37. doi:10.1097/BLO.0b013e318073c2d9
- Liu, L.Y., Jin, C.L., Cao, D.H., Zhao, N., Lin, C.K. and Sun, K.L. (2007) Analysis of association between COL9A1 gene and idiopathic congenital Talipes equinovarus. Hereditas, 29, 427-432. doi:10.1360/yc-007-0427
- Dietz, F.R., Cole, W.G., Tosi, L.L., Carroll, N.C., Werner, R.D., Comstock, D., et al. (2005) A search for the gene(s) predisposing to idiopathic clubfoot. Clinical Genetics, 67, 361-362. doi:10.1111/j.1399-0004.2005.00407.x
- Ippolito, E., De Maio, F., Mancini, F., Bellini, D. and Orefice, A. (2009) Leg muscle atrophy in idiopathic congenital clubfoot: Is it primitive or acquired? Journal of Children’s Orthopaedics, 3, 171-178. doi:10.1007/s11832-009-0179-4
- Ippolito, E. and Ponseti, I.V. (1980) Congenital club foot in the human fetus. A histological study. Journal of Bone and Joint Surgery, 62, 8-22.
- Poon, R., Li, C. and Alman, B.A. (2009) Beta-catenin mediates soft tissue contracture in clubfoot. Clinical Orthopaedics and Related Research, 467, 1180-1185. doi:10.1007/s11999-008-0692-7
- Isaacs, H., Handelsman, J.E., Badenhorst, M. and Pickering, A. (1977) The muscles in club foot—A histological, histochemical and electron microscopic study. Journal of Bone & Joint Surgery, British Volume, 59-B, 465-472.
- Fukuhara, K., Schollmeier, G. and Uhthoff, H.K. (1994) The pathogenesis of club foot. A histomorphometric and immunohistochemical study of fetuses. Journal of Bone & Joint Surgery, British Volume, 76, 450-457.
- Handelsman, J.E. and Badalamente, M.A. (1981) Neuromuscular studies in clubfoot. Journal of Pediatric Orthopaedics, 1, 23-32. doi:10.1097/01241398-198101010-00004
- Aoki, T., Okada, N., Ishida, M., Yogosawa, S., Makino, Y. and Tamura, T.A. (1999) TIP120B: A novel TIP120- family protein that is expressed specifically in muscle tissues. Biochemical and Biophysical Research Communications, 261, 911-916. doi:10.1006/bbrc.1999.1147
- Gosztonyi, G., Dorfmüller-Küchlin, S., Sparmann, M. and Eisenschenk, A. (1989) Morphometric study of muscle in congenital idiopathic club foot. Pathology-Research and Practice, 185, 790-794.
- Martinsson, T., Oldfors, A., Darin, N., Berg, K., Tajsharghi, H., Kyllerman, M., et al. (2000) Autosomal dominant myopathy: Missense mutation (Glu-706 -> Lys) in the myosin heavy chain IIa gene. Proceedings of the National Academy of Sciences, 97, 14614-14619. doi:10.1073/pnas.250289597
- Veugelers, M., Bressan, M., McDermott, D.A., Weremowicz, S., Morton, C.C., Mabry, C.C., et al. (2007) Mutation of perinatal myosin heavy chain associated with a Carney complex variant. The New England Journal of Medicine, 351, 460-469. doi:10.1056/NEJMoa040584
- Toydemir, R.M., Rutherford, A., Whitby, F.G., Jorde, L.B., Carey, J.C. and Bamshad, M.J. (2006) Mutations in embryonic myosin heavy chain (MYH3) cause FreemanSheldon syndrome and Sheldon-Hall syndrome. Nature Genetics, 38, 561-565. doi:10.1038/ng1775
- Laziz, I., Armand, A.S., Pariset, C., Lecolle, S., Della Gaspera, B., Charbonnier, F., et al. (2007) Sprouty gene expression is regulated by nerve and FGF6 during regeneration of mouse muscles. Growth Factors, 25, 151-159. doi:10.1080/08977190701723166
- De Alvaro, C., Martinez, N., Rojas, J.M. and Lorenzo, M. (2005) Sprouty-2 overexpression in C2C12 cells confers myogenic differentiation properties in the presence of FGF2. Molecular Biology of the Cell, 16, 4454-4461. doi:10.1091/mbc.E05-05-0419
- Garcia, R., Grindlay, J., Rath, O., Fee, F. and Kolch, W. (2009) Regulation of human myoblast differentiation by PEBP4. EMBO Reports, 10, 278-284. doi:10.1038/embor.2009.4
- Gredinger, E., Gerber, A.N., Tamir, Y., Tapscott, S.J. and Bengal, E. (1998) Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. The Journal of Biological Chemistry, 273, 10436-10444. doi:10.1074/jbc.273.17.10436
- Pajcini, K.V., Pomerantz, J.H., Alkan, O., Doyonnas, R. and Blau, H.M. (2008) Myoblasts and macrophages share molecular components that contribute to cell-cell fusion. The Journal of Cell Biology, 180, 1005-1019. doi:10.1083/jcb.200707191
- Holaska, J.M., Rais-Bahrami, S. and Wilson, K.L. (2006) Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Human Molecular Genetics, 15, 3459-3472. doi:10.1093/hmg/ddl423
- Semenova, E., Wang, X., Jablonski, M.M., Levorese, J. and Tilghman, S.M. (2003) An engineered 800 kilobase deletion of Uchl3 and Lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Human Molecular Genetics, 12, 1301-1312. doi:10.1093/hmg/ddg140
- Sambrook, J., Fritsch, F. and Maniatis, T. (1989) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, New York.
- Keret, D., Ezra, E., Lokiec, F., Hayek, S., Segev, E. and Wientroub, S. (2002) Efficacy of prenatal ultrasonography in confirmed club foot. Journal of Bone & Joint Surgery, British Volume, 84, 1015-1019. doi:10.1302/0301-620X.84B7.12689
- Cooper, D.M. and Dietz, F.R. (1995) Treatment of congenital idiopathic clubfoot. A thirty-year follow-up note. Journal of Bone and Joint Surgery, 77, 1477-1489.
- Morcuende, J.A., Dolan, L.A., Dietz, F.R. and Ponseti, I.V. (2004) Radical reduction in the rate of extensive corrective surgery for clubfoot using the Ponseti method. Pediatrics, 113, 376-380. doi:10.1542/peds.113.2.376