Open Journal of Molecular and Integrative Physiology
Vol.4 No.2(2014), Article ID:45972,9 pages DOI:10.4236/ojmip.2014.42002
Mitochondrial Dysregulation in Skeletal Muscle from Patients Diagnosed with Alzheimer’s Disease and Sporadic Inclusion Body Myositis
Elika Shabrokh, John Kavanaugh, Ryan McMillan, Josh Pittman, Matt Hulver, Madlyn Frisard*
Department of Human Nutrition, Foods, and Exercise, Virginia Tech, Blacksburg, USA
Email: *frisardm@vt.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 5 March 2014; revised 5 April 2014; accepted 12 April 2014
ABSTRACT
Mitochondrial dysfunction is implicated in Alzheimer’s disease (AD) and disruption of mitochondrial dynamic pathways has been documented in brains from patients diagnosed with AD; although it is unclear whether other tissues are also affected. Much less is known about the mitochondria in patients diagnosed with sporadic Inclusion Body Myositis (sIBM). The current study examined mitochondrial biology in skeletal muscle from AD and sIBM patients compared to healthy, elderly individuals. Skeletal muscle samples were obtained from the National Disease Research Interchange and mRNA, protein content, and enzyme activity was used to assess mitochondrial parameters. Patients diagnosed with AD or sIBM demonstrated reduced mitofusin 2 and optic atrophy protein 1 protein. AD patients also displayed increased mRNA of superoxide dismutase 2, catalase, and uncoupling protein 3. Amyloid ( precursor protein mRNA was higher in sIBM patients only compared to both AD patients and elderly individuals. Both total and phosphorylated AMPK protein content, an upstream regulator of mitochondrial dynamics and biogenesis, were also reduced in sIBM patients. The current study demonstrates a disruption in signaling pathways regulating mitochondrial dynamics in both AD and sIBM patients, although the underlying causes may differ.
Keywords:Mitochondrial Dynamics, Autophagy, Mitochondrial Biogenesis, Amyloid Beta Precursor Protein, 5’-AMP-Activated Protein Kinase

1. Introduction
Mitochondria are dynamic organelles that play a pivotal role in cellular function, not only as a major site of ATP production, but also as an organelle regulating energy metabolism, protein turnover, cellular proliferation, and apoptosis [1] [2] . Not surprisingly, defects in this organelle can have a profound impact on cellular function and are implicated in the development of metabolic and neurological disease [3] -[5] . Mitochondrial function is dependent on a number of factors including mitochondrial biogenesis, mitochondrial autophagy, and dynamics [6] [7] . Mitochondrial dynamics is a concept that includes mitochondrial movement within the cell and mitochondrial interactions controlled by fusion/fission events [8] . The importance of these events has recently become evident with the identification of genes responsible for fusion (mitofusin 1 and 2, optic atrophy protein 1) and fission (dynamin Related Protein 1 and fission 1) [9] -[12] . Pharmacologic and/or genetic manipulation of these genes can result in gross mitochondrial abnormalities including, altered substrate metabolism, reduced oxygen consumption, a decline in ATP synthesis, and mtDNA nucleotide dyshomeostasis [8] . Furthermore, impairment in mitochondrial function is detrimental to myofiber health and can result in fiber death and muscle atrophy [2] .
Disruption of mitochondrial dynamic pathways has been documented in the brains of Alzheimer’s patients and occurred in conjunction with mitochondrial network fragmentation, increased reactive oxygen species (ROS) production, and accumulation of pathological protein fragments including amyloid β and tau [13] -[16] , although it is not known if other tissues are affected. Much less is known about the role of the mitochondria in the development of sporadic Inclusion Body Myositis (sIBM), however, mtDNA dyshomeostas along with structural and functional abnormalities have been observed in patients diagnosed with the disease [17] -[19] . Furthermore, Boncompagni, et al. [20] observed structural and functional alterations in mitochondria from MCK-βAPP mice, which over express amyloid b precursor protein (AβPP) in skeletal muscle and display characteristics of the disease. Importantly, these alterations are observed preceding the reported appearance of histopathological and clinical features and may represent a key early event in disease pathology [17] [20] [21] .
Both AD and sIBM are characterized by the accumulation of protein fragments such as amyloid β (Aβ), a peptide fragment processed from the AβPP, in the brain and skeletal muscle, respectively [22] [23] . In fact, the presence of Aβ in both diseases originally suggested a common pathology. However, while the role of Aβ in the development of AD has been established, whether Aβ accumulation in skeletal muscle from sIBM patients is a cause or secondary side effect is not known [24] -[26] .
Recent work has demonstrated that AβPP can interact with both the outer and inner mitochondrial membrane import channels and prevent import of de novo synthesized nuclear-encoded mitochondrial proteins [27] -[29] . Furthermore, intramitochondrial amyloid β may directly impair mitochondrial function by disrupting mitochondrial dynamic pathways [16] [30] . This provides a potential mechanism for the observed impairments in mitochondrial function and suggests a role for mitochondrial dynamic pathways in the development of sIBM. The purpose of the current paper was to examine pathways regulating mitochondrial fusion and fission as well as regulators of mitochondrial biogenesis and autophagy in skeletal muscle from patients diagnosed with AD and sIBM compared with healthy, elderly individuals.
2. Methods
Human Samples. Human skeletal muscle samples were obtained from the National Register Disease Interchange (New York, New York). Samples were obtained from patients diagnosed with AD (n = 5; age = 79 ± 3; 2 males and 3 females), sIBM (n = 2; age = 69 ± 4; all males), and elderly controls with no evidence of muscle disease (n = 6; age = 66 ± 4; all males). Samples were collected from the vastus lateralis muscle within 12 hours post mortem. Samples were shipped overnight on dry ice and processed for assessment of mRNA, protein, and enzyme activity.
Gene Expression. RNA was extracted using an RNeasy Mini Kit (Qiagen) and DNase I treatment (Qiagen, Valencia, CA), according to the manufacturer’s instructions. qRT-PCR was performed using an ABI 7900 Sequence Detection System instrument and Taq Man Universal PCR Master Mix according to the manufacturer’s specifications (Applied Biosystems, Foster City, CA) and as previously described [31] . mRNA of uncoupling protein three (UCP3), manganese superoxide dismutase (SOD2), catalase, beclin, peroxisome proliferator-activated receptor gamma coactivator 1-alpha, myosin heavy chain 7B, myosin heavy chain 2, and myosin heavy chain 4 were assessed. Primers and 5# FAM-labeled TaqMan probes were purchased as prevalidated assays (Applied Biosystems, Foster City, CA). Relative quantification of target genes was calculated using the 2−ΔCT method. Derivation of the 2−ΔCT equation has been described in Applied Biosystems User Bulletin No. 2 (P/N 4303859). Target gene expression was normalized against GAPDH.
Protein Content. Western analysis was performed as previously described using cell lysates harvested in Mammalian Cell Lysis Buffer (Sigma Aldrich) [31] . Proteins (30 μg) were separated using a 10% CriterionTris·HCl gel (Bio-Rad, Hercules, CA) and subsequently transferred to a polyvinylidene difluoride membrane (Bio-Rad). Blots were probed with primary antibodies against GAPDH (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), peroxisome proliferator-activated receptor (PPARα) (1:1000; Abcam, Cambridge, MA), mitofusin 2 (1:1000; Cell Signaling, Danvers, MA), optic atrophy protein 1 (1:1000; Abcam, Cambridge, MA), dynamin related protein 1 (1:1000; Abcam, Cambridge, MA) phosphorylated 5’-AMP-activated protein kinase (AMPK) and total AMPK (1:1000; both Abcam, Cambridge, MA) followed by anti-rabbit, mouse, or goat secondary antibodies (1:10000; Jackson Immuno Research Laboratories, West Grove, PA). Proteins were visualized using Super-Signal Chemiluminescent Substrate (Pierce, Rockville, IL) and a ChemiDoc XRS Imaging System (Bio-Rad). Protein content was normalized to GAPDH and phosphorylated AMPK was adjusted for total AMPK and normalized to GAPDH.
Enzyme Activity. Enzyme activities were assessed in muscle homogenates (20-fold dilution). Sample buffer consisted of 0.1 mol/l KH2PO4/Na2PHO4 and 2 mmol/l EDTA, pH 7.2. Phosphofructokinase (PFK), citrate synthase (CS), malate dehydrogenase (MDH) and beta hydroxyacyl-CoAdehydrogenase (β-HAD) activities were determined spectrophotometrically as previously described [31] [32] .
Citrate synthase catalyzes the formation of citrate and coenzyme A (CoASH) from acetyl-CoA and oxaloactetate. CoASH reduces DTNB and CS activity was determined from the reduction of DTMB over time. Ten microliters of a 1:5 diluted muscle homogenate was added, in triplicate, to 170 μl of a solution containing Tris buffer (0.1 M, pH 8.3), DNTB (1 mM, in 0.1 M in Tris buffer) and oxaloacetate (0.01 M, in 0.1 M Tris buffer). Following a 2-minute background reading, the spectrophotometer (SPECTRAmax ME, Molecular Devices Corporation, Sunnyvale California) was calibrated and 30 μl of 3 mM acetyl CoA was added to initiate the reaction. Absorbance was measured at 405 nm at 37˚C every 12 seconds for 7 minutes. Maximum CS activity was calculated and reported as μmol/min/mg.
For the determination of β-hydroxyacyl-CoA dehydrogenase, oxidation of NADH to NAD was measured. In triplicate, 35 μl of whole muscle homogenate was added to 190 μl of a buffer containing 0.1 M liquid triethanolamine, 5 mM EDTA tetrasodium salt dihydrate, and 0.45 mM NADH. The spectrophotometer (SPECTRAmax PLUS 384, Molecular Devices Corporation, Sunnyvale California) was calibrated and 15 μl of 2 mM acetoacetyl CoA was added to initiate the reaction. Absorbance was measured at 340 nm every 12 seconds for 6 minutes at 37C. Maximum BHAD activity was calculated and reported as μmol/min/mg.
Malate dehydrogenase reversibly catalyzes the oxidation of malate to oxaloacetate using the reduction of NAD+ to NADH. The rate of the disappearance of NADH was measured spectrophotometrically at 340 nm at 37˚C. Briefly, 10ul of sample were pipetted in triplicate in wells. Then, 290 ul of reaction media (0.1 M potassium phosphate buffer, pH 7.4 plus 0.006 M oxaloacetic acid, prepared in potassium phosphate buffer plus 0.00375 M NADH, prepared in potassium phosphate buffer) was added to the wells and samples were read for 5 minutes at 340 nm. The rate of disappearance of NADH was analyzed and expressed relative to protein content. Data is expressed as means ± S.E.M.
Phosphofructokinase phosphorylates fructose-6-phosphate to fructose-1,6-bisphosphate, and is a key regulatory step in glycolysis. Phosphofructokinase activity was measured spectrophotometrically at 340 nm at 37˚C. Briefly, 30 ul of sample homogenate were pipetted in triplicate. Assay buffer (12 mM MgCl2 , 400 mM KCL, 2 mM AMP, 1 mM ATP, 0.17 mM NADH, 0.0025 mg/mL, Antimycin 0.05 mg/mL Aldolase 0.05 mg/mL GAPDH, in 100 mM Tris buffer, pH = 8.2) was then added to each well. After a 2-minute background reading, 3 mM fructose-6-phosphate was added to each sample well and followed by a 7-minute kinetic reading. Changes in absorbance across time were recorded and expressed relative to protein content. Data is expressed as means ± S.E.M.
Statistical Analysis. Results were analyzed with non-parametric Kruskal-Wallis one-way analysis of variance analysis. Comparisons between groups were assessed using a Mann-Whitney-Wilcoxon (MWW) test. The level of significance was set at P < 0.05.
3. Results
Transcriptional regulation in skeletal muscle from patients diagnosed with sIBM and Alzheimer’s disease. mRNA analysis revealed that AβPP was significantly higher in patients diagnosed with sIBM compared to AD patients and controls (Figure 1(A)). Despite this, mRNA of UCP3 and the antioxidants catalase and SOD2 were all significantly higher in AD patients compared to elderly controls (Figures 1(B)-(D)). While there were no significant differences in UCP3 concentrations between sIBM patients and controls, there were trends for higher and lower catalase and SOD2 mRNA, respectively. Beclin, a regulator of autophagy, was significantly higher in the AD patients with a trend for higher mRNA observed in sIBM patients compared to controls (Figure 1(E)). Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) expression, a regulator of mitochondrial biogenesis, was significantly higher in both sIBM and AD compared to controls (Figure 2(A)). To determine whether changes in expression patterns were associated with differences in skeletal muscle fiber type,
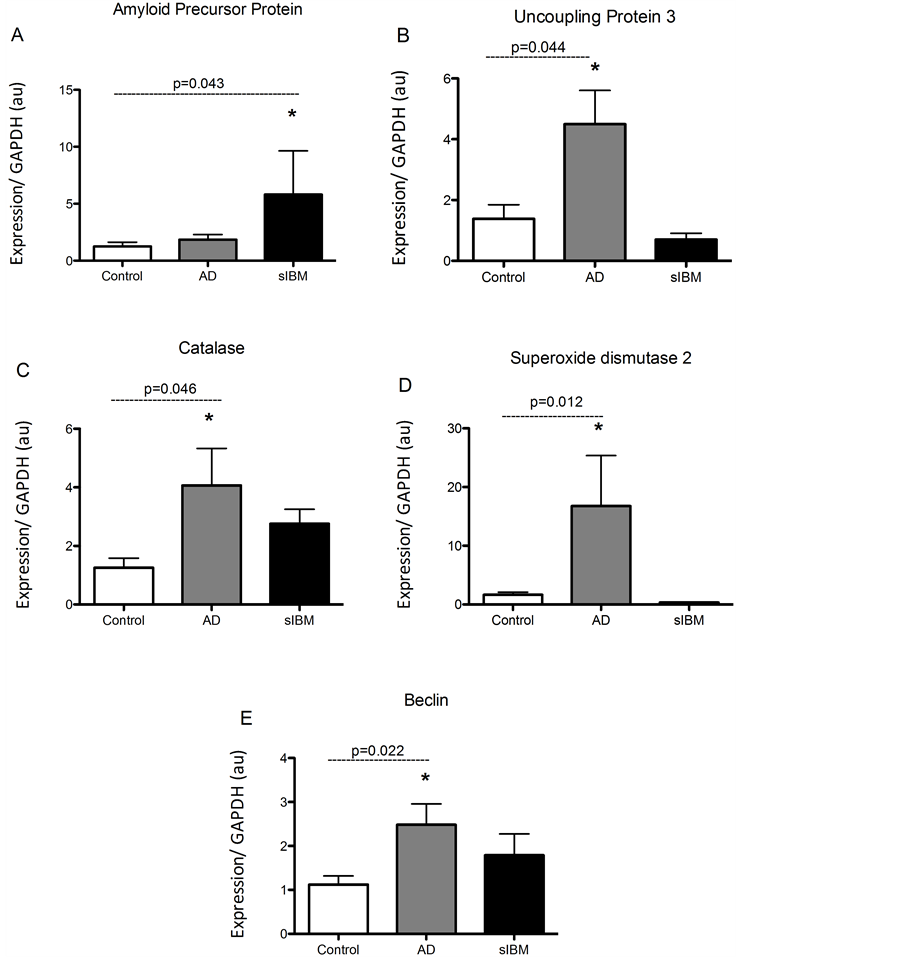
Figure 1. Transcriptional regulation in skeletal muscle from patients diagnosed with AD and sIBM. Relative mRNA content of amyloid precursor protein (APP) (A), uncoupling protein 3 (UCP3) (B), catalase (C), manganese superoxide dismutase (SOD2) (D), and beclin (E) in skeletal muscle from patients diagnosed with AD (n = 5), sIBM (n = 2), and elderly individuals (n = 6). Data are presented as means ± S.E.M. and normalized to GAPDH mRNA, *P < 0.05.

Figure 2. Transcriptional regulation in skeletal muscle from patients diagnosed with AD and sIBM. Relative mRNA content of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a) (A), myosin heavy chain 7B (MHC7B) (B), myosin heavy chain 2 (MHC2) (C), and myosin heavy chain 4 (MHC4) (D) in skeletal muscle from patients diagnosed with AD (n = 5), sIBM (n = 2) for PGC1a, MHC7B, and MHC2 and n = 1 for MHC4), and elderly individuals (n = 6). Data are presented as means ± S.E.M. and normalized to GAPDH mRNA. *P < 0.05, compared to controls.
mRNA of myosin heavy chain 7b, 2, and 4, markers of type 1, type 2A, and type 2B, respectively, were assessed (Figure 2(B)-(D)). There were no differences in expression of genes responsible for fiber type suggesting that observed differences may not be due to differences in fiber type. We were however only able to measure MHC4 in one sIBM sample, and therefore only limited conclusions can be made.
Protein content in skeletal muscle from patients diagnosed with sIBM and Alzheimer’s disease. While there were no significant differences in PGC1a protein content, MFN2, OPA1, and DRP1 were all significantly lower in both patient populations compared to elderly controls (Figure 3(B)-(D)). Additionally, both phosphorylated and total 5’-AMP-activated protein kinase (AMPK) protein content was significantly lower in sIBM patients compared to both AD and controls, respectively (Figure 3(E)).
Enzyme activity in skeletal muscle from patients diagnosed with sIBM and AD. Due to lack of sample, we were only able to measure enzyme activity in one patient diagnosed with sIBM, although we were able to measure enzyme activity in all of the AD and control samples. While there were no significant differences in enzyme activity of phosphofructokinase, malate dehydrogenase, or citrate synthase (Figure 4(A), Figure 4(B) and Figure 4(D)), there was a trend for reduced activity of beta hydroxyacyl CoA dehydrogenase activity, the primary regulator of beta-oxidation in AD patients compared to elderly controls (Figure 4(C)).
4. Discussion
The current study supports previous findings and provides a potential mechanism of mitochondrial dysfunction by demonstrating reduced protein content of MFN2 and OPA1 in patients diagnosed with AD and sIBM [33] . Additionally, there was a similar decline in DRP1 protein in sIBM patients, although it did not reach statistical significance in AD patients. Mitochondrial dynamics, the repetitive cycles of fusion and fission, have been recognized as a critical process in the maintenance of mitochondrial homeostasis. These opposing processes determine the architecture of the entire mitochondrial population of the cell with fission events segregating dysfunctional mitochondria from the network to be processed for autophagy and fusion events allowing for equilibration of matrix metabolites and membrane components [7] [34] [35] . A down regulation of the primary regulators of
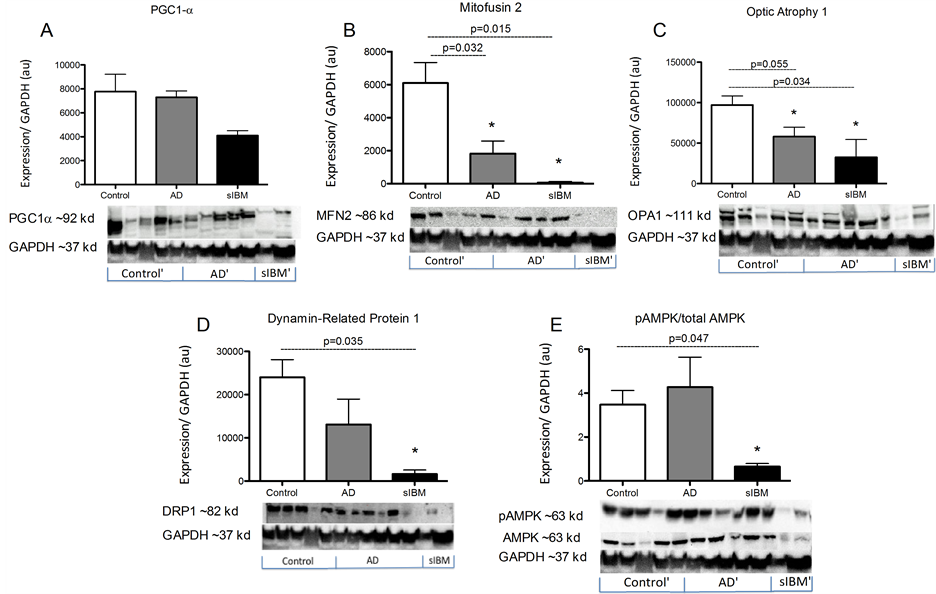
Figure 3. Metabolic enzyme activity in skeletal muscle from patients diagnosed with AD and sIBM. Protein content of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (A), mitofusin 2 (B), optic atrophy protein 1 (C), dynamin related protein 1 (D), and phosphorylated 5’-AMP-activated protein kinase (AMPK) and total AMPK (E) in skeletal muscle from patients diagnosed with AD (n = 5), sIBM (n = 2), and elderly individuals (n = 6). Data are presented as means ± S.E.M. and normalized to GAPDH protein content. *P < 0.05, compared to controls.
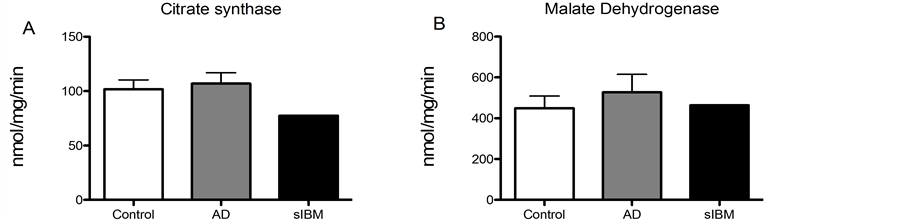
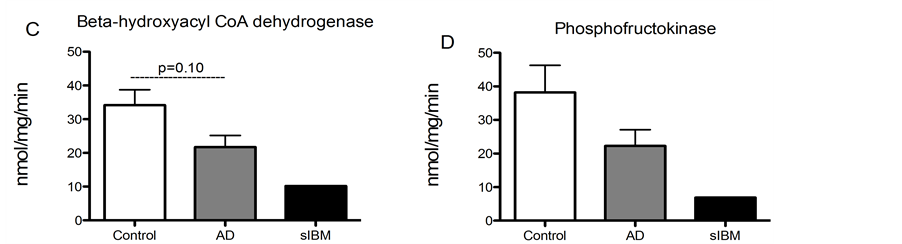
Figure 4. Metabolic enzyme activity was measured in skeletal muscle from patients diagnosed with sIBM, AD and healthy controls. Maximal enzymatic activities of citrate synthase (A), malate dehydrogenase (B), beta hydroxyacyl-Co A dehydrogenase (C), and phosphofructokinase-1 (D) in skeletal musclefrom patients diagnosed with AD (n = 5), sIBM (n = 1), and elderly individuals (n = 6). Data presented at means ± S.E.M. and presented as nmol of mg protein per minute. *P < 0.05, compared to controls.
these pathways could account for the structural and functional alterations previously observed in sIBM as well as demonstrating for the first time, alterations in mitochondrial signaling pathways in skeletal muscle from patients diagnosed with AD.
The above effects are observed in conjunction with a significant increase in PGC1a mRNA in both patient populations, although there were no significant differences in PGC1 protein content. PGC1a is regulated at a number of levels and it has been previously demonstrated that PGC1a mRNA does not reflect protein content [36] [37] . The observed increase in PGC1amRNA along with increased expression of regulators of mitochondrial autophagy, may indicate a compensatory effect to remove and replace damaged, dysfunctional mitochondria in the face of dysregulated mitochondrial dynamic pathways [38] .
While these findings indicate a potential disruption of mitochondrial dynamics and function, the mechanisms underlying the observed similarities between the phenotypes may differ. While AbPP mRNA was higher in sIBM patients, which supports previous findings [39] ; there were no differences between AD patients and elderly controls. The lack of an increase in APP mRNA in AD patients in the current study does not support a link between mitochondrial defects and increased AbPP expression in skeletal muscle in AD patients. Additionally, there was reduced content of both phosphorylated and total AMPK in sIBM patients compared to elderly controls. It has been demonstrated that increased AMPK activity results in an up regulation of MFN2, OPA1, and DRP1 in murine skeletal muscle [40] . Therefore, a down regulation of AMPK may explain the down regulation of MFN2, OPA1, and DRP1 observed in the patients with sIBM. The fact that there was reduced AMPK protein content may also suggest a potential therapeutic strategy for treatment of individuals afflicted with this disease.
While there was reduced phosphorylated and total AMPK content in sIBM patients, there were no significant differences between AD patients and controls. This indicates that something other than AMPK may be responsible for the decline in MFN2 and OPA1 protein content in AD patients. Interestingly, there was an increase in UCP3, SOD2, and Catalase mRNA. This transcriptional up regulation of antioxidant genes may be a protective mechanism in response to an increase in intracellular reactive oxygen species concentrations in skeletal muscle from AD patients [41] [42] . ROS has been demonstrated to regulate expression of both fusion and fission proteins and may therefore also be responsible for the alterations observed in the current work [43] [44] . On the other hand, genetic or pharmaceutical alterations of mitochondrial fusion or fission pathways has been shown to result in increased production of reactive oxygen species [8] . Unfortunately, it is not possible to determine cause and effect from the current study.
There are limitations to the current study. The very low N of our sIBM patients is a concern. However sIBM, although considered the most common myopathy in older individuals, still has a low prevalence rate with only 0.002% of individuals over 50 in the US diagnosed with the disease [45] . Additionally, the AD patients were significantly older than the controls and sIBM patients, making it difficult to exclude an age effect. However, all of the individuals studied were over the age of 60 and the observed similarities along with differences between the two patients populations would suggest that the observations in the current work are not simply due to age.
5. Conclusion
In conclusion, we are reporting for the first time that regulators of mitochondrial fusion and fission are down regulated in skeletal muscle from patients diagnosed with AD and sIBM. These findings suggest a potential for mitochondrial dysfunction in skeletal muscle in both disease states although the underlying causes may differ. This work highlights the need for the future study in an effort to identify new opportunities for prevention or treatment of these life-altering diseases.
References
- McBride, H.M., Neuspiel, M. and Wasiak, S. (2006) Mitochondria: More than Just a Powerhouse. Current Biology, 16, R551-R560. http://dx.doi.org/10.1016/j.cub.2006.06.054
- Romanello, V. and Sandri, M. (2010) Mitochondrial Biogenesis and Fragmentation as Regulators of Muscle Protein Degradation. Current Hypertension Reports, 12, 433-439. http://dx.doi.org/10.1007/s11906-010-0157-8
- Boudina, S. and Abel, E.D. (2006) Mitochondrial Uncoupling: A Key Contributor to Reduced Cardiac Efficiency in Diabetes. Physiology, 21, 250-258. http://dx.doi.org/10.1152/physiol.00008.2006
- de Castro, I.P., Martins, L.M. and Tufi, R. (2010) Mitochondrial Quality Control and Neurological Disease: An Emerging Connection. Expert Reviews in Molecular Medicine. http://dx.doi.org/10.1017/S1462399410001456
- Lanza, I.R. and Nair, K.S. (2010) Mitochondrial Function as a Determinant of Life Span. Pflugers Archiv: European Journal of Physiology, 459, 277-289. http://dx.doi.org/10.1007/s00424-009-0724-5
- Berman, S.B., Pineda, F.J. and Hardwick, J.M. (2008) Mitochondrial Fission and Fusion Dynamics: The Long and Short of It. Cell Death and Differentiation, 15, 1147-1152. http://dx.doi.org/10.1038/cdd.2008.57
- Karbowski, M. and Youle, R.J. (2003) Dynamics of Mitochondrial Morphology in Healthy Cells and during Apoptosis. Cell Death and Differentiation, 10, 870-880. http://dx.doi.org/10.1038/sj.cdd.4401260
- Liesa, M., Palacin, M. and Zorzano, A. (2009) Mitochondrial Dynamics in Mammalian Health and Disease. Physiological Reviews, 89, 799-845. http://dx.doi.org/10.1152/physrev.00030.2008
- Chen, H., Vermulst, M., Wang, Y.E., Chomyn, A., Prolla, T.A., McCaffery, J.M., et al. (2010) Mitochondrial Fusion is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell, 141, 280-289. http://dx.doi.org/10.1016/j.cell.2010.02.026
- Cipolat, S., Martins de Brito, O., Dal Zilio, B. and Scorrano, L. (2004) OPA1 Requires Mitofusin 1 to Promote Mitochondrial Fusion. Proceedings of the National Academy of Sciences of the United States of America, 101, 15927-15932. http://dx.doi.org/10.1073/pnas.0407043101
- Pitts, K.R., Yoon, Y., Krueger, E.W. and McNiven, M.A. (1999) The Dynamin-Like Protein DLP1 Is Essential for Normal Distribution and Morphology of the Endoplasmic Reticulum and Mitochondria in Mammalian Cells. Molecular Biology of the Cell, 10, 4403-4417.
- Rojo, M., Legros, F., Chateau, D. and Lombes, A. (2002) Membrane Topology and Mitochondrial Targeting of Mitofusins, Ubiquitous Mammalian Homologs of the Transmembrane GTPase Fzo. Journal of Cell Science, 115, 1663- 1674.
- Blass, J.P. (2000) The Mitochondrial Spiral. An Adequate Cause of Dementia in the Alzheimer’s Syndrome. Annals of the New York Academy of Sciences, 924, 170-183.
- Cardenas, A.M., Ardiles, A.O., Barraza, N., Baez-Matus, X. and Caviedes, P. (2012) Role of Tau Protein in Neuronal Damage in Alzheimer’s Disease and Down Syndrome. Archives of Medical Research, 43, 645-654. http://dx.doi.org/10.1016/j.arcmed.2012.10.012
- Keller, J.N., Guo, Q., Holtsberg, F.W., Bruce-Keller, A.J. and Mattson, M.P. (1998) Increased Sensitivity to Mitochondrial Toxin-Induced Apoptosis in Neural Cells Expressing Mutant Presenilin-1 Is Linked to Perturbed Calcium Homeostasis and Enhanced Oxyradical Production. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 18, 4439-4450.
- Wang, X., Su, B., Siedlak, S.L., Moreira, P.I., Fujioka, H., Wang, Y., et al. (2008) Amyloid-Beta Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proceedings of the National Academy of Sciences of the United States of America, 105, 19318-19323. http://dx.doi.org/10.1073/pnas.0804871105
- Moslemi, A.R., Lindberg, C. and Oldfors, A. (1997) Analysis of Multiple Mitochondrial DNA Deletions in Inclusion Body Myositis. Human Mutation, 10, 381-386. http://dx.doi.org/10.1002/(SICI)1098-1004(1997)10:5<381::AID-HUMU8>3.0.CO;2-I
- Rifai, Z., Welle, S., Kamp, C. and Thornton, C.A. (1995) Ragged Red Fibers in Normal Aging and Inflammatory Myopathy. Annals of Neurology, 37, 24-29. http://dx.doi.org/10.1002/ana.410370107
- Schroder, J.M. and Molna, R.M. (1997) Mitochondrial Abnormalities and Peripheral Neuropathy in Inflammatory Myopathy, Especially Inclusion Body Myositis. Molecular and Cellular Biochemistry, 174, 277-281.
- Boncompagni, S., Moussa, C.E., Levy, E., Pezone, M.J., Lopez, J.R., Protasi, F., et al. (2012) Mitochondrial Dysfunction in Skeletal Muscle of Amyloid Precursor Protein-Overexpressing Mice. The Journal of Biological Chemistry, 287, 20534-20544. http://dx.doi.org/10.1074/jbc.M112.359588
- Oldfors, A., Moslemi, A.R., Jonasson, L., Ohlsson, M., Kollberg, G. and Lindberg, C. (2006) Mitochondrial Abnormalities in Inclusion-Body Myositis. Neurology, 66, S49-S55. http://dx.doi.org/10.1212/01.wnl.0000192127.63013.8d
- Askanas, V., Engel, W.K. and Alvarez, R.B. (1993) Enhanced Detection of Congo-Red-Positive Amyloid Deposits in Muscle Fibers of Inclusion Body Myositis and Brain of Alzheimer’s Disease Using Fluorescence Technique. Neurology, 43, 1265-1267.
- Askanas, V. and Engel. W.K. (2011) Sporadic Inclusion-Body Myositis: Conformational Multifactorial Ageing-Related Degenerative Muscle Disease Associated with Proteasomal and Lysosomal Inhibition, Endoplasmic Reticulum Stress, and Accumulation of Amyloid-Beta42 Oligomers and Phosphorylated Tau. Presse médicale, 40, e219-e235. http://dx.doi.org/10.1016/j.lpm.2010.11.024
- Greenberg, S.A. (2009) Inclusion Body Myositis: Review of Recent Literature. Current Neurology and Neuroscience Reports, 9, 83-89.
- Zhu, X., Perry, G., Moreira, P.I., Aliev, G., Cash, A.D., Hirai, K., et al. (2006) Mitochondrial Abnormalities and Oxidative Imbalance in Alzheimer Disease. Journal of Alzheimer’s Disease, 9, 147-153.
- Cardoso, S.M., Santana, I., Swerdlow, R.H. and Oliveira, C.R. (2004) Mitochondria Dysfunction of Alzheimer’s Disease Cybrids Enhances a Beta Toxicity. Journal of Neurochemistry, 89, 1417-1426.
- Anandatheerthavarada, H.K., Biswas, G., Robin, M.A. and Avadhani, N.G. (2003) Mitochondrial Targeting and a Novel Transmembrane Arrest of Alzheimer’s Amyloid Precursor Protein Impairs Mitochondrial Function in Neuronal Cells. The Journal of Cell Biology, 161, 41-54. http://dx.doi.org/10.1083/jcb.200207030
- Devi, L., Prabhu, B.M., Galati, D.F., Avadhani, N.G. and Anandatheerthavarada, H.K. (2006) Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. The Journal of Neuroscience, 26, 9057-9068. http://dx.doi.org/10.1523/JNEUROSCI.1469-06.2006
- Mancuso, M., Orsucci, D., LoGerfo, A., Calsolaro, V. and Siciliano, G. (2010) Clinical Features and Pathogenesis of Alzheimer’s Disease: Involvement of Mitochondria and Mitochondrial DNA. Advances in Experimental Medicine and Biology, 685, 34-44.
- Mark, R.J., Pang, Z., Geddes, J.W., Uchida, K. and Mattson, M.P. (1997) Amyloid Beta-Peptide Impairs Glucose Transport in Hippocampal and Cortical Neurons: Involvement of Membrane Lipid Peroxidation. The Journal of Neuroscience, 17, 1046-1054.
- Frisard, M.I., McMillan, R.P., Marchand, J., Wahlberg, K.A., Wu, Y., Voelker, K.A., et al. (2010) Toll-Like Receptor 4 Modulates Skeletal Muscle Substrate Metabolism. American Journal of physiology Endocrinology and Metabolism, 298, E988-E998. http://dx.doi.org/10.1152/ajpendo.00307.2009
- Hulver, M.W., Berggren, J.R., Carper, M.J., Miyazaki, M., Ntambi, J.M., Hoffman, E.P., et al. (2005) Elevated Stearoyl-CoA Desaturase-1 Expression in Skeletal Muscle Contributes to Abnormal Fatty Acid Partitioning in Obese Humans. Cell Metabolism, 2, 251-261. http://dx.doi.org/10.1016/j.cmet.2005.09.002
- Twig, G., Hyde, B. and Shirihai, O.S. (2008) Mitochondrial Fusion, Fission and Autophagy as a Quality Control Axis: The Bioenergetic View. Biochimica et Biophysica Acta, 1777, 1092-1097.
- Busch, K.B., Bereiter-Hahn, J., Wittig, I., Schagger, H. and Jendrach, M. (2006) Mitochondrial Dynamics Generate Equal Distribution but Patchwork Localization of Respiratory Complex I. Molecular Membrane Biology, 23, 509-520. http://dx.doi.org/10.1080/09687860600877292
- Wikstrom, J.D., Twig, G. and Shirihai, O.S. (2009) What Can Mitochondrial Heterogeneity Tell Us about Mitochondrial Dynamics and Autophagy? The International Journal of Biochemistry & Cell Biology, 41, 1914-1927. http://dx.doi.org/10.1016/j.biocel.2009.06.006
- McGee, S.L. and Hargreaves, M. (2004) Exercise and Myocyte Enhancer Factor 2 Regulation in Human Skeletal Muscle. Diabetes, 53, 1208-1214.
- Watt, M.J., Southgate, R.J., Holmes, A.G. and Febbraio, M.A. (2004) Suppression of Plasma Free Fatty Acids Upregulates Peroxisome Proliferator-Activated Receptor (PPAR) Alpha and Delta and PPAR Coactivator 1α in Human Skeletal Muscle, but Not Lipid Regulatory Genes. Journal of Molecular Endocrinology, 33, 533-544. http://dx.doi.org/10.1677/jme.1.01499
- Romanello, V. and Sandri, M. (2013) Mitochondrial Biogenesis and Fragmentation as Regulators of Protein Degradation in Striated Muscles. Journal of Molecular and Cellular Cardiology, 55, 64-72. http://dx.doi.org/10.1016/j.yjmcc.2012.08.001
- Sarkozi, E., Askanas, V., Johnson, S.A., Engel, W.K. and Alvarez, R.B. (1993) Beta-Amyloid Precursor Protein mRNA Is Increased in Inclusion-Body Myositis Muscle. Neuroreport, 4, 815-818. http://dx.doi.org/10.1097/00001756-199306000-00055
- Garcia-Roves, P.M., Osler, M.E., Holmstrom, M.H. and Zierath, J.R. (2008) Gain-of-Function R225Q Mutation in AMP-Activated Protein Kinase Gamma3 Subunit Increases Mitochondrial Biogenesis in Glycolytic Skeletal Muscle. The Journal of Biological Chemistry, 283, 35724-35734. http://dx.doi.org/10.1074/jbc.M805078200
- Shull, S., Heintz, N.H., Periasamy, M., Manohar, M., Janssen, Y.M., Marsh, J.P., et al. (1991) Differential Regulation of Antioxidant Enzymes in Response to Oxidants. The Journal of Biological Chemistry, 266, 24398-24403.
- Warner, B.B., Stuart, L., Gebb, S. and Wispe, J.R. (1996) Redox Regulation of Manganese Superoxide Dismutase. The American Journal of Physiology, 271, L150-L158.
- Benard, G., Bellance, N., James, D., Parrone, P., Fernandez, H., Letellier, T., et al. (2007) Mitochondrial Bioenergetics and Structural Network Organization. Journal of Cell Science, 120, 838-848. http://dx.doi.org/10.1242/jcs.03381
- Distelmaier, F., Valsecchi, F., Forkink, M., van Emst-de Vries, S., Swarts, H.G., Rodenburg, R.J., et al. (2012) Trolox-Sensitive Reactive Oxygen Species Regulate Mitochondrial Morphology, Oxidative Phosphorylation and Cytosolic Calcium Handling in Healthy Cells. Antioxidants & Redox Signaling, 17, 1657-1669. http://dx.doi.org/10.1089/ars.2011.4294
NOTES

*Corresponding author.