Open Journal of Molecular and Integrative Physiology
Vol.4 No.1(2014), Article ID:42732,10 pages DOI:10.4236/ojmip.2014.41001
Effects of delayed estrogen treatment and 20-HETE synthesis inhibition on postischemic pial artery response to acetylcholine in rats
![]()
1Department of Anesthesiology/Critical Care Medicine, School of Medicine, Johns Hopkins University, Baltimore, USA
2Daniel K. Inouye Graduate School of Nursing, Uniformed Services University of the Health Sciences, Bethesda, USA
Email: marguerite.littleton-kearney@usuhs.edu
Copyright © 2014 Chikao Miyazaki et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Chikao Miyazaki et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 5 December 2013; revised 5 January 2014; accepted 12 January 2014
Keywords:Hypoestrogenicity; Estrogen Replacement; Cerebral Ischemia/Reperfusion; Cerebral Microvessels; Estrogen Receptor Alpha; 20-HETE; HET0016
ABSTRACT
Relatively little is known about the effects of estrogen on postischemic cerebral vasomotor dynamics after ischemic injury. Emerging hypotheses suggest that the timing after menopause at which hormone replacement is initiated might be important and might modulate the potential benefits of estrogen on brain rescue once a cerebral ischemic event occurs. Therefore, we sought to determine if protracted hypoestrogenicity modifies estrogen’s protective effects on postischemic pial artery dilatory dysfunction and if the arachidonic acid metabolite 20-hydroxyeicosatetraeonic (20-HETE) contributes to the dysfunction. Pial artery dilation to acetylcholine was examined before and 1 hour after 15 minutes forebrain ischemia. The rat study groups included: sexually mature males (M), naïve (N), OVX (OV), estrogen-treated OVX females (E1; estrogen started 1 week post ovariectomy) and delayed estrogen-treated (E10; started 10 weeks post ovariectomy) females. Postischemic responses were assessed before and after superfusion of the 20-HETE synthesis inhibitor N-hydroxy-N’-(4-butyl-2-methylphenyl)-formamidine (HET0016). Postischemic acetylcholine dilation was depressed in M, OV and E10 compared to N and E1 rats. Compared to E1, delayed estrogen replacement worsened acetylcholine-induced dilation. Postischemic microvascular estrogen receptor alpha (ERα) density was similar in the OV, E1 and E10 rats. Postischemic application of HET0016 failed to improve acetylcholine dilation. Continuous infusion of HET0016 during and after ischemia did not reverse postischemic pial vasodilatory dysfunction. Timing of estrogen replacement may be critical for vascular health after cerebral ischemic injury. Postischemic loss of acetylcholine reactivity does not appear to involve mechanisms related to 20-HETE synthesis or microvascular ERα expression.
1. INTRODUCTION
Numerous animal models of acute stroke injury demonstrate the protective effects of estrogen, yet data from the Women’s Health Initiative (WHI) and others have led to questions regarding the benefit of chronic hormone replacement therapy [1-3]. One limitation in translating the findings from animal models to clinical applications is that the end-points of the studies and treatment regimens differ. Clinical studies such as the WHI examined stroke incidence, but did not address the neuroprotective or beneficial vascular effects of estrogen demonstrated in acute stroke models. Further, clear effects of estrogen on cerebral vascular recovery after ischemic insult have not been established. Our previous studies indicate that early estrogen repletion is vasoprotective in the postischemic pial arterial bed and it protects both vasodilatory [4] and vasoconstrictive [5] function in these vessels. Such vasomotor dysfunction may reflect loss of pial arteriolar ability to adjust nutrient blood flow to downstream penetrating arterioles and subsequent perfusion of underlying neurons [6]. How pial artery vasomotor dysregulation impacts ischemic brain injury recovery remains undetermined, but impaired postischemic pial dilatory capacity may intensify neuronal injury by disruption of neurovascular coupling.
Recent data in rodent focal stroke models indicate that chronic estrogen replacement can reduce infarct volume [7,8] and synthesis of proinflammatory cytokines [8,9], but this effect diminishes after a period of prolonged estrogen deprivation. These studies suggest that commencing hormone replacement after a protracted period of hypoestrogenicity might negatively impact brain rescue once a cerebral ischemic event occurs. The effects of delayed estrogen replacement on pial arteriole dilatory capacity after ischemic injury have not been well investigated.
Estrogen has been demonstrated to modulate vessel function via regulation of nitric oxide production [10] and estrogen increases endothelial nitric oxide synthase (eNOS) expression in cerebral blood vessels [11,12]. Data indicate that the arachidonic acid metabolite 20- hydroxyeicosatetraenoic acid (20-HETE) may uncouple endothelial nitric oxide (NO) synthase (eNOS) and contribute to endothelial dysfunction [13]. 20-HETE constricts cerebral vessels and accumulating evidence suggests that 20-HETE might worsen reperfusion deficits [14,15], possibly by augmenting reactive oxygen species production [16] and depression of pial artery capacity to respond to vasodilators [17,18]. Produced by cytochrome P450 (CYP) 4A [19], 20-HETE increases after stroke [16,20]. Because NO can inhibit CYP production of 20- HETE [17], estrogen might modulate cerebrovascular reactivity to an NO and endothelial-dependent dilators such as acetylcholine (Ach) via repression of 20-HETE following ischemia. Estrogen could thereby maintain a higher basal level of NO production, greater coupling of eNOS and reduce endothelial dysfunction.
In the present study we first sought to determine if estrogen replacement after protracted hypoestrogenicity attenuates pial arteriole dilatory dysfunction to Ach after ischemia and reperfusion in a manner similar to that observed with early estrogen replacement post-ovariectomy [4]. Secondly, we investigated the possibility that delayed estrogen replacement alters estrogen receptor protein expression in the cerebral microvasculature. Next, we determined if postischemic application of the 20- HETE synthesis inhibitor N-hydroxy-N’-(4-butyl-2-methylphenyl)-formamidine (HET0016) improves Ach reactivity in low estrogen states. Finally, we determined if continuous application of HET0016 throughout ischemia and early reperfusion preserves Ach reactivity during hypoestrogenicity.
2. MATERIALS AND METHODS
All study protocols were approved by the Johns Hopkins University Animal Care and Use Committee. Sexually mature Fisher 344 rats (Harlan, Indianapolis, IN) were used. In the first experiment with postischemic application of HET0016, five groups (n = 6) were studied: males (M), naïve females (N) ovariectomized females (OV), females with estrogen replacement started one week (E1) or ten weeks (E10) after ovariectomy was performed. We employed the ten-week delay in estrogen treatment after ovariectomy to model the effects of estrogen treatment after a protracted time of hormone depletion. This ten-week delay was demonstrated to reverse estrogen’s protective effects on brain injury and cytokine production in the mouse model of stroke [8]. In addition, postischemic brains were harvested from the OV, E1 and E10 for cortical microvessel isolation and determination of estrogen receptor (ERα) density and eNOS. In another experiment with pre-, intra-, and postischemic HET0016 application, a group of OV rats were used. Finally, an additional group of naïve female rats were studied with HET0016 application and no ischemia. Slow-release pellets (21 day; Innovative Research of America, Sarasota, FL) of 17β-estradiol (E2; 0.05 mg) were used for estrogen replacement. This method produces stable physiologic estrogen levels between 7 and 21 days after implantation [5]. Terminal blood samples were collected for radioimmunoassay (Coat-a-Count; DPC, Los Angeles CA) measurement of estrogen.
2.1. Transient Forebrain Ischemia
The four-vessel occlusion model was used to induce transient global ischemia as previously described [4]. One day after coagulation of the vertebral arteries, ischemia was induced for 15 min by occluding the carotid arteries and tightening the cervical ties (to reduce collateral blood flow from extra cranial sources). Reduction of cerebral blood flow was confirmed by pupillary dilation and by visualization of blood flow stasis in the cranial microvessels. At end-ischemia, the ties were released and blood flow was reestablished.
2.2. Pial Arteriole Diameter Measurement
Using the closed cranial window technique, pial arteriolar responses were determined via measurement of the inner diameter as previously described [11]. The intrawindow pressure was maintained at 5 - 8 mmHg, and temperature was controlled with a warming lamp. In all groups anesthesia was maintained with isoflurane (1% - 1.5%) throughout the experiments.
Pial arteriolar responses were visualized through the sealed cranial window using a microscope coupled to a computer video recording system (Molecular Devices Corporation; Sunnyvale, CA). For each rat, pial arteriolar responses were measured on 1 - 2 main pial vessels and 1 - 2 daughter branches (21 - 50 μm). Vessel diameters were averaged for each rat (resolution, approximately 2 - 3 μm) and were expressed as the percentage of the baseline diameter prior to infusion of each drug. We observed no differences in the magnitude of response to Ach when vessel diameters were between 20 μm and 60 μm; therefore, the percent change in diameter measurements on the daughter vessels was averaged with those of the parent vessel for analysis for each rat. The responses in approximately 12 vessels were analyzed for each group.
2.3. Experimental Protocols
After cranial window construction, the animals were recovered for 30 min before obtaining baseline pial arteriole diameter measurements. Ach (10 μmol/L) was infused (0.2 mL/min) into the window and allowed to dwell for 5 min. The vessel diameters were re-measured, and percent change in vessel diameter was calculated. Because we tested the response to Ach three times in each rat and also tested the response to the non-endothelium dependent dilator papaverine, time constraints prevented assessment of a full Ach dose-response curve in each animal. The submaximal dose of 10 μmol/L was chosen because the dilation to this dose was sufficiently large to statistically detect partial decrements in the response and we previously observed a robust protective effect on postischemic pial artery dilation to this concentration of Ach [4]. After determination of the preischemic response, the window was rinsed for 10 min with artificial cerebral spinal fluid, and ischemia was induced. The animals were allowed to reperfuse for 60 min, basal diameters were measured to establish a new postischemic baseline, and pial arteriolar responses to Ach were retested. After Ach washout, HET0016 (1 μmol/L; Cayman Chemicals, Ann Arbor, MI) was infused into the cranial window for 5 min and allowed to dwell in the window for an additional 20 min prior to measurement of vessel diameters to establish a new baseline under the influence of HET0016. Responses to Ach were repeated in the presence of HET0016 and expressed as the percent change based on the new baseline established in the influence of HET0016. This concentration of HET0016 blocks 20-HETE synthesis in isolated cerebral arteries [16] and cerebral arteriole constriction [21,22] in a closed cranial window preparation, as well as inhibits 20-HETE dependent cerebrovascular responses [23]. After washing out the Ach, responses to 1 mmol/L papaverine were tested.
To determine if continuous inhibition of 20-HETE throughout ischemia and reperfusion can ameliorate or rescue the response to Ach; HET0016 was administered as a continuous infusion into the cranial window in an additional group of OV rats. We tested the effect of continuous infusion only in OV rats because of the prominent postischemic pial artery dysfunction associated with estrogen depletion. We first assessed the baseline response to Ach (10 μmol/L) and then infused a loading dose of HET0016 at 0.2 ml/min (1 μmol/L) for 10 minutes. The rate was subsequently decreased to a continuous infusion rate of 0.05 ml/min. The vessel response to Ach was reassessed in the presence of HET0016 and ischemia was then induced. The HET0016 infusion was maintained throughout ischemia and during the entire reperfusion period. At 60 minutes reperfusion, the response to Ach was re-tested.
2.4. Isolation of Cerebral Microvessels
Postischemic cortices from the OV, E1 and E10 groups (n = 5/group) were harvested and microvessels isolated to determine if there were differences in the density of ERα protein. Because data suggest that ERα is the receptor isoform that is predominately expressed in cerebral microvessel smooth muscle and endothelium [24,25], we examined ERα rather than ERβ. Microvessels were isolated according to the method of Silbergeld and AliOsma [26]. Briefly, the animals were sacrificed with an overdose of isoflurane, the thoracic cavity was opened, and the heart was perfused with ice-cold saline to remove red blood cells. The brain was harvested, and the cortex was separated from the rest of the brain. Cortices were minced with a scalpel, suspended in 12 mL of ice-cold minimal essential medium (MEM, Gibco Laboratories, Grand Island, NY), and then homogenized at low speed for 40 seconds. The homogenate was centrifuged at 250 g for 10 minutes at 4˚C. The supernatant was discarded; the pellet was resuspended in 25% dextran (molecular weight, 100,000 - 200,000 Daltons; Sigma-Aldrich Company, St. Louis, MO) in MEM, and centrifuged at 2000 g for 20 minutes at 4˚C. After discarding the supernatant, the pellet was resuspended in MEM and run three times through a nylon mesh sieve (60 µm). The microvessel fraction, which contained small arterioles, venules, and capillaries trapped on the mesh, was collected and stored at –80˚C. A small aliquot of the microvessels was evaluated using light microscopy to confirm the purity of the preparation.
2.5. Western Blots
To determine the levels of ERα protein expression, the microvessel samples were thawed and suspended in an ice-cold RIPA buffer (Sigma-Aldrich) and one minipellet of protease inhibitors (Roche Applied Science, Indianapolis, IN) for every 10 mL of buffer. After homogenization, the sample was centrifuged (12,000 g; 20 minutes; 4˚C) and the supernatant was collected for determination of total protein concentration using the Bradford method. For each sample, 2 μg of microvessel protein was loaded and separated on NuPAGE Novex 4% - 12% Bis-Tris Gel (Invitrogen, Carlsbad, CA). After electrophoresis separation, proteins were transferred to a nitrocellulose membrane (Invitrogen) and the membrane was incubated (20˚C) with blocking buffer (5% nonfat dry milk; 0.1% Tween; 1% BSA). The membrane was then incubated overnight (4˚C) with either monoclonal mouse anti-ERα (C311) (Santa Cruz biotechnology Inc.: Santa Cruz, CA; 1:150 dilution in blocking buffer) or monoclonal mouse anti-actin antibodies that recognize the C-terminal of all isoforms (Sigma; 1:10,000 dilution in blocking buffer). Following overnight incubation, the membrane was rinsed with Tween-PBS for 45 minutes and incubated (20˚C) for 90 minutes with anti-mouse IgG antibody conjugated to horseradish peroxidase (Amersham Biosciences, Piscataway, NJ; 1:5000). The membrane was rinsed with Tween-PBS for 45 minutes, incubated with electrochemiluminescence reagent (Amersham) for 60 seconds, and opposed to hyperfilm (Amersham) to visualize ERα expression. This procedure results in enrichment of von Willebrand factor immunoreactivity.
We tested the purity of our microvessel preparation by comparing the expression of the endothelial marker von Willebrand factor in the cortical microvessel fraction to expression in the remaining cortical tissue. Human endothelial cell lysate and expression in aorta were used as positive controls. A polyclonal rabbit anti-human Von Willebrand Factor (1:200), (Dako, Carpentaria, CA 93013, USA) was used as the primary antibody and goat anti-rabbit IgG (H + L)-HRP Conjugate (1:10,000: BioRad Laboratories, Hercules, CA94547, USA) as the secondary antibody. Actin was used as a protein loading control.
2.6. Data Analyses
All data are reported as mean ± SD. Physiologic parameters and vessel diameters were evaluated using oneway or two-way analysis of variance or a student’s t-test as appropriate. In the case of a significant F-value, Neuman-Keuls post-hoc tests for multiple comparisons (Sigma Stat 12) were performed to isolate the differences. A p-value ≤ 0.05 was considered significant.
3. RESULTS
3.1. Physiologic Parameters
Preand postischemic physiologic variables were similar among the groups as shown in Table 1. In N rats, preischemic mean arterial blood pressure (MABP) was within normal ranges, but slightly higher compared to postischemic levels. In these studies, the estrogen replacement regimens resulted in mean plasma estrogen levels of 13.2 ± 7.5 pg/mL (E1-15.6 ± 6 pg/mL; E10- 10.3 ± 9.4 pg/mL) as compared to levels of 0.7 ± 1.5 pg/mL in the OV group. These physiologically relevant levels are consistent with those reported in the literature for naïve rats (range 5 - 20 pg/mL) [27].
3.2. Effects of Delayed Estrogen Treatment
The diameter of pial arterioles before and after ischemia was similar in all groups prior to HET0016 treatment (Figure 1). Preischemic Ach-evoked responses were comparable among all of the female groups. In contrast, in males preischemic dilation to Ach was less (p < 0.027) compared to naive females, but not significantly different from the other female groups (Figure 2). As expected, pial arteriole dilation to Ach was depressed during early reperfusion in all groups. However, we observed that the naïve female rats (20.5% ± 1.3%) or the rats who received estrogen early estrogen replacement (20.7% ± 1.3%) demonstrated a more robust Ach-induced pial arterial vasodilation in the postischemic period compared to males (3.0% ± 0.8%) or females with a 10 week delay in estrogen replacement (4.6 ± 0.9 vs. 3.0% ± 0.8%) as shown in Figure 2.
3.3. Effects of Delayed Estrogen Treatment on Cerebral Microvascular ERα and eNOS
Because subsequent estrogen replacement after prolonged hypoestrogenicity reversed the hormone’s protective effect on postischemic pial arteriolar dilatory capacity, we examined postischemic microvascular ERα expression in OV, E1 and E10 (5/group) rats. ERα protein expression was similar in all rats irrespective of hormone status or timing of estrogen replacement (Figure 3).
3.4. Postischemic Effects of HET0016
Compared to the initial postischemic pial arterial diame-
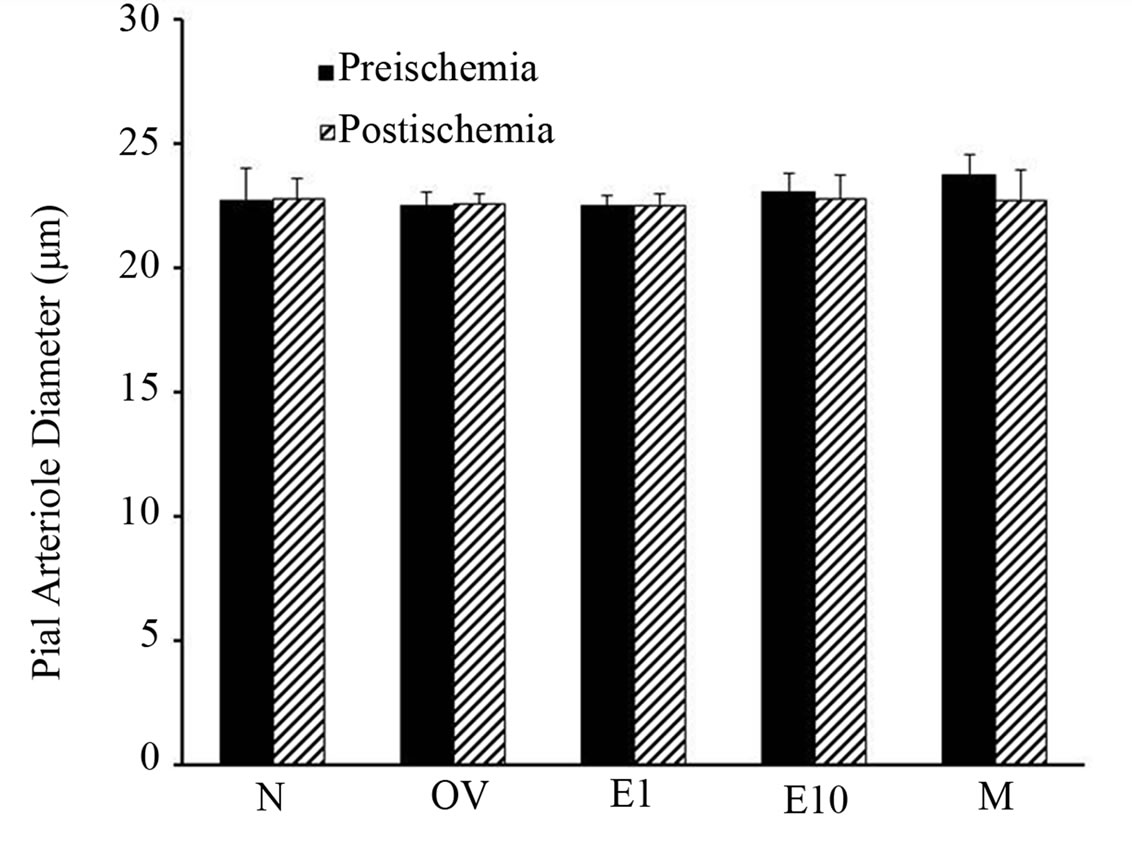
Figure 1. Preand postischemic diameters of pial arterioles in sexually mature (3-month-old) males (M), naïve females (N), ovariectomized females (OV), and estrogen-replaced females 1 week after OVX (E1) or 10 weeks after OVX (E10). Data are expressed as mean ± SD.

Table 1. Physiologic parameters pre and post ischemia.
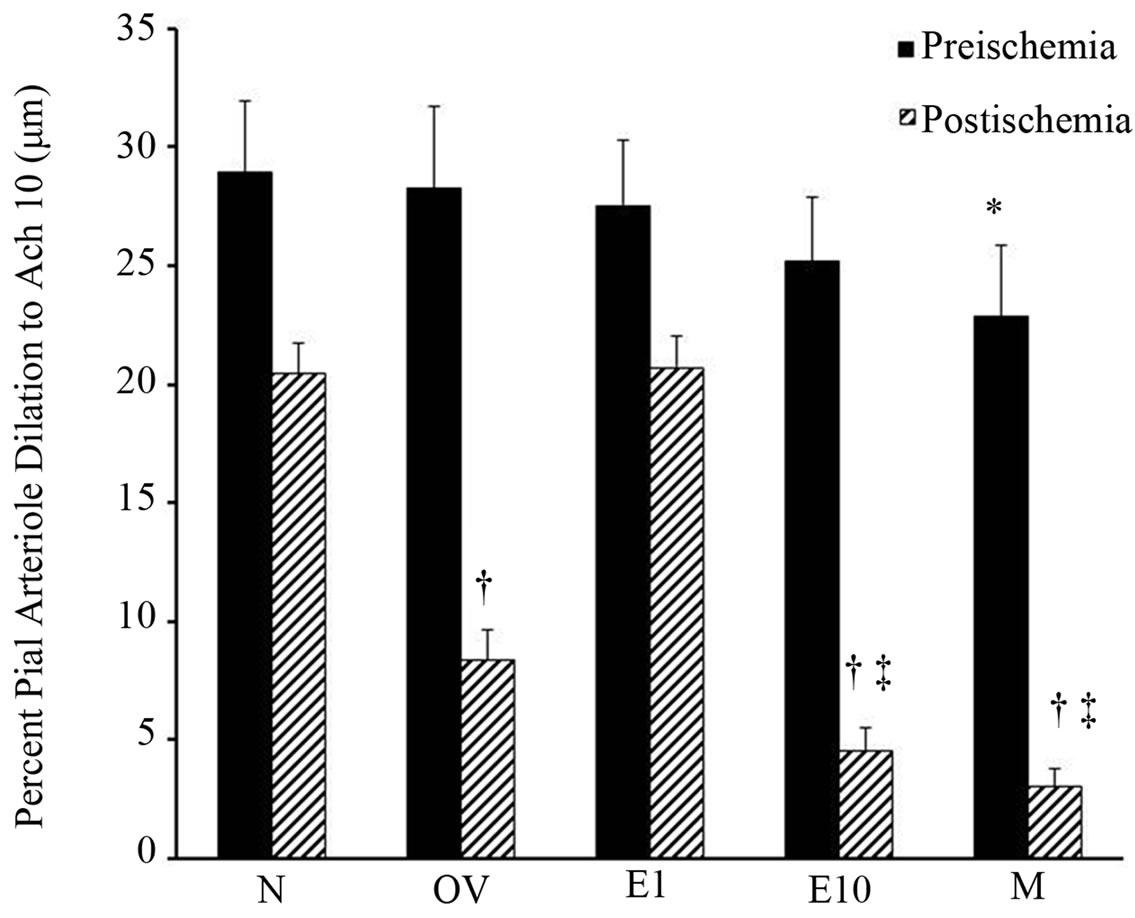
Figure 2. Preand postischemic pial arteriole dilation to Ach (10 μmol/L) in sexually mature (3-month-old) males (M), naïve females (N), ovariectomized females (OV), and estrogen-replaced females 1 week after OVX (E1) or 10 weeks after OVX (E10) ; *p < 0.027 from preischemic N; †p < 0.001 from postischemic N, E1; ‡p < 0.01 from postischemic OV. Data are expressed as mean ± SD.
ters intrawindow application of HET0016 modestly dilated the vessels in all groups as shown in Figure 4 (range 5.9% - 12.6% increase). We then determined if estrogen status altered the pial arteriole postischemic vasodilatory response to Ach when 20-HETE was inhibited by HET0016 (Figure 5). When the percent pial artery dilation to Ach + HET0016 were compared among groups we found a greater vasodilatory response to Ach in the N (10.1 ± 1.5) and E1 (10.0 ± 1.1) versus the OV (6.8 ± 0.5), E10 (4.8 ± 1.3) and M (4.0 ± 0.5) rats (Figure 5). Although, dilatation to Ach + HET0016 was similar between the M and E10 groups, responses were sig-
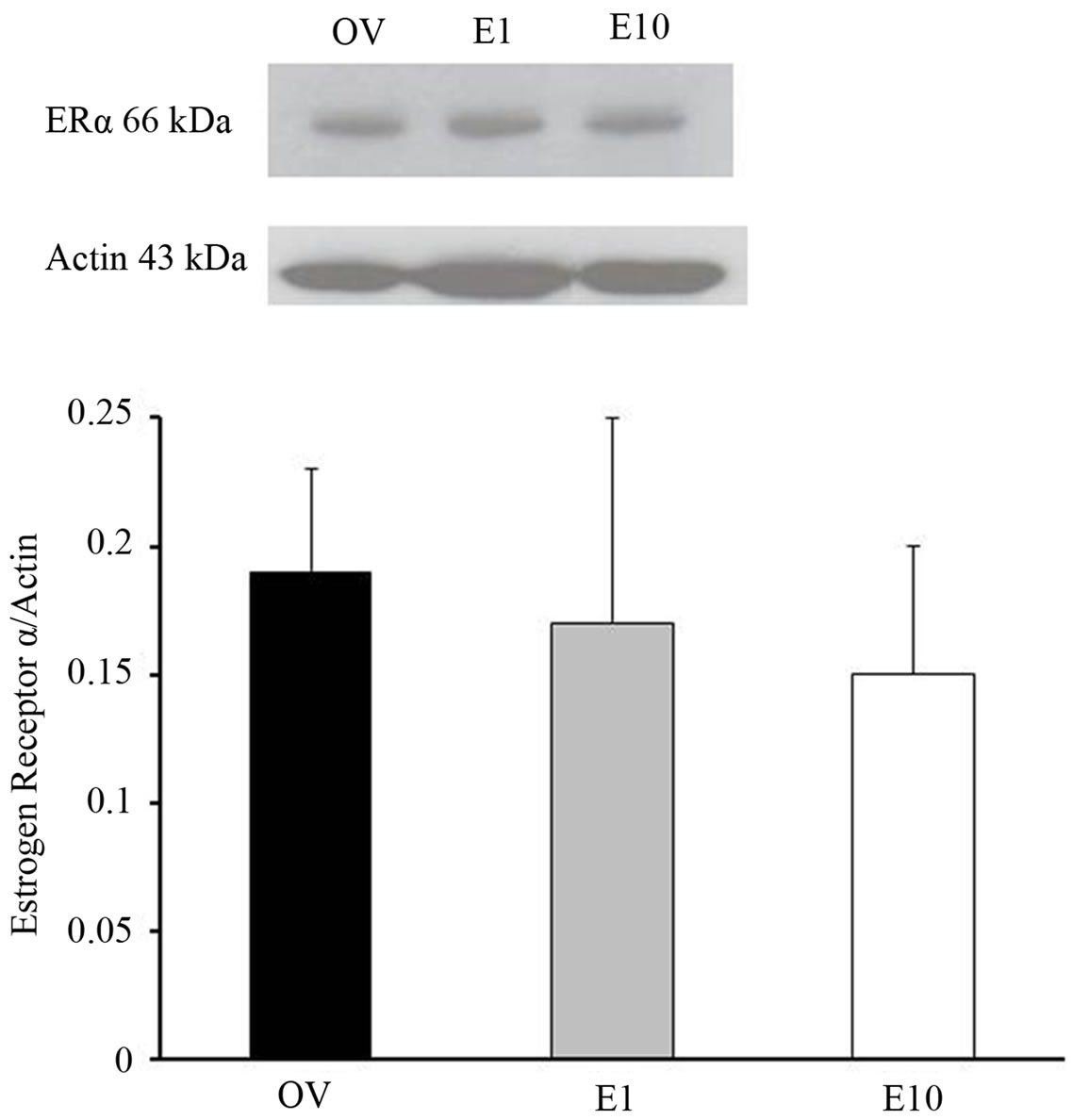
Figure 3. Western blots of postischemic ERα expression in isolated cortical microvessels from OV3, E1 and E10 rats. Data are expressed as a ratio of ERα to actin and include a representative blot of ERα expression. Data are expressed as mean ± SD.
nificantly greater in the OV group compared to the M and E10 group (p < 0.001) as shown in Figure 5.
In order to assure that differences in postischemic pial arteriolar responses to Ach + HET0016 were not affected by time or cranial window failure, we tested pial artery to the non-endothelium dependent vasodilator, papaverine, after we tested earlier postischemic vasodilatory responses. Superfusion of 1 mmol/L of papaverine at the end of the experiment elicited similar dilation in all
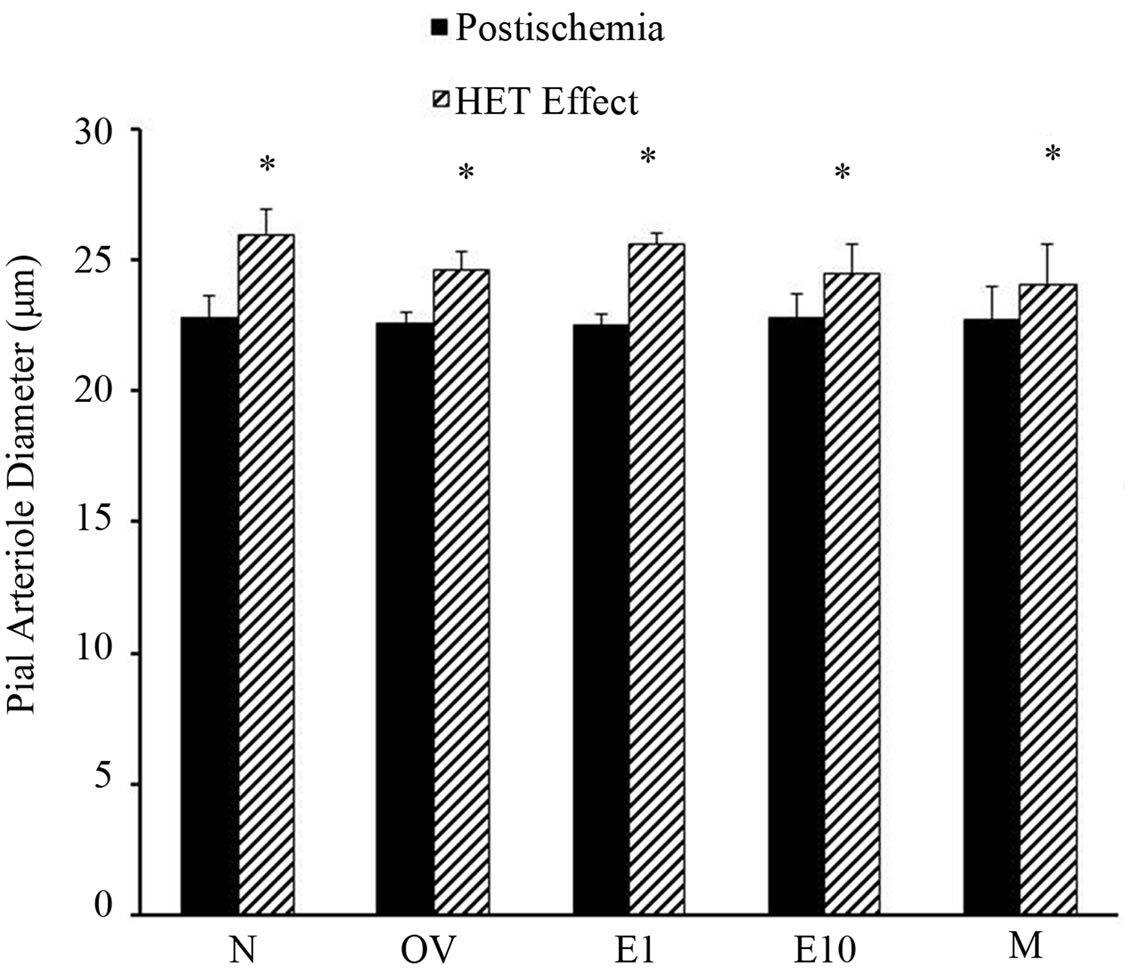
Figure 4. Diameters of postischemic pial arterioles prior to and after superfusion of HET0016 (1 µmol/L) in sexually mature (3-month-old) males (M), naïve females (N), ovariectomized females (OV), and estrogen-replaced females 1 week after OVX (E1) or 10 weeks after OVX (E10); *p < 0.05 from preischemic pial diameter. Data are expressed as mean ± SD.
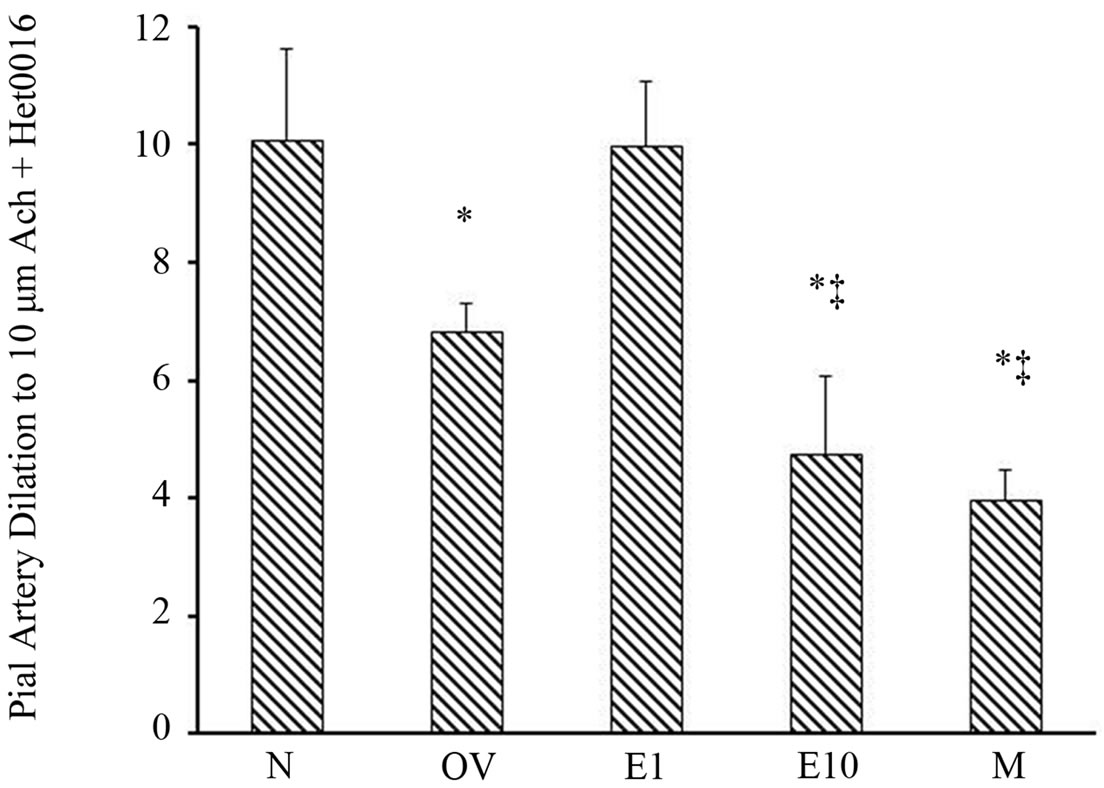
Figure 5. Postischemic pial arteriole dilation to Ach (10 µM/L) + HET0016 (1 µM/L) in sexually mature (3-month-old) males (M), naïve females (N), ovariectomized females (OV), and estrogen-replaced females 1 week after OVX (E1) or 10 weeks after OVX (E10). *p < 0.0001 from N and E1; ‡p < 0.001 from OV. Data are expressed as mean ± SD.
groups (Figure 6).
Because of the marked postischemic depression of Ach reactivity in estrogen-depleted rats, we tested if inhibition 20-HETE synthesis with HET0016 throughout ischemia and reperfusion would preserve vascular function in OV rats. However, postischemic Ach reactivity remained depressed after continuous superfusion of HET0016 started before ischemia (Figure 7).
4. DISCUSSION
This study presents new information demonstrating that extended hypoestrogenicity, prior to initiating estrogen replacement therapy, reverses the protective effect of early institution of estrogen treatment after ovariectomy on postischemic pial arteriolar dilation to Ach. This effect is not caused by decreased postischemic microvascular ERα expression when estrogen repletion is delayed. In contrast to our hypothesis, topical application of the 20-HETE synthesis inhibitor HET0016 after 1 h of reperfusion or throughout ischemia and reperfusion failed to preserve postischemic Ach reactivity in hypoestrogen states or after delayed estrogen replacement.
Timely estrogen replacement after ovariectomy improves postischemic cerebral microvascular dilatory dysfunction in rat pial arteries [4,11]. Our present data confirm and extend these earlier findings. We demonstrate that the efficacy of estrogen in restoring postischemic pial arteriolar sensitivity to Ach-stimulated vasodilation depends on the time when hormone replace-
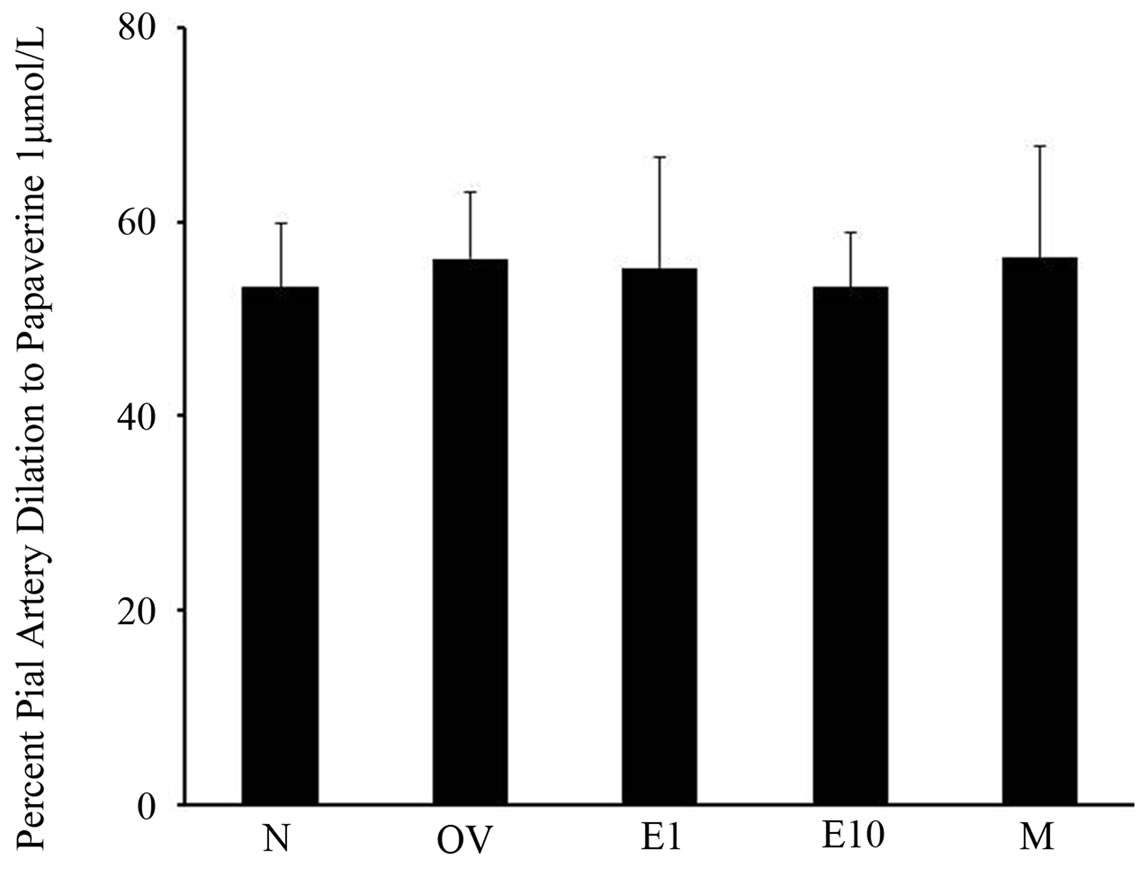
Figure 6. Postischemic pial arteriole dilation to papaverine (1 µmol/L) in sexually mature (3-month-old) males (M), naïve females (N), ovariectomized females (OV), and estrogen-replaced females 1 week after OVX (E1) or 10 weeks after OVX (E10). Data are expressed as mean ± SD.
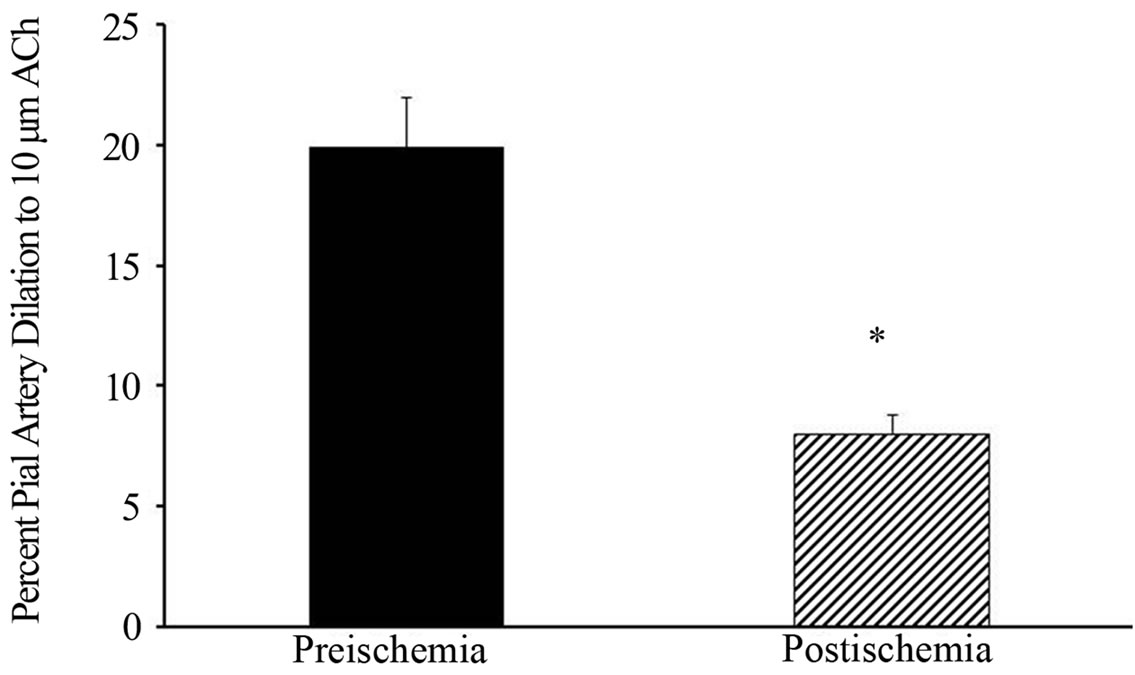
Figure 7. Preand postischemic pial arteriole dilation to Ach with continuous HET0016 infusion in ovariectomized 3-monthold female rats. *p < 0.001 from preischemic level. Data are expressed as mean ± SD.
ment begins. Other evidence also suggests a differential effect between early and delayed estrogen replacement in healthy and injured brain [7,8,28]. In normal brain estrogen replacement after prolonged hypoestrogenicity fails to increase hippocampal choline acetyltransferase protein in middle-aged rats [28] otherwise seen with early estrogen replacement. Furthermore, extended estrogen deprivation with subsequent hormone replacement reduces estrogen’s neuroprotection after focal stroke in young [8] and reproductively senescent rodents [7]. Early estrogen replacement prevents brain accumulation of such proinflammatory markers as monocyte chemoattractant protein-1 and interleukin-6 after focal stroke, whereas estrogen replacement after a 10-week delay was ineffective [8]. Others have also demonstrated that delayed estrogen replacement increases gene expression of proinflammatory mediators at tumor necrosis factor alpha and of iNOS [9]. Thus, our data extend previous work in brain by demonstrating loss of estrogen efficacy on the brain’s vascular responses after ischemic injury.
In aortic rings from healthy rats, estrogen replacement after 8 months of estrogen-deprivation resulted in lost sensitivity to Ach-evoked dilation, while estrogen replacement after 1 or 4 month deprivation restored sensitivity [29]. We found no published reports of the effects of delayed estrogen replacement on the intact normal or postischemic pial vasculature. Our data from rats subjected to 10-week estrogen deprivation followed by estrogen replacement show no apparent effect on healthy pial vessel dilatory reactivity prior to ischemic injury. This lack of effect is consistent with the study in isolated rat aorta [29]. In healthy blood vessels, estrogen replacement after a 1 - 4 month hypoestrogenic period may augment eNOS expression sufficiently to maintain normal coupling with muscarinic receptor activation, but insufficiently to maintain coupling after ischemia [29]. In pial arteries, as in other vessels such as aorta, Ach evokes vasodilation via NO and estrogen promotes NO synthesis by its actions on eNOS [30,31]. We and others have shown that estrogen replacement within 1 week of OVX increases eNOS expression in isolated cerebral microvessels [11,12]. Estrogen-induced augmentation of eNOS immunoreactivity persists for at least 2 h of reperfusion after global ischemic injury [11] and may preserve pial artery dilatory capacity during early reperfusion. The cerebral microvascular endothelium is damaged after ischemia [32] and estrogen may ameliorate postischemic endothelial damage and preserve the ability of eNOS to remain coupled to muscarinic receptor activation. Data suggest that estrogen may be important for endothelial health because 6 months of estrogen depletion increases endothelial TUNEL-positive cells, whereas estrogen replacement early post-ovariectomy markedly reduces this effect in non-cerebral arteries [33]. In cerebral microvessels chronic eNOS reduction resulting from protracted estrogen deprivation may give rise to an endothelium that is more susceptible to ischemia-induced injury such that restoration of normal estrogen levels may be unable to reverse a long-term functional deficit.
We reasoned that the lack of effect of delayed estrogen treatment on postischemic dilatory capacity may be caused by decreased number of available receptors. However, our data do not support this hypothesis since we could find no difference in ERα protein expression in OV, E1 or E10 rats. It is possible that functional estrogen receptors are altered by estrogen depletion or by ischemia, resulting receptors that are less responsive to signaling. Alternatively, the possibility also exits that ERβ and not ERα are altered by long-term estrogen depletion. Further studies are warranted to determine precisely why delayed estrogen treatment is ineffective in protecting pial artery postischemic vasodilator capacity.
Current data suggest that 20-HETE negatively impacts cerebral vascular recovery after ischemic events [34]. However, the effects of 20-HETE on postischemic dilatory capacity are poorly understood. 20-HETE is capable of interfering with the calcium ionophore-induced association of HSP90 with eNOS, thereby increasing superoxide production and decreasing NO production [13]. In isolated cerebral arteries from spontaneously hypertensive stroke-prone rats, HET0016 was found to decrease basal superoxide production, increase Ach-induced relaxation [16] and to reduce markers of oxidative stress [35]. Because estrogen has antioxidant effects and is known to increase cerebral microvascular eNOS expression [11,12] it is possible that estrogen status may modulate postischemic 20-HETE effects on the cerebral vasculature after ischemic injury.
In the current study, we investigated the possibility that inhibition of 20-HETE production diminishes the loss of sensitivity to the NO-dependent vasodilator Ach early during postischemic reperfusion. We found that neither pretreatment combined with post-treatment nor post-treatment alone with HET0016 reversed postischemic pial artery dilatory dysfunction during early reperfusion. Because 20-HETE synthesis by CYP can be inhibited by NO [17], derived from neurons during ischemia and early reperfusion may have been sufficient to inhibit excess 20-HETE synthesis. Nevertheless, the increase in basal diameter seen with HET0016 superfusion either before or after ischemia suggests the presence of tonic 20-HETE synthesis under the conditions of this experiment. This postulated basal level of 20-HETE does not appear to be responsible for the decreased Ach reactivity after ischemia in the hypoestrogen state.
We found that postischemic superfusion of HET0016 blunted estrogen’s protective effect on Ach-evoked pial artery dilation in naïve and E1 rats. Estrogen may enhance eNOS thus augmenting NO synthesis. Because higher 20-HETE levels subsequent to increased CYP4A expression attenuate arterial responsiveness to Ach as a consequence of reduced NO availability [13,36,37], we expected that inhibiting 20-HETE synthesis might fully restore the Ach response rather than blunt it further. We have no simple explanation for this unexpected finding although it should be noted that a time control group was not performed with a second postischemic exposure to Ach in the absence of HET0016 to exclude a possible detrimental effect of additional reperfusion duration or repeated Ach exposure. Nevertheless, the vessels did remain responsive to papaverine. Recent reports suggest 20-HETE may have counter regulatory actions in the cerebrovasculature since 20-HETE or its conversion product 20-COOH-AA relaxes mouse basilar artery via a cyclooxygenase-dependent processes [38]. This counter regulatory effect may be important in the postischemic cerebral vasculature and estrogen may potentiate the effect.
In conclusion, the vasomotor dysfunction that ordinarily occurs after ischemic brain injury can be mitigated by early estrogen replacement, but estrogen repletion after a period of protracted hypoestrogenicity fails to have the same protective effect. The differences in the postischemic response to Ach-evoked pial artery dilation does not appear to be a function of hormone-induced changes in microvascular ERα density since we could detect no differences among the estrogen-depleted and the estrogen-treated groups. Moreover, it is unlikely that 20-HETE synthesis contributes to pial arteriolar dilatory dysfunction during early reperfusion after cerebral ischemic injury in hypoestrogen states.
Abnormally low CBF accompanies stroke and cardiac arrest, with subsequent damage to the function of the neurovascular unit. It is conceivable that persistently depressed cerebrovascular responsiveness might compound tissue injury. Relatively little is known about the effects of estrogen on postischemic cerebral vasomotor dynamics after ischemic injury. Emerging hypotheses suggest that the timing after menopause at which hormone replacement is initiated might be important and might modulate the potential benefits of estrogen on brain rescue once a cerebral ischemic event occurs. Our current studies along with others support the concept that the timing of estrogen therapy may be critical to augment estrogen’s protective effect on the brain microvasculature after cerebral ischemic injury by partially preserving pial arteriolar dilatory capacity.
ACKNOWLEDGEMENTS
The work for these studies was supported by NIH grants NR5339 and NS38684.
DISCLAIMER
The views expressed are those of the authors and do not reflect the official policy/position of the Uniformed Services University of the Health Sciences, the Department of the Defense, or the United States government.
REFERENCES
- Wassertheil-Smoller, S., Hendrix, S., Heiss, G. and Limacher, M. (2003) Effect of estrogen plus progestin on stroke in the women’s health initiative. Stroke, 34, 244.
- Hendrix, S.L., Wassertheil-Smoller, S, Johnson, K.C., Howard, B.V., Kooperberg, C., Rossouw, J.E., Trevisan, M., et al. (2006) Effects of conjugated equine estrogen on stroke in the women’s health initiative. Circulation, 113, 2425-2434. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.594077
- Grodstein, F., Manson, J.E., Stampfer, M.J. and Rexrode, K. (2008) Postmenopausal hormone therapy and stroke: Role of time since menopause and age at initiation of hormone therapy. Archives of Internal Medicine, 168, 861-866. http://dx.doi.org/10.1001/archinte.168.8.861
- Watanabe, Y., Littleton-Kearney, M.T., Traystman, R.J. and Hurn, P.D. (2001) Estrogen restores postischemic pial microvascular dilation. American Journal of Physiology Heart and Circulation Physiology, 281, H155-H160.
- Qin, X., Hurn, P.D. and Littleton-Kearney, M.T. (2005) Estrogen restores postischemic sensitivity to the thromboxane mimetic U46619 in rat pial artery. Journal of Cerebral Blood Flow and Metabolism, 25, 1041-1046. http://dx.doi.org/10.1038/sj.jcbfm.9600105
- Pelligrino, D.A. (2006) Regulation of the cerebral circulation. Journal of Applied Physiology, 100, 3-4. http://dx.doi.org/10.1152/japplphysiol.01254.2005
- Selvamani, A. and Sohrabji, F. (2008) Reproductive age modulates the impact of focal ischemia on the forebrain as well as the effects of estrogen treatment in female rats. Neurobiology of Aging, 31, 1618-1628. http://dx.doi.org/10.1016/j.neurobiolaging.2008.08.014
- Suzuki, S., Brown, C.M., Dela Cruz, C.D., Yang, E., Bridwell, D.A. and Wise, P.M. (2007) Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proceedings of the National Academy of Sciences of the United States of America, 104, 6013-6018. http://dx.doi.org/10.1073/pnas.0610394104
- Pechenino, A.S., Lin, L., Mbai, F.N., Lee, A.R., He, X.M., Stallone, J.N. and Knowlton, A.A. (2010) Impact of aging vs. estrogen loss on cardiac gene expression: Estrogen replacement and inflammation. Physiologic Genomics, 43, 1065-1073. http://dx.doi.org/10.1152/physiolgenomics.00228.2010
- Guo, X., Lu, X., Ren, H., Levin, E.R. and Kassab, G.S. (2006) Estrogen modulates the mechanical homeostasis of mouse arterial vessels through nitric oxide. American Journal of Physiology Heart and Circulation Physiology, 290, H1788-H1797. http://dx.doi.org/10.1152/ajpheart.01070.2005
- Li, M., Zeynalov, E., Li, X., Miyazaki, C., Koehler, R.C. and Littleton-Kearney, M.T. (2009) Effects of estrogen on postischemic pial artery reactivity to ADP. Microcirculation, 16, 403-413. http://dx.doi.org/10.1080/10739680902827738
- McNeill, A.M., Kim, N., Duckles, S.P., Krause, D.N. and Kontos, H.A. (1999) Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke, 30, 2186-2190. http://dx.doi.org/10.1161/01.STR.30.10.2186
- Cheng, J., Ou, J.S., Singh, H., Falck, J.R., Narsimhaswamy, D., Pritchard Jr., K.A., et al. (2008) 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. American Journal of Physiology Heart and Circulation Physiology, 294, H1018-H1026. http://dx.doi.org/10.1152/ajpheart.01172.2007
- Renic, M., Klaus, J.A., Omura, T., Kawashima, N., Onishi, M., Miyata, N., et al. (2009) Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. Journal of Cerebral Blood Flow and Metabolism, 29, 629-639. http://dx.doi.org/10.1038/jcbfm.2008.156
- Poloyac, S.M., Zhang, Y., Bies, R.R., Kochanek, P.M. and Graham, S.H. (2006) Protective effect of the 20- HETE inhibitor HET0016 on brain damage after temporary focal ischemia. Journal of Cerebral Blood Flow and Metabolism, 26, 1551-1561. http://dx.doi.org/10.1038/sj.jcbfm.9600309
- Dunn, K.M., Renic, M., Flasch, A.K., Harder, D.R., Falck, J.R. and Roman, R.J. (2008) Elevated production of 20- HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. American Journal of Physiology Heart and Circulation Physiology, 295, H2455-2465. http://dx.doi.org/10.1152/ajpheart.00512.2008
- Alonso-Galicia, M., Maier, K.G., Greene, A.S., Cowley Jr., A.W. and Roman, R.J. (2002) Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. American Journal of Regulatory, Integrative and Comparative Physiology, 283, R60-R68.
- Yu, M., Cambj-Sapunar, L., Kehl, F., Maier, K.G., Takeuchi, K., Miyata, N., et al. (2004) Effects of a 20- HETE antagonist and agonists on cerebral vascular tone. European Journal of Pharmacology, 486, 297-306. http://dx.doi.org/10.1016/j.ejphar.2004.01.009
- Gebremedhin, D., Lange, A.R., Lowry, T.F., Taheri, M.R., Birks, E.K., Hudetz, A.G., et al. (2000) Production of 20-HETE and its role in autoregulation of cerebral blood flow Circulation Research, 87, 60-65. http://dx.doi.org/10.1161/01.RES.87.1.60
- Omura, T., Tanaka, Y., Miyata, N., Koizumi, C., Sakurai, T., Fukasawa, M., et al. (2006) Effect of a new inhibitor of the synthesis of 20-HETE on cerebral ischemia reperfusion injury. Stroke, 37, 1307-1313. http://dx.doi.org/10.1161/01.STR.0000217398.37075.07
- Cao, S., Wang, L.C., Kwansa, H., Roman, R.J., Harder, D.R. and Koehler, R.C. (2009) Endothelin rather than 20- HETE contributes to loss of pial arteriolar dilation during focal cerebral ischemia with and without polymeric hemoglobin transfusion. American Journal of Regulatory, Integrative and Comparative Physiology, 296, R1412- R1418. http://dx.doi.org/10.1152/ajpregu.00003.2009
- Liu, X., Li, C., Gebremedhin, D., Hwang, S.H., Hammock, B.D., Falck, J.R., Roman, R.J., Harder, D.R. and Koehler, R.C. (2011) Epoxyeicosatrienoic acid-dependent cerebral vasodilation evoked by metabotropic glutamate receptor activation in vivo. American Journal of Physiology Heart and Circulation Physiology, 301, H373-H381.
- Qin, X., Kwansa, H., Bucci, E., Roman, R.J. and Koehler, R.C. (2006) Role of 20-HETE in the pial arteriolar constrictor response to decreased hematocrit after exchange transfusion of cell-free polymeric hemoglobin. Journal of Applied Physiology, 100, 336-342. http://dx.doi.org/10.1152/japplphysiol.00890.2005
- Stirone, C., Boroujerdi, A., Duckles, S.P. and Krause, D.N. (2005) Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: Rapid and long-term effects. Molecular Pharmacology, 67, 105-113. http://dx.doi.org/10.1124/mol.104.004465
- Razmara, A., Sunday, L., Stirone, C., Wang, X.B., Krause, D.N. and Duckles, S.P., et al. (2008) Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. Journal of Pharmacology and Experimental Theraputics, 325, 782-790.
- Silbergeld, D.L. and Ali-Osman, F. (1991) Isolation and characterization of microvessels from normal brain and brain tumors. Journal of Neuro-Oncology, 11, 49-55. http://dx.doi.org/10.1007/BF00166997
- Welschen, R., Osman, P., Dullaart, J., de Greef, W.J., Uilenbroek, J.T. and de Jong, F.H. (1975) Levels of follicle-stimulating hormone, luteinizing hormone, oestradiol-17 beta and progesterone, and follicular growth in the pseudopregnant rat. Journal of Endocrinology, 64, 37-47. http://dx.doi.org/10.1677/joe.0.0640037
- Bohacek, J., Bearl, A.M. and Daniel, J.M. (2008) Longterm ovarian hormone deprivation alters the ability of subsequent oestradiol replacement to regulate choline acetyltransferase protein levels in the hippocampus and prefrontal cortex of middle-aged rats. Journal of Neuroendocrinology, 20, 1023-1027. http://dx.doi.org/10.1111/j.1365-2826.2008.01752.x
- Pinna, C., Cignarella, A., Sanvito, P., Pelosi, V. and Bolego, C. (2008) Prolonged ovarian hormone deprivation impairs the protective vascular actions of estrogen receptor alpha agonists. Hypertension, 51, 1210-1217. http://dx.doi.org/10.1161/HYPERTENSIONAHA.107.106807
- Pelligrino, D.A., Ye, S., Tan, F., Santizo, R.A., Feinstein, D.L. and Wang, Q. (2000) Nitric-oxide-dependent pial arteriolar dilation in the female rat: Effects of chronic estrogen depletion and repletion. Biochemical and Biophysical Research Communications, 269, 165-171. http://dx.doi.org/10.1006/bbrc.2000.2206
- Krause, D.N., Duckles, S.P. and Pelligrino, D.A. (2006) Influence of sex steroid hormones on cerebrovascular function. Journal of Applied Physiology, 101, 1252-1261. http://dx.doi.org/10.1152/japplphysiol.01095.2005
- Fisher, M. (2008) Injuries to the vascular endothelium: Vascular wall and endothelial dysfunction. Review of Neurolological Diseases, 5, S4-S11.
- Moien-Afshari, F., Kenyon, E., Choy, J.C., Battistini, B., McManus, B.M. and Laher, I. (2003) Long-term effects of ovariectomy and estrogen replacement treatment on endothelial function in mature rats. Maturitas, 45, 213- 223. http://dx.doi.org/10.1016/S0378-5122(03)00149-X
- Imig, J.D., Simpkins, A.N., Renic, M. and Harder, D.R. (2011) Cytochrome P450 eicosanoids and cerebral vascular function. Expert Reviews in Molecular Medicine, 13, e7. http://dx.doi.org/10.1017/S1462399411001773
- Toth, P., Csiszar, A., Sosnowska, D., Tucsek, D., Cseplo, P., Springo, Z., et al. (2013) Treatment with cytochrome P450 ω-hydrolase inhibitor HET0016 attenuates cerebrovascular inflammation, oxidative stress and improves vasomotor function in spontaneously hypertensive rats. British Journal of Pharmacology, 168, 1878-1888. http://dx.doi.org/10.1111/bph.12079
- Wang, J.S., Singh, H., Zhang, F., Ishizuka, T., Deng, H., Kemp, R., et al. (2006) Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circulation Research, 98, 962-969. http://dx.doi.org/10.1161/01.RES.0000217283.98806.a6
- Singh, H., Cheng, J., Deng, H., Kemp, R., Ishizuka, T., Nasjletti, A., et al. (2007) Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension, 50, 123-129. http://dx.doi.org/10.1161/HYPERTENSIONAHA.107.089599
- Fang, X., Faraci, F.M., Kaduce, T.L., Harmon, S., Modrick, M.L., Hu, S., et al. (2006) 20-hydroxyeicosatetraenoic acid is a potent dilator of mouse basilar artery: Role of cyclooxygenase. American Journal of Physiology Heart and Circulation Physiology, 291, H2301-H2307. http://dx.doi.org/10.1152/ajpheart.00349.2006