Open Journal of Molecular and Integrative Physiology
Vol.05 No.01(2015), Article ID:53743,16 pages
10.4236/ojmip.2015.51001
Kinetic Characterization of Na,K-ATPase Inhibition by the Acetaminophen Metabolite N-Acetylbenzoquinoneimine
Jeff B. Helms1,2*, Lauren P. Saunders2, Jamie Meyer2, Charles J. Costa2, Eric Plowman2, Nick Williford1, Matthew Corbitt1, Jeremy P. Holden3, Craig Gatto2
1Department of Biology, Upper Iowa University, Fayette, IA, USA
2Division of Neuroscience and Physiology, Department of Biological Science, Illinois State University, Normal, IL, USA
3Department of Internal Medicine, University of Wisconsin-Madison Medical Center, Madison, WI, USA
Email: *jbhelms@ilstu.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 11 January 2015; accepted 26 January 2015; published 3 February 2015
ABSTRACT
N-acetylbenzoquinoneimine (NABQI) is a toxic metabolite of the common analgesic acetaminophen (APAP). NABQI is an electrophilic intermediate formed via the oxidation of APAP within the cytochrome P450 system. Within the normally recommended low-dose use of APAP, NABQI is a minor metabolite which is either quickly reduced back to APAP or conjugated to Glutathione (GSH) producing an innocuous by-product. However, with overdose or prolonged high-dose usage of ace- taminophen, GSH levels can become depleted and the bioactive NABQI is thought to form adducts with proteins and oxidize protein sulfhydryls producing intra- and intermolecular disulfide brid- ges in proteins. In this work we investigated the effect of NABQI on purified kidney Na,K-ATPase to see if the clinical renal insufficiencies seen in APAP overdose may be linked to inhibition of the Na,K-ATPase. Our work has shown that NABQI does indeed inhibit the Na,K-ATPase in a dose dependent (IC50 = 19.8 ± 2.9 µM) and irreversible manner. Interestingly, brief storage of NABQI at −20˚C eliminates the irreversible effects of the compound, and leads to a product that remains a potent reversible inhibitor of the Na,K-ATPase (IC50 = 58.7 ± 19.5 µM). Further, the reversible inhibition produced by stored NABQI competes with para-nitrop.
Keywords:
Na,K-ATPase, Paracetamol, N-Acetylbenzoquinoneimine, Overdose, Renal, Electrophillic

1. Introduction
Acetaminophen (APAP) is a common and widely used mild analgesic and antipyretic drug. Long-term chronic use of APAP, as well as high single dosages have been shown to be hepatotoxic and may lead to renal insufficiency or failure even in the absence of hepatic failure [1] [2] . APAP in therapeutic doses is metabolized primarily by glucuronidation and sulfation (30% and 55% respectively) [3] [4] , however potentially toxic metabolic products also result in [1] . One such metabolic product is N-acetylbenzoquinoneimine (NABQI). NABQI is formed by the oxidation of APAP (see Figure 1 for structures) by the cytochrome P450 system [5] . It is generally accepted that NABQI is the electrophilic intermediate responsible for the toxicity of APAP [6] [7] . At normal therapeutic doses this minor metabolite is quickly reduced in the presence of reduced glutathione (GSH) back to APAP or covalently linked to GSH to form a non-toxic conjugate [8] . However, with high doses of APAP available GSH is rapidly depleted and bioactive NABQI has been shown to form adducts with proteins which can subsequently lead to the formation of intra- and intermolecular disulfide bridges in proteins [9] . It has further been shown that toxic doses of APAP both in vivo and in situ lead to bioactivation and toxicity consistent with NABQI formation [10] [11] . Hoivik et al. [10] [11] demonstrate that following NABQI formation, there is a preferential arylation of a 58 kDa protein which was initially thought to mediate cellular changes that lead to the toxicity of NABQI. More recent research has clearly shown that within the liver NABQI formation followed by protein modification is an initiating event which propagates and ultimately leads to mitochondrial dysfunction and ultimately hepatic cell death (for a review see [12] . Moreover, the electrophilicity of NABQI makes it susceptible to nucleophilic attack by many different proteins, which could ultimately lead to a disruption of cellular homeostasis when critical proteins become modified and intensive efforts were made to identify those critical proteins [9] [12] -[14] . However, with a few exceptions, such as glutamine synthetase and carbamyl pho- sphate synthetase-1 for example, enzyme activity of the covalently bound proteins was only inhibited to a small extent so it would appear that these modifications were not the direct cause of necrotic cell death in APAP induced hepatocyte death. In a minority of APAP overdose situations (1% - 2%) there is renal insufficiency and potentially renal failure and this has been shown to occur even in the absence of liver toxiciaty [2] [15] . While the APAP induced liver necrosis has been comprehensively studied the mechanisms of renal insufficiency and nephrotoxicicity remain less clear [15] [16] . Histopathological study of nephrotoxicity has shown damage to the proximal tubule, which is an area that relies on the function of Na,K-ATPase to drive transport in the kidney [15] .
The Na,K-ATPase (sodium pump) is a ubiquitous transmembrane protein found in the plasma membrane of animal cells which plays a key role in maintaining ionic homeostasis. The minimum functional unit of the sodium pump appears to be a heterodimer of 1 alpha and 1 beta subunit (for a review see [17] ). The alpha subunit is a protein of approximately 110 kDa with 10 membrane spanning domains with both N- and C-termini located cytoplasmically. The large cytoplasmic loop between transmembrane regions M4 and M5 contains the amino acids necessary for nucleotide binding and the aspartic acid residue which is phosphorylated during the catalytic

Figure 1. Chemical structures of acetaminophen (top left) and N-acetylebenzoquinoneimine (NABQI). NABQI is the bioactive, toxic metabolite created by the oxidation of acetaminophen in the cytochrome P450 system.
cycle [18] . The beta subunit is a single pass transmembrane protein with the N-terminus located cytoplasmically and the C-terminus extracellularly. The beta subunit is glycosylated at three conserved sites and contains three disulfide bridges. The beta subunit is thought to play a role in trafficking of the alpha-beta dimer to the plasma membrane [19] -[22] and may also serve a regulatory role in K+-coordination [22] -[24] .
The catalytic cycle of the sodium pump is described in a model first described by Post and Albers (Figure 2), in which the pump alternates between two major conformations designated E1 and E2 [25] -[27] . In the E1 conformation MgATP binds to the M4M5 loop while 3 Na+ ions bind to the pump from the cytosol. The pump is then phosphorylated (E1-P) leading to Na+ occlusion (the ions are not accessible from either the cytoplasmic or extracellular space) and ADP is released. A conformational change then occurs, leading to the release of Na+ to the extracellular space (E2-P) and subsequent loading of two extracellular K+ ions. Upon binding of K+ the pump is de-phosphorylated (E2) and K+ ions are occluded; incidentally, the enzyme in this form is known to also hydrolyze other phosphate compounds, such as p-nitrophenylphosphate and 3-O-methylfluorescein phosphate [28] . A subsequent conformational change takes place releasing K+ into the cytoplasm and returning the pump to the E1 state.
Given the critical homeostatic role of the Na,K-ATPase, inhibition of this enzyme by APAP and its bioactive metabolites has been investigated [29] -[34] . In this report, we extend these investigations and demonstrate for the first time that APAP itself does not inhibit the Na,K-ATPase (up to 5 mM APAP). Rather, the APAP metabolite, NABQI is a potent irreversible inhibitor of the purified Na,K-ATPase. Interestingly, storing NABQI solutions at −20˚C eliminates the irreversible characteristics of the drug, but NABQI nonetheless remains a reversible inhibitor of the purified Na,K-ATPase. A preliminary report of these findings was presented to the annual meeting of the Biophysical Society [35] .
2. Methods
2.1. Reagents
Ammonium molybdate, acetaminophen, hydrochloric acid, sodium phosphate, potassium chloride, sucrose, oua- bain, ascorbic acid, EDTA, Folin’s reagent, imidazole, magnesium chloride, Na2ATP, sodium bicarbonate, sodium chloride, p-nitrophenyl phosphate, dimethylsulfoxide (DMSO) and Trizma base were from Sigma-Aldrich (St. Louis, MO). Ethanal, p-benzoquinone and acetamide were from Thermo Fischer Scientific (Waltham, MA).

Figure 2. Post/Albers model of the catalytic cycle of the sodium pump. The E1 form of the sodium pump binds ATP and 3 Na+ from the cytoplasm. The pump is then phosphorylated with the release of ADP and moves to the E1-P form. Conformational change opens the transport site with Na+ bound to the extracellular space and Na+ is then released (E2-P). Two K+ bind to the transport site from the extracellular space followed by conformational change leading to the occlusion of the K+ ions. It is in this K+ occluded E2-K state that K+ mediated phosphatase activity takes place. NABQI competes with pNPP for phosphates activity and the generalized location of such inhibition is shown. Following release of K+ to the cytoplasm the pump undergoes a conformational change back to the E1 form of the pump and is ready for the next catalytic cycle.
NABQI was from Toronto Research Chemicals (Toronto, ONT Canada). Frozen sheep whole kidneys were purchased from Pel-Freez (Rogers, AR).
2.2. Na,K-ATPase Enzyme Purification from Ovine Kidney
Na,K-ATPase was purified from sheep kidney as described by Jorgensen [36] . Briefly, the outer medulla was dissected from three sheep kidneys and then homogenized in a blender in a medium containing: 250 mM sucrose, 25 mM imidazole, and 1 mM EDTA (pH = 7.2). The homogenate was filtered through four layers of cheesecloth and the filtrate centrifuged at 10,000 × g, 15 min. The supernatant liquid was collected and again centrifuged at 10,000 × g, 15 min. The supernatant liquid was collected from the second spin and the microsomal fraction sedimented at 65,000 × g, 1 hour. The pellets were collected and resuspended in the above homogenizing medium with the addition of 3 mM ATP to a protein concentration of 6 mg/ml and this microsomal preparation was incubated for 10 min at room temperature. Following the incubation, the microsomal preparation was diluted to a final concentration of 2 mg/ml with 0.4% SDS (w/w in ATP-containing homogenizing medium) and incubated an additional 10 min at room temperature. The detergent-treated microsomes were layered on top of a three-step sucrose gradient (all densities prepared in homogenizing medium) of 15%, 25%, and 45% and centrifuged in a swinging bucket rotor (Beckman SW-28) at 100,000 × g, 2.5 hours. The membrane fractions collected at the 25% - 45% interface were used for these experiments. Protein concentration was determined by the method of Lowry et al. [37] .
2.3. Na,K-ATPase Activity
Ouabain-sensitive Na,K-ATPase activity was measured using a previously described colorimetric assay that detects inorganic phosphate [38] . For all ATPase experiments 1 µg of Na,K-ATPase protein was incubated with the appropriate assay for 15 minutes at 37˚C.
For NABQI ATPase IC50 experiments, Na pump ATPase activity was assayed in the presence of varying NABQI ranging from 1 - 1000 µM NABQI or in the absence of NABQI (as indicated in figures). Final reaction volumes were brought up to 600 µl by the addition of 500 µl complete assay solution containing (in mM): 60 Imidazole, 0.6 EGTA, 156 NaCl, 24 KCl, 3.6 of both MgCl2 and ATP with 10 NaN3 as a preservative (pH 7.2).
For reversibility experiments, 100 µl of Na,K-ATPase at a concentration of 1 mg/ml was incubated at 37˚C for 15 minutes with either 50 µM final NABQI (200 µM for 72 hour NABQI), p-benzoquinone or Tris-HCl buffer. Following incubation the enzyme was diluted to a final concentration of 24 µg/ml in a two-step dilution using 50 mM Tris-HCl pH 7.2. ATPase reactions were run using 25 µl of the 24 µg/ml enzyme leaving the final concentration of free NABQI in the reaction at approximately 50 nM (200 nM for 72 hrs NABQI).
The p-benzoquinone time course experiments were conducted using 3, 7, 32 and 48 µM concentrations of p-benzoquinone incubated with 1 mg/ml of Na,K-ATPase protein. At times indicated (Figure 14) aliquots from the reaction were diluted 41-fold and Na,K-ATPase activity was then determined. The data were then plotted as a single exponential.
In each case, the Na,K-ATPase activity was determined as ouabain-sensitive ATP hydrolysis. The ATPase reaction was stopped by addition of 1 mL of an ice-cold stop solution (540 mM HCl, 1.29 g ascorbic acid, 2.2 ml of a freshly prepared 10% Ammonium molybdate solution). Samples were incubated with the stop solution for 10 minutes on ice. After the 10 minutes, 1.5 mL of ACG solution (150 mM Na-m-arsenite, 70 mM Na-citrate, 350 mM acetic acid), was added to each sample to intensify the color change. Samples were then incubated for 5 minutes at 37˚C followed by 5 minutes at room temperature. A 300 µl volume of each tube was transferred to a 96-well plate (Fisher, St. Louis, MO) and the absorbance at 850 nm was recorded using a Versa Max micro-plate reader (Molecular Devices Corporation, Sunnyvale, California).
2.4. Para-Nitrophenylphosphatase Activity
All pNPPase assays were conducted essentially as described in Drapeau & Blostein [39] with the exception that liberated PO4, rather than pNP, was measured to eliminate complications due to the yellow color of NABQI. In all pNPPase experiments, a 5 µg quantity of purified Na,K-ATPase was used. In brief, the assay buffer contained 50 mM MOPS (pH 7.4 with Tris), 3 mM MgCl2, 2 mM KCl, with a final concentration of 5 mM di-trispNPP. For IC50 experiments, the concentration of NABQI ran from 31.3 µM to 1 mM in a six-step dilution series. For reversibility experiments 200 µl solutions of 0.6 mg/ml Na,K-ATPase in 50 mM Tris pH 7.4 were made either with or without the inhibitor (50 µM NABQI). These solutions were incubated at room temperature for 30 minutes. Following the incubation the volume of the tube was brought up to 1 ml with 50 mM Tris. To determine the effect of pre-incubation with NABQI, 25 µl of the diluted solutions (5 µg/ml final Na,K-ATPase concentration) were then used for pNPPase assays as previously described. In order to investigate whether or not there was a differential effect on pNPPase activity following NABQI storage, the pNPPase experiments were conducted with both freshly prepared and 72 hour stored NABQI, and final concentrations of either 50 µM final NABQI or 200 µM for 72 hour stored NABQI. Following incubation with NABQI, the enzyme was diluted to a final concentration of 24 µg/ml in a two-step dilution using 50 mM Tris-HCl (pH 7.2). pNPPase reactions were run using 25 µL of the 24 µg/ml enzyme leaving the final concentration of free NABQI in the reaction at approximately 50 nM (200 nM for 72 hrs NABQI). For pNPP competition experiments NABQI was held constant at 60 µM while the pNPP concentration was varied from 312 µM to 10 mM. Each reaction tube was incubated at 37˚C for 15 minutes. Reactions were then stopped, developed and measured using the same stopping and ACG solutions as with the ATPase assays described above.
3. Results
The focus of this investigation was to determine whether NABQI interacts directly with purified renal Na,K- ATPase. Since it has been reported that both APAP [1] [2] [5] and its reactive metabolite NABQI [6] [7] may affect Na,K-ATPase activity in situ, our investigation focused on elucidating the direct nature of these effects in un-sided preparations of Na,K-ATPase (i.e., both extracellular and cytoplasmic surfaces are freely accessible). To determine the effect of APAP on the pump, ouabain-sensitive ATPase activity was measured in the presence of increasing concentrations of the drug. Figure 3 shows that even with concentrations up to 5 mM, acetaminophen does not inhibit the Na,K pump. In contrast, NABQI inhibits Na,K-ATPase activity in a dose-dependent manner, with an apparent IC50 of 58.7 ± 19.49 µM, i.e., at least four orders of magnitude more potent than APAP (Figure 4).
Next, we wanted to determine if NABQI inhibition of Na,K-ATPase was a matter of permanent modification as had been reported in the literature [30] [31] [40] . Initially inhibition by NABQI appeared reversible, rather than the result of permanent pump modification (Figure 5) in contrast to in vivo and in situ experiments reported by other laboratories [30] [31] . Further investigations revealed that freshly prepared NABQI dramatically reduc- ed the apparent IC50 value nearly three-fold to 19.8 ± 2.87 µM (Figure 6). Thus, we wanted to check whether

Figure 3. The effect of increasing concentrations of Acetaminophen (APAP) on sodium pump ATPase activity. No significant difference in the activity of the sodium pump, as measured by ATP hydrolysis (○) is demonstrated at concentrations of APAP up to 5 mM. Activity from three different experiments were normalized to the activity in the absence of APAP.

Figure 4. The effect of increasing concentrations of NABQI on sodium pump ATPase activity. NABQI inhibits the sodium pump in a dose dependent manner. NABQI, which had been stored for 48 - 72 hours is a potent inhibitor of sodium pump ATPase activity. IC50 = 58 ± 19.4 µM. Activity is normalized to the activity in the absence of inhibitor from three separate experiments. Points represent means ± standard error.

Figure 5. Na,K-ATPase activity following pre-treatment with 200 µM NABQI A solution containing 1 mg/ml Na,K-ATPase was pre-treated with either 200 µM NABQI which had been stored for 24 hrs at −20˚C (dark bar) or 60 µl of buffer (light gray). Following incubation for 30 min. at RT the solutions were diluted to 24 µg/ml and ATPase assays conducted. The pre-treated sodium pump (activity 0.97 ± 0.029) demonstrated no significant difference (t = 0.07, df 6, p 0.47) in ATPase activity as compared to the control (activity 1.0 ± 0.04). Data are normalized duplicate values from three separate experiments.

Figure 6. Dose-dependence of NABQI inhibition of sodium pump ATPase activity. NABQI inhibits the sodium pump in a dose dependent manner. Freshly prepared NABQI is approximately 3 times more potent an inhibitor of sodium pump ATPase activity (IC50 = 19.8 ± 2.87 µM) than stored NABQI. Activity is normalized to the activity in the absence of inhibitor from three separate experiments. Points represent means ± standard error.
storage of NABQI in solution altered its inhibitory potency; indeed, the same NABQI solutions after 48 - 72 hours of storage at −20˚C had apparent IC50 values similar to our original findings (Figure 4).
As the strength of NABQI is affected by solution age, the role of solution age on the reversibility of inhibition was measured. We found that freshly prepared NABQI permanently inactivated the Na,K-ATPase (Figure 7), consistent with previous reports (Figure 7) [30] [31] [40] .
3.1. Reversible Component of NABQI ATPase Inhibition
Although freshly prepared NABQI irreversibly inactivated the Na,K-ATPase, stored stocks of NABQI produced a reversible inhibition. This change in inhibition type must mirror a chemical change in the solution-phase NABQI, and indicate that NABQI is not stable in aqueous solution. The properties of this bioactive breakdown product were measured in both an old NABQI solution as well as from fresh solutions of NABQI breakdown products.
In an old NABQI solution, we determined the mode of reversible inhibition of NABQI on the Na,K-ATPase reaction cycle with various substrates to elucidate the enzyme conformation that interacts with NABQI. In order to determine if NABQI preferentially bound to the E2P conformation, we measured the [K+] dose-dependence of NABQI inhibition. NABQI inhibits [K+]-activated ATPase activity in an uncompetitive manner with a Ki of 65.2 ± 14.8 µM (Figure 8). NABQI also demonstrates mixed type inhibition with ATP for sodium pump ATPase activity (Figure 9). Since there was no competition with K+, we decided not to test Na+ competition as it seems unlikely to compete with the cation-site inside facing conformation.
3.2. NABQI Inhibition of pNPP Phosphatase Activity
It has been known for several decades that the Na,K-ATPase, like many P-type ATPases, possesses a non- transporting phosphatase activity [39] [41] [42] . In the Na,K-ATPase this activity has been characterized by measuring K+-dependent para-nitrophenylphosphate (pNPP) hydrolysis [39] [41] . Since NABQI is similar in structure to pNPP we determined whether NABQI was an equally potent inhibitor of K+-dependent pNPPase activity. Figure 10 demonstrates that NABQI, whether freshly made or stored, inhibited the pNPPase activity of

Figure 7. Na,K-ATPase activity following pre-treatment with 200 µM NABQI. A solution containing 1 mg/ml Na,K-ATPase was pre-treated with either 20 µM freshly prepared NABQI (dark bar) or buffer alone (light gray). Following incubation for 30 min. at room temperature the solutions were diluted to 24 µg/ml and ATPase assays conducted. Activity for the freshly prepared NABQI (0.16 ± 0.01) was significantly lower (t = 6.02, df = 8, p = 0.0003) than the activity for the control (1.0 ± 0.14) demonstrating that freshly prepared NABQI is a potent irreversible inhibitor of the sodium pump.

Figure 8. Dose-dependence of [K+] on sodium pump inhibition by NABQI. NABQI does not compete with K+ for sodium pump ATPase activity. ATPase activity was measured in the presence (○) of 30 µM NABQI or in the absence (□) of NABQI. NABQI uncompetitively inhibits ATPase activity with a Ki of 65.2 ± 14.8 µM. Points represent means ± standard error. Data shown are normalized from three different experiments and were fit to the Michaelis-Menten and uncompetitive inhibition equations.

Figure 9. Dose-dependence of [ATP] on NABQI inhibition. NABQI demonstrates mixed type inhibition with ATP for sodium pump ATPase activity. ATPase activity was measured in the presence of increasing concentrations of ATP (up to 1 mM) in the presence of no inhibitor (○), 30 µM NABQI (□) or 100 µM NABQI (◊). Changes to both Km and Vmax where shown in the presence of NABQI. For the control Vmax = 1.07 ± 0.07 with a Km of 0.19 ± 0.04 µM. In the presence of 30 µM NABQI Vmax dropped to 0.55 ± 0.06 while Km decreased to 0.10 ± 0.06 µM. In the presence of 100 µM NABQI Vmax −0.36 ± 0.06 while Km decreased to 0.07 ± 0.05 µM. Data for three experiments were normalized to activity in maximum activity in the absence of inhibitor. Data points represent mean ± standard error.

Figure 10. Dose-dependence of [NABQI] on pNPPase activity. The K+ dependent phosphatase activity of the sodium pump was measured with freshly prepared NABQI (○), 1 day stored (□) or two day stored (◊). Since NABQI solutions are yellow in color liberated Pi rather than pNP were measured. As was the case for ATPase activity freshly prepared NABQI was a more potent inhibitor (IC50 = 168.33 ± 46.95 µM) as compared to either NABQI stored for one day (IC50 = 446.59 ± 64.99 µM) or two day stored NABQI (IC50 = 421.69 ± 62.0 µM). At concentrations up to 1 mM NABQI did not fully inhibit sodium pump pNPPase activity. Each point represents mean ± standard error from three different experiments.
the Na,K-ATPase in a dose-dependent manner. Not surprisingly, freshly prepared NABQI is approximately 2.5-fold more potent than stored solutions of NABQI, presumably due to the irreversible component associated with freshly prepared NABQI (IC50 168.33 ± 46.95 µM versus 446.59 ± 64.99 µM for NABQI which had been stored for 1 day and 421.69 ± 62.03 µM for NABQI stored 2 days).
Since NABQI was indeed able to inhibit pNPPase activity, we wanted to also investigate if inhibition was the result of permanent modification, as had been observed with freshly prepared NABQI in this study and others (Figure 7) [30] [31] [40] . Figure 11 clearly shows that, as with ATPase activity, freshly prepared NABQI irreversibly inhibited pNPPase activity, whereas stored NABQI (72 hours, −20˚C) was clearly a reversible inhibitor.
Given the similar structures of NABQI and pNPP, we measured the pNPP concentration dependence of activity in the presence and absence of NABQI (Figure 12). NABQI competitively inhibits pNPP activity with a Ki of 29.4 ± 3.19 µM.

Figure 11. Reversibility of freshly prepared and stored NABQI on sodium pump pNPPase activity. As with ATPase activity we investigated whether or not the inhibitory effect of NABQI was reversible. (a) As observed with ATPase, freshly prepared NABQI (dark gray bar) inhibited the sodium pump in an irreversible manner. Pre-treatment with NABQI lead to slightly less than a 50% reduction in activity (0.93 ± 0.02 control versus 0.50 ± 0.03 pre-treated). The difference in activity was significant (t = 4.5, df 24, p ≤ 0.0001); (b) However, when the sodium pump was pre-treated with NABQI which had been stored at −20˚C for up to 72 hrs there was no significant difference (t = 0.26, df = 22, p = 0.39) in the activity between the pre-treated (dark gray―Vmax 1.00 ± 0.045) and the control (light gray―Vmax 1.06 ± 0.044). This result demonstrated that the stored NABQI while still a potent inhibitor of the sodium pump did so in a reversible manner.

Figure 12. The effect of increasing concentrations of pNPP on NABQI inhibition of sodium pump pNPPase activity. pNPPase activity was measured in the absence of NABQI (□) or presence of 60 µM NABQI (○) with increasing concentrations of pNPP up to 10 mM. (a) Fitting the data to the Michaelis-Menten and competitive inhibition equations demonstrates that NABQI competes with pNPP with a Ki of 29.4 ± 3.19 µM; (b) To better illustrate that both conditions produced the same Vmax while the presence of NABQI increased the Km a reciprocal plot (1/V plotted against 1/[pNPP]) was then done. Data points represent mean ± standard error.
3.3. Inhibition of Na,K-ATPase by Potential NABQI Breakdown Products
It has been proposed that NABQI breaks down into p-benzoquinone and acetamide [43] so these compounds were tested to determine if they contribute to the Na,K-ATPase inhibition observed with NABQI. Acetamide does not appear to affect Na,K-ATPase activity at concentrations up to 100 mM (data not shown). The reversibility of inhibition by p-benzoquinone was tested as for NABQI, and p-benzoquinone also irreversibly inhibited Na,K-ATPase. Time courses of p-benzoquinone inactivation were determined by measuring the loss of ATPase activity over the course of p-benzoquinone exposure (Figure 14(a)). Consequently, it appears that at least some (if not all) of the irreversible inhibition observed with NABQI may be due to its further conversion to p-benzo- quinone.
4. Discussion
A large body of work exists showing that APAP in large single doses may result in hepatotoxicity or renal toxicity in both animals [10] [29] [31] [33] [34] [44] -[46] and humans [2] [6] [30] [32] [46] [47] . There is also general agreement that one of the key metabolites responsible for much of the APAP toxicity is NABQI [1] [6] [48] -[50] . Oxidation of APAP to the reactive metabolite NABQI occurs via the cytochrome P450 system in both liver and kidney (for review see [49] ). At high APAP dosages the kidney cytochrome P450 system is a critical pathway in NABQI formation leading to renal toxicity [1] [11] . For instance, Trumper et al. [46] demonstrated that there was no difference in the renal damage caused by APAP in bile-canulated and non-canulated rats, thus indicating that the renal toxicity was not dependent on hepatic bioactivation of APAP. Additionally, Hoivik et al. [11] demonstrated in mouse kidney slices that APAP bioactivation lead to both formation of a 58-kDa acetaminophen binding protein, as well as, cellular depletion of ATP and potassium indicative of NABQI toxicity. Interestingly, high APAP exposure in tissue slices showed a continued increase in protein arylation for the first 12 hours of exposure; in contrast in vivo maximum arylation is usually complete in the first few hours [10] . These differences in binding were attributed to in vivo clearing of the APAP from blood and tissue. Given our in vitro observations, it is likely that at least some Na,K-ATPase inhibition observed after hours of exposure to APAP is due to a mixture of both irreversible arylation and direct reversible inhibition via NABQI (or a further breakdown product).
The Na,K-ATPase is a critical enzyme in both hepatic and renal cells for maintaining low intracellular Na+ levels, which establishes the driving force for many secondary active transport systems. In particular, it has been speculated that increased cellular Na+ caused by APAP-induced Na,K-ATPase damage may increase cytosollic Ca2+ and leads to irreversible cell damage; [51] increased cytosollic Ca2+ presumably results from reverse mode Na/Ca exchange in the plasma membrane [52] . Subsequent studies in rat liver plasma membrane preparations demonstrated that 3 hours after exposure to toxic levels of APAP, Na,K-ATPase activity decreased 52% compared to non-treated control animals [29] . Similarly, Tukel [32] reported a decrease in activity of the erythrocyte Na,K-ATPase with a maximal effect 2-3 hours after APAP overdose resulting in an approximate 50% reduction in Na,K-ATPase activity as compared to the control. However, neither study investigated whether the mode of action of APAP, or its metabolic by-products, was directly on Na,K-ATPase or indirect via other cellular processes.
In our current study, we directly compared whether APAP or the APAP metabolite NABQI was responsible for the Na,K-ATPase inhibition reported in vivo. Additionally, we performed inhibition experiments directly on purified Na,K-ATPase protein from mammalian kidney (ovine) to eliminate other possible cellular components from contributing to our observations. In this study, APAP was found to have no direct effect on Na,K-ATPase activity even at extremely high concentrations of up to 5 mM (Figure 3). This finding is consistent with previous work showing bioactivation is required for the hepatotoxic and nephrotoxic effects of APAP overdose. Although, Gu et al. [52] did show a sustained mild APAP toxicity in kidney tissue from hepatic P450 knock-out mice; these observations do not rule out an effect of extrahepatic P450 bioactivation. Given that APAP is completely innocuous at extremely high doses (Figure 3) our data also supports earlier reports that APAP needs to be bioactivated either by hepatic or extrahepatic mechanisms to become toxic.
Na,K-ATPase Inhibition by the Acetaminophen Metabolite, NABQI
In these experiments it was found that freshly prepared NABQI inhibited the Na,K-ATPase in a dose-dependent manner (Figure 6 and Figure 10). Further, our data indicate that fresh NABQI inhibits both ATPase and pNPPase activities of the enzyme irreversibly (Figure 7 and Figure 11(a)). The three most likely explanations for irreversible inhibition are: 1) formation of an adduct between NABQI and the Na,K-ATPase alpha subunit, 2) NABQI catalytically activating the formation of intra- or intermolecular disulfide cross-bridges within the Na,K- ATPase alpha subunit, or 3) NABQI is further metabolized to a compound which produces irreversible inhibition. Although modification of the beta subunit cannot be ruled out, the six predominately accessible cysteine residues in the beta subunit endogenously exist as three disulfide bridges. Our finding that NABQI irreversibly inhibits the Na,K-ATPase, may explain the observations by Chibalin et al. [53] where NABQI inhibition of the Na,K pump was correlated with the appearance of internalized Na,K pump subunits in endosomes. That is, conjugated, non-functional pumps would be targeted for removal and degradation via the ubiquitin-proteosome pathway [54] [55] .
On the other hand, after brief storage of NABQI at −20˚C, there was no significant difference observed in either Na,K-ATPase or pNPPase activities with pre-treatment with up to 200 µM NABQI (Figure 5 and Figure 11(b)). This result indicates that NABQI had undergone a change which renders it incapable of covalently attaching to the protein. Acetamide and p-benzoquinone have been suggested as possible breakdown products of NABQI (Figure 13); [43] however, as p-benzoquinone is also an irreversible inhibitor (Figure 14), it cannot be the reversible breakdown product observed in older NABQI solutions. Para-benzoquinone has been used to attach Na,K-ATPase to ELISA plates [56] and thus does clearly react with the Na,K-ATPase and here we show that this interaction irreversibly inhibits the enzyme (Figure 14).

Figure 13. The hydrolysis of NABQI to acetamide and p-benzoquinone. Koymans et al. (1989) proposed that NABQI may be hydrolyzed to acetamide and p-benzoquinone. Both of these have shown to be either inhibitory in high concentration (acetamide) or to bind to the sodium pump (p-benzoquinone).

Figure 14. Irreversible inhibition of Na,K-ATPase by p-benzoquinone. (a) Time courses of p-benzoquinone inactivation of Na,K-ATPase. Na,K-ATPase (1 mg/mL) was incubated with p-benzoquinone. At times indicated on the x-axis aliquots were removed and diluted 41-fold and the amount of functioning Na,K-ATPase was then quantified via ouabain-sensitive ATPase activity. Concentrations of p-benzoquinone were 3 (○), 7 (□), 32 (◊), and 48 (Δ) µM. Time courses are fit to single exponentials; (b) Rate of p-benzoquinone inactivation of Na,K-ATPase. Rates from (a) were linear with respect to [PBQ] and result in an inactivation rate of 0.0056 ± 0.0007 min−1.
However, the NABQI breakdown product that mediates reversible Na,K-ATPase inhibition remains elusive, but may explain the lack of APAP-protein adducts in kidney tissue seen by Gu et al. [52] . In this work we demonstrated that half-maximal inhibition was achieved at approximately 20 µM with freshly prepared NABQI, which is in agreement with Nicotera et al. [30] . In experiments with human erythrocytes Nicotera et al. [30] demonstrated a half-maximal inhibition of the Na pump with NABQI concentrations between 20 and 36 µM. Interestingly, even at high concentrations (200 µM) they never observed more than 20% inhibition. In our work, a 200 µM concentration of NABQI inhibited Na,K-ATPase by nearly 65%. Consequently, our results fit more closely with the observations of Corcoran et al. [29] and Tukel [32] in rat and human erythrocytes, respectively showing Na,K-ATPase inhibition of greater than 50% in APAP overdose situations. When investigating the dose-dependent inhibition of “stored” NABQI we found that this compound had an IC50 value of approximately 60 µM compared to 20 µM for the freshly prepared NABQI.
We now understand that the increased potency of freshly prepared NABQI is likely due to the irreversible component of inhibition. Therefore, we sought to determine if the irreversibility might be caused by the further breakdown into either acetamide or p-benzoquinone (or both) (Figure 13). We observed no inactivation of Na,K-ATPase by acetamide (up to 100 mm, data not shown). In contrast, p-benzoquinone was a potent irreversible inhibitor of Na,K-ATPase. In order to determine whether p-benzyoquinone was specifically targeting a certain site on the Na,K-ATPase, we measured the rate of enzyme inactivation at several p-benzoquinone concentrations (Figure 14(a)). The time courses were exponential, as expected for an irreversible inhibitor. When the observed inactivation rates for p-benzoquinone were plotted against p-benzoquinone concentration there was a linear dependence (Figure 14(b)), consistent with one-step, irreversible inhibition according to the scheme:
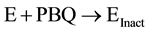
where E is the Na,K-ATPase, PBQ is p-benzoquinone, and Einact is the damaged inactivated Na,K-ATPase. The lack of an equilibrium preceding the irreversible step implies that the inhibition is non-specific and may suggest that this metabolite can modify other proteins in addition to the Na,K-ATPase. The rate of inactivation is 0.0056 ± 0.0007 min−1. The y-intercept corresponds to the reverse rate of reaction and is approximately zero, consistent with irreversible inhibition.
For completeness, we also investigated the reversible inhibition observed with stored solutions of NABQI. A likely explanation for reversible inhibition is that the reactive breakdown product p-benzoquinone has a finite half-life that eliminates its reactivity and the resultant compound (possibly the di-enolate) has reversible inhibitory properties. The reversible inhibition observed is not competitive with either ATP or K+ for ATPase activity. Therefore, the inhibitor does not bind to either the E2P or the E1 conformations of Na,K-ATPase. Interestingly, the reversible NABQI metabolite does compete with pNPP in the K+-stimulated phosphatase activity of the pump (Figure 12). While the identity of the molecular intermediates involved in Na,K-ATPase phosphatase activity have not been completely elucidated, it has been shown that phosphatase activity occurs after K+ and pNPP bind intracellularly [39] [41] . Thus, given the competition between NABQI and pNPP, our work suggests that the reversible inhibitory effects of NABQI may take place on the cytoplasmic face of the enzyme. These findings may extrapolate to imply that in vivo NABQI crosses the plasma membrane. However, we cannot rule out the possibility of adduct formation on the extracellular surface which prevents critical cytoplasmic conformational changes.
5. Conclusion
Acetaminophen has been the most commonly used over the counter pain reliever and fever reducer in the United States for the past 50 years, with nearly a quarter of the adult population using the drug in a given week [57] . APAP is safe drug when used as directed (i.e., <4000 mg/day). However, prolonged use at higher concentrations can cause liver and kidney damage. In this study, we directly tested the toxicity of the predominant metabolite of APAP, NABQI on Na,K-ATPase function, an enzyme that plays a critical role in the transport processes of liver and kidney. We observed that NABQI is a potent inhibitor of the Na,K-ATPase, with both reversible and irreversible components. The subsequent NABQI metabolites (i.e., p-benzoquinone and acetamide) likely contribute to the complexities involved in the inhibition of this enzyme. The irreversible inhibition by NABQI observed is consistent with protein arylation. Such arylation has been proposed for various mitochondrial proteins and the Ca-ATPase in cell death within the liver (or a review see [12] ) and our kinetic characterization of NABQI on renal Na,K-ATPase demonstrates that this may contribute to the pathophysiology of renal toxicicty via APAP overdose. Given the ubiquitous reliance of renal function on proper Na,K-ATPase operation, these results provide a plausible molecular mechanism for the damaging effects of excessive APAP use and why in some instances patients suffer from renal insufficiency in the absence of hepatic failure.
Acknowledgements
The research presented within this paper was funded by NIH grant GM061583 to Craig Gatto and AHA Predoctoral Fellowship 0315236 to Jeff B Helms.
References
- Bessems, J.G. and Vermeulen N.P. (2001) Paracetamol (Acetaminophen)-Induced Toxicity: Molecular and Biochemical Mechanisms, Analogues and Protective Approaches. Critical Reviews in Toxicology, 31, 55-138. http://dx.doi.org/10.1080/20014091111677
- Eguia, L. and Materson, B.J. (1997) Acetaminophen-Related Acute Renal Failure without Fulminant Liver Failure. Pharmacotherapy, 17, 363-370.
- Prescott, L.F. (1980) Kinetics and Metabolism of Paracetamol and Phenacetin. British Journal of Clinical Pharmacology, 10, 291S-298S. http://dx.doi.org/10.1111/j.1365-2125.1980.tb01812.x
- Tone, Y., Kawamata, K., Murakami, T., Higashi, J. and Yata, N. (1990) Dose-Dependent Pharmacokinetics and First- Pass Metabolism of Acetaminophen in Rats. Journal of Pharmacobio-Dynamics, 13, 327-335. http://dx.doi.org/10.1248/bpb1978.13.327
- Vermeulen, N.P., Bessems, J.G. and Van de Stratt, R. (1992) Molecular Aspects of Paracetamol-Induced Hepatotoxicity and Its Mechanism-Based Prevention. Drug Metabolism Reviews, 24, 367-407. http://dx.doi.org/10.3109/03602539208996298
- Albano, E., Rundgren, M., Harvison, P.J., Nelson, S.D. and Moldeus, P. (1985) Mechanisms of N-Acetyl-p-benzoqui- nonimine Cytotoxicity. Molecular Pharmacology, 28, 306-311.
- Van de Stratt, R., Vromans, R.M., Bosman, P., De Vries, J. and Vermeulen, N.P. (1988) Cytochrome P-450-Mediated Oxidation of Substrates by Electron-Transfer; Role of Oxygen Radicals and of 1- and 2-Electron Oxidation of Parace- tamol. Chemico-Biological Interactions, 64, 267-280. http://dx.doi.org/10.1016/0009-2797(88)90102-0
- Van de Stratt, R., De Vries, J., Kulkens, T., Debets, A.J. and Vermeulen, N.P. (1986) Paracetamol, 3-Monoalkyl- and 3,5-Dialkyl Derivatives. Comparison of Their Microsomal Cytochrome P-450 Dependent Oxidation and Toxicity in Freshly Isolated Hepatocytes. Biochemical Pharmacology, 35, 3693-3699.
- Nelson, S.D. (1995) Mechanisms of the Formation and Disposition of Reactive Metabolites That Can Cause Acute Liver Injury. Drug Metabolism Reviews, 27, 147-177. http://dx.doi.org/10.3109/03602539509029821
- Hoivik, D.J., Manautou, J.E., Tverr, A., Mankowski, D.C., Khairallah, D.A. and Cohen, S.D. (1996) Evidence Suggesting the 58-kDa Acetaminophen Binding Protein Is a Preferential Target for Acetaminophen Electrophile. Fundamental and Applied Toxicology, 32, 79-86. http://dx.doi.org/10.1093/toxsci/32.1.79
- Hoivik, D.J., Fisher, R.L., Brendel, K., Gandolfi, A.J., Khairallah, E.A. and Cohen, S.D. (1996) Protein Arylation Precedes Acetaminophen Toxicity in a Dynamic Organ Slice culture of Mouse Kidney. Fundamental and Applied Toxicology, 34, 99-104. http://dx.doi.org/10.1006/faat.1996.0180
- Jaeschke, H. and Bajt, M.L. (2005) Review: Intracellular Signaling Mechanisms of Acetaminophen-Induced Liver Cell Death. Toxicology Sciences, 89, 31-41. http://dx.doi.org/10.1093/toxsci/kfi336
- Hinson, J.A., Roberts, D.W., Halmes, N.C., Gibson, J.D. and Pumford, N.R. (1996) Immunochemical Detection of Drug-Protein Adducts in Acetaminophen Hepatotoxicity. Biological Reactive Intermediates V, 387, 47-55. http://dx.doi.org/10.1007/978-1-4757-9480-9_7
- Holtzman, J.L. (1995) The Role of Covalent Binding to Microsomal Proteins in the Hepatotoxicity of Acetaminophen. Drug Metabolism Reviews, 27, 277-297. http://dx.doi.org/10.3109/03602539509029827
- Mazer, M. and Perrone, J. (2008) Acetaminophen-Induced Nephrotoxicity: Pathophysiology, Clinical Manifestations, and Management. Journal of Medical Toxicology, 4, 2-6. http://dx.doi.org/10.1007/BF03160941
- Mour, G., Feinfeld, D.A., Caraccia, T. and McGuigan, M. (2005) Acute Renal Dysfunction in Acetaminophen Poisoning. Renal Failure, 27, 381-383.
- Kaplan, J.H. (2002) Biochemistry of Na,K-ATPase. Annual Review of Biochemistry, 71, 511-535. http://dx.doi.org/10.1146/annurev.biochem.71.102201.141218
- Gatto, C., Wang, A.X. and Kaplan, J.H. (1998) The M4M5 Cytoplasmic Loop of the Na,K-ATPase, Overexpressed in Escherichia coli, Binds Nucleoside Triphosphates with the Same Selectivity as the Intact Native Protein. Journal of Biological Chemistry, 273, 10578-10585. http://dx.doi.org/10.1074/jbc.273.17.10578
- Gatto, C., McLoud, S.M. and Kaplan, J.H. (2001) Heterologous Expression of Na+-K+-ATPase in Insect Cells: Intracellular Distribution of Pump Subunits. American Journal of Physiology―Cell Physiology, 281, C982-C992.
- Geering, K. (1991) The Functional Role of the Beta-Subunit in the Maturation and Intracellular Transport of Na,K- ATPase. FEBS Letters, 285, 189-193.
- Geering, K., Beggah, A., Good, P., Girardet, S., Roy, S., Schaer, D. and Jaunin, P. (1996) Oligomerization and Maturation of Na,K-ATPase: Functional Interaction of the Cytoplasmic NH2 Terminus of the Beta Subunit with the Alpha Subunit. Journal of Cell Biology, 133, 1193-1204. http://dx.doi.org/10.1083/jcb.133.6.1193
- Geering, K. (2001) The Functional Role of β Subunits in Oligomeric P-Type ATPases. Journal of Bioenergetics and Biomembranes, 33, 425-438. http://dx.doi.org/10.1023/A:1010623724749
- Geering, K. (2008) Functional Roles of Na,K-ATPase Subunits. Current Opinion in Nephrology and Hypertension, 17, 526-532. http://dx.doi.org/10.1097/MNH.0b013e3283036cbf
- Shinoda, T., Ogawa, H., Cornelius, F. and Toyoshima, C. (2009) Crystal Structure of the Sodium-Potassium Pump at 2.4 Å Resolution. Nature, 459, 446-450. http://dx.doi.org/10.1038/nature07939
- Albers, R.W., Fahn, S. and Koval, G.J. (1963) The Role of Sodium Ions in the Activation of Electrophorus Electric Organ Adenosine Triphosphatase. Proceedings of the National Academy of Sciences of the United States of America, 50, 474-481. http://dx.doi.org/10.1073/pnas.50.3.474
- Albers, R.W., Koval, G.J. and Siegel, G.J. (1968) Studies on the Interaction of Ouabain and Other Cardio-Active Steroids with Sodium-Potassium-Activated Adenosine Triphosphatase. Molecular Pharmacology, 4, 324-336.
- Post, R.L., Hegyvary, C. and Kume, S. (1972) Activation by Adenosine Triphosphate in the Phosphorylation Kinetics of Sodium and Potassium ion Transport Adenosine Triphosphatase. Journal of Biological Chemistry, 247, 6530-6540.
- Huang, W. and Askari, A. (1975) (Na+, K+)-Activated Adenosinetriphosphatase: Fluorimetric Determination of the Associated K+-Dependent 3-O-Methylfluorescein Phosphatase and Its Use for the Assay of Enzyme Samples with Low Activities. Analytical Biochemistry, 66, 265-271. http://dx.doi.org/10.1016/0003-2697(75)90745-9
- Corcoran, G.B., Chung, S.J. and Salazar, D.E. (1987) Early Inhibition of the Na+/K+-ATPase Ion Pump during Acetaminophen-Induced Hepatotoxicity in Rat. Biochemical and Biophysical Research Communications, 149, 203-207. http://dx.doi.org/10.1016/0006-291X(87)91624-X
- Nicotera, P., Hinds, T.R., Nelson, S.D. and Vincenzi, F.F. (1990) Differential Effects of Arylating and Oxidizing Analogs of N-Acetyl-p-Benzoquinoneimine on Red Blood Cell Membrane Proteins. Archives of Biochemistry and Biophysics, 283, 200-205. http://dx.doi.org/10.1016/0003-9861(90)90631-8
- Smolarek, T.A., Higgins, C.V. and Amacher, D.E. (1990) Metabolism and Cytotoxicity of Acetaminophen in Hepatocyte Cultures from Rat, Rabbit, Dog, and Monkey. Drug Metabolism and Disposition, 18, 659-663.
- Tukel, S.S. (1995) Effects of Acetaminophen on Methemoglobin, Superoxide Dismutase and Na+-K+ ATPase Activities of Human Erythrocytes. Biochemistry & Molecular Biology International, 35, 719-724.
- Trumper, L., Coux, G. and Elias, M.M. (2000) Effect of Acetaminophen on Na+, K+ ATPase and Alkaline Phosphatase on Plasma Membranes of Renal Proximal Tubules. Toxicology and Applied Pharmacology, 164, 143-148. http://dx.doi.org/10.1006/taap.2000.8889
- Trumper, L., Coux, G., Monasterolo, L.A., Molinas, S., Garcia, V.M. and Elias, M.M. (2005) Effect of Acetaminophen on the Membrane Anchoring of Na+, K+ATPase of Rat Renal Cortical Cells. Biochimica et Biophysica Acta (BBA) ―Molecular Basis of Disease, 1740, 332-339. http://dx.doi.org/10.1016/j.bbadis.2004.09.011
- Helms, J.B., Meyer, J., Costa, C.J., Plowman, E., Holden, J.P. and Gatto, C. (2007) Inhibition of the Na,K-ATPase by the Acetaminophen Metabolite N-Acetylbenzoquinoneimine. Biophysical Journal, 145A.
- Jorgensen, P.L. (1974) Purification and Characterization of (Na+ Plus K+)-ATPase. IV. Estimation of the Purity and of the Molecular Weight and Polypeptide Content Per Enzyme Unit in Preparations from the Outer Medulla of Rabbit Kidney. Biochimica et Biophysica Acta (BBA)―Molecular Basis of Disease, 356, 53-67. http://dx.doi.org/10.1016/0005-2736(74)90293-4
- Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J. (1951) Protein Measurement with the Folin Phenol Reagent. Journal of Biological Chemistry, 193, 265-275.
- Brotherus, J. (1981) Membrane Transport of Sodium and Potassium. Duodecim, 97, 1560-1570.
- Drapeau, P. and Blostein, R. (1980) Interactions of K+ with (Na,K)-ATPase Orientation of K+-Phosphatase Sites Studied with Inside-Out Red Cell Membrane Vesicles. Journal of Biological Chemistry, 255, 7827-7834.
- Josephy, P.D. (2005) The Molecular Toxicology of Acetaminophen. Drug Metabolism Reviews, 37, 581-594. http://dx.doi.org/10.1080/03602530500205200
- Gatto, C., Arnett, K.L. and Milanick, M.A. (2007) Divalent Cation Interactions with Na,K-ATPase Cytoplasmic Cation Sites: Implications for the Para-Nitrophenyl Phosphatase Reaction Mechanism. Journal of Membrane Biology, 216, 49-59. http://dx.doi.org/10.1007/s00232-007-9028-x
- Robinson, J.D., Levine, G.M. and Robinson, L.J. (1983) A Model for the Reaction Pathways of the K+-Dependent Phosphatase Activity of the (Na+ + K+)-Dependent ATPase. Biochimica et Biophysica Acta (BBA)―Molecular Basis of Disease, 731, 406-414. http://dx.doi.org/10.1016/0005-2736(83)90035-4
- Koymans, L., van Lenthe, J.H., Van de, S.R., Donne-Op den Kelder, G.M. and Vermeulen, N.P. (1989) A Theoretical Study on the Metabolic Activation of Paracetamol by Cytochrome P-450: Indications for a Uniform Oxidation Mechanism. Chemical Research in Toxicology, 2, 60-66. http://dx.doi.org/10.1021/tx00007a011
- Trumper, L., Monasterolo, L.A., Ochoa, E. and Elias, M.M. (1995) Tubular Effects of Acetaminophen in the Isolated Perfused Rat Kidney. Archives of Toxicology, 69, 248-252. http://dx.doi.org/10.1007/s002040050166
- Trumper, L., Girardi, G. and Elias, M.M. (1992) Acetaminophen Nephrotoxicity in Male Wistar Rats. Archives of Toxicology, 66, 107-111. http://dx.doi.org/10.1007/BF02342503
- Trumper, L., Monasterolo, L.A. and Elias, M.M. (1996) Nephrotoxicity of Acetaminophen in Male Wistar Rats: Role of Hepatically Derived Metabolites. Journal of Pharmacology and Experimental Therapeutics, 279, 548-554.
- Nicotera, P., Rundgren, M., Porubek, D.J., Cotgreave, I., Moldeus, P., Orrenius, S. and Nelson, S.D. (1989) On the Role of Ca2+ in the Toxicity of Alkylating and Oxidizing Quinone Imines in Isolated Hepatocytes. Chemical Research in Toxicology, 2, 46-50. http://dx.doi.org/10.1021/tx00007a008
- Bessems, J.G., Gaisser, H.D., TeKoppele, J.M., Van Bennekom, W.P., Commandeur, J.N. and Vermeulen, N.P. (1995) 3,5-Disubstituted Analogues of Paracetamol. Synthesis, Analgesic Activity and Cytotoxicity. Chemico-Biological Interactions, 98, 237-250. http://dx.doi.org/10.1016/0009-2797(95)03649-0
- Bessems, J.G., TeKoppele, J.M., Van Dijk, P.A., Van Stee, L.L., Commandeur, J.N. and Vermeulen N.P. (1996) Rat Liver Microsomal Cytochrome P450-Dependent Oxidation of 3,5-Disubstituted Analogues of Paracetamol. Xenobiotica, 26, 647-666.
- Gu, J., Cui, H., Behr, M., Zhang, L., Zhang, Q.Y., Yang, W., Hinson, J.A. and Ding, X. (2005) In Vivo Mechanisms of Tissue-Selective Drug Toxicity: Effects of Liver-Specific Knockout of the NADPH-Cytochrome P450 Reductase Gene on Acetaminophen Toxicity in Kidney, Lung, and Nasal Mucosa. Molecular Pharmacology, 67, 623-630. http://dx.doi.org/10.1124/mol.104.007898
- Trump, B.F. and Berezesky, I.K. (1985) The Role of Calcium in Cell Injury and Repair: A Hypothesis. Survey and Synthesis of Pathology Research, 4, 248-256.
- Schanne, F.A. and Moore, L. (1986) Liver Plasma Membrane Calcium Transport. Evidence for a Na+-Dependent Ca2+ Flux. Journal of Biological Chemistry, 261, 9886-9889.
- Chibalin, A.V., Katz, A.I., Berggren, P.O. and Bertorello, A.M. (1997) Receptor-Mediated Inhibition of Renal Na+-K+-ATPase Is Associated with Endocytosis of Its Alpha- and Beta-Subunits. American Journal of Physiology, 273, C1458-C1465.
- Coppi, M.V. and Guidotti, G. (1997) Ubiquitination of Na,K-ATPase Alpha1 and Alpha2 Subunits. FEBS Letters, 405, 281-284. http://dx.doi.org/10.1016/S0014-5793(97)00182-8
- Wilkinson, K.D. (2000) Ubiquitination and Deubiquitination: Targeting of Proteins for Degradation by the Proteasome. Seminars in Cell & Developmental Biology, 11, 141-148. http://dx.doi.org/10.1006/scdb.2000.0164
- Urayama, O., Nagamune, H., Nakao, M., Hara, Y., Sugiyama, H., Sato, K. and Nakao, T. (1985) Isolation and Characterization of a Monoclonal Antibody against Pig Kidney Sodium- and Potassium-Activated ATPase. Biochemical Journal, 98, 209-217.
- Kaufman, D.W., Kelly, J.P., Rosenberg, L., Anderson, T.E. and Mitchell, A.A. (2002) Recent Patterns of Medication Use in the Ambulatory Adult Population of the United States. The Journal of the American Medical Association, 287, 337-344. http://dx.doi.org/10.1001/jama.287.3.337
NOTES

*Corresponding author.