Open Journal of Molecular and Integrative Physiology
Vol.3 No.1(2013), Article ID:27851,5 pages DOI:10.4236/ojmip.2013.31001
HMG CoA reductase inhibition by Simvastatin gets rat β-Myosin heavy chain disappeared: A statin paradox
![]()
1Department of Science, University of Rome, Rome, Italy
2Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland
Email: valentina.pallottini@uniroma3.it
Received 18 January 2013; revised 18 February 2013; accepted 26 February 2013
Keywords: Heart; Myosin Heavy Chains; Statins; Ubiquinone
ABSTRACT
3-hydroxy-3methylglutaryl Coenzyme A reductase, the rate limiting enzyme of mevalonate pathway, generates, in addition to cholesterol, a range of products involved in several biological functions: oligoprenyl groups, dolichol and ubiquinone. The latter, in particular, participates in electron transport chain and, in turn, in tissue energy supply. The enzyme is inhibited by statins that, besides lowering cholesterolemia, seem to impair human energy-dependent myocardial functions (e.g. stroke volume, cardiac output, and contractile index). The modulation of heart contractile properties could be explained by the decrease of ventricle ubiquinone content and/or by putative changes in proportion of the different myosin heavy chain isoforms. Since we previously demonstrated that chronic statin treatment modifies myosin heavy chain isoform pattern in skeletal muscle impairing its functional properties, this work was aimed at investigating the effects of statin chronic treatment on both ventricle ubiquinone content and myosin heavy chain isoforms. Our results showed that simvastatin treatment leads to a reduced amount of rat ventricle ubiquinone and to β myosin heavy chain disappearance. Thus, statins which are prescribed to prevent cardiovascular disease, might induce cardiac metabolic and structural modifications whose functional implications on contractility are still to be established and carefully considered.
1. INTRODUCTION
The 3-hydroxy-3-methylglutaryl Coenzyme A (HMGR) is the key and rate-limiting enzyme of mevalonate pathway which provides cells with vital bioactive compounds such as cholesterol, ubiquinone (CoQ), dolichol and prenyls [1]. In skeletal muscle, in particular, cholesterol is a structural entity of the T-tubular system [2]; CoQ is involved in electron transport during oxidative phosphorylation, assuring the production of ATP needed for muscle contraction; oligoprenyl groups are necessary for post-translational modification of proteins involved in signal transduction pathway; lastly, dolichol is essential for N-linked glycosylation of proteins, such as the fusogenic proteins responsible for myoblast fusion into multinucleated syncitia [3].
As central regulator of cholesterol homeostasis HMGR is highly regulated: it undergoes shortand longterm regulations, the latter concerns the modulation of HMGR protein levels by several factors which include, among others, the transcription factor SREBP (Sterol Regulatory Element Binding Protein) and Insig (Insulin induced gene), able to affect the enzyme transcription and degradation as a function of intracellular sterol amount and in dependence on cholesterol uptake by LDLr (Low Density Lipoprotein receptor). In addition to being involved in the regulation of lipogenic genes, SREBP-1a and -1c were demonstrated to inhibit myoblast-to-myotube differentiation and to induce in vivo and in vitro muscle atrophy through the expression of the transcriptional repressors BHLHB2 and BHLHB3. Both the proteins can affect the expression of several genes involved in muscle contraction and development [4] .
The well-established link between plasma cholesterol levels and coronary artery disease and the contribution of elevated plasma cholesterol, specifically Low Density Lipoprotein (LDL)-cholesterol, to other diseases including cancer, obesity and diabetes, have made control of plasma cholesterol a major health aim [5].
The decrease of cellular cholesterol synthesis leads to a homeostatic response involving up-regulation of cellsurface receptors that bind atherogenic LDL and Very Low Density Lipoproteins (VLDL). The reduction of plasma LDL-cholesterol accounts for the clinical utility of statins that competitively inhibit HMGR [6].
Besides the benefits exerted on plasma lipid profile, statin treatment can lead to severe adverse effects on muscle tissues going from fatigue, weakness, and pain to symptoms associated to rhabdomyolysis, which is a lifethreatening condition [7].
Although less investigated, statin side effects were detected also on cardiac muscle. Pisarenko and co-workers demonstrated that 1-month lovastatin therapy impairs human energy-dependent myocardial functions by decreasing stroke volume, cardiac output, and contractile index. The authors ascribed the impairment of myocardium energy supply to the suppression of CoQ synthesis caused by statin treatment [8]. Although studies have repeatedly demonstrated a reduction of circulating CoQ concentrations with statin therapy, it is unclear whether tissue levels of CoQ are significantly affected [9].
The modulation of the contractile properties of the heart could also be explained by putative changes in proportion of the different Myosin Heavy Chain (MHC) isoforms, α and β, that seem to be related to the level of mechanical performance of the heart [10]. Indeed, our previous work demonstrates that simvastatin chronic treatment (1.5 mg/kg/die) induces in rats functional impairment because of a shift of MHC isoforms from the fastest IIb to the slower IIa/x in fast skeletal muscle (Extensor digitorum longus) [3].
Myosin is a major protein component of heart and skeletal muscles and is the element responsible for energy transduction and force development in these tissues. Each myosin molecule contains four light chains and two heavy chains with ATPase activity. Ventricular muscle has three myosin isoforms that are designated, V1-3, in order of decreasing electrophoretic mobility and ATPase activity [11]. These isoforms differ only in their MHC composition. The V1 form is comprised of two α-MHCs, V3 has two β-MHCs, and V2 has one MHC of each type [10]. Differences in speed of contraction are well documented for the ventricles and are correlated with variations in the ratio of α to β-MHCs (reviewed in Swynghedauw, 1986 [12]). Fast-contracting ventricles (e.g., mice and rats) contain predominately the V1 form (αα); ventricles with intermediate speed (rabbit and guinea pig) are predominately V3 (ββ) but possess small amounts of the V1 and V2 (αβ) forms; slow ventricles (human and bovine) contain less than 10%V1 [10].
Thus, owing to the ability of statins to impair skeletal muscle performance through a MHC isoform shift and to the inadequate knowledge about their capacity to reduce ventricular CoQ content, the aim of our research was to evaluate whether HMGR inhibition by simvastatin could modify ventricular CoQ9 (the predominant ubiquinone form in rats) content and could affect cardiac fiber phenotype.
2. MATERIALS AND METHODS
2.1. Reagents
All chemicals were obtained from commercial sources and were of the highest quality available; when the source is not specified they were obtained from SigmaAldrich (Milan, Italy).
2.2. Animals
3-months-old male Wistar Rattus norvegicus (Harlan Nossan, S. Pietro al Natisone, Italy) were housed under controlled temperature (20˚C ± 1˚C), humidity (55% ± 10%), and illumination (lights on for 12 hours daily, from 7 a.m. to 7 p.m.). Food and water were provided ad libitum. The experiments were performed according to the ethical guidelines for the conduct of animal research (Ministero della Salute, Official Italian Regulation No. 116/92, Communication to Ministero della Salute no. 391/121). Rats were divided in two groups of 7 animals each. The first group was treated daily with intraperitoneal injection of 1.5 mg/kg simvastatin in vehicle (dimethyl sulfoxide—DMSO—1 ml/kg) for 3 weeks. Control animals received daily an equal volume of vehicle. At the end of the treatment rats were anaesthetized with urethane (1.2 g/kg) and plasma was obtained from blood collected into EDTA (1 mg/ml blood).
The cardiac ventricles of the animals from each group were dissected and immediately frozen in liquid nitrogen for subsequent biochemical assays.
2.3. Biochemical Analysis
Plasma cholesterol and triglyceride analysis. Plasma cholesterol content of rats was assessed through the colorimetric CHOD-POD kit according to manufacturer’s instructions (Assel, Rome, Italy). The amount of plasma triglycerides was measured by standardized commercial methods on a fully automated system (Modular, Roche, Basel Switzerland).
Protein evaluation. Total lysates and Western blot analysis were performed as already reported [3]. Primary antibodies used were β-MHC NOQ7.5.4D (Sigma-Aldrich), α-MHC BA-G5 (Abcam, Cambridge, UK), SREBP-1 (Santa Cruz Biotechnology, USA), vinculin and α-tubulin (Sigma-Aldrich).
Lipid extraction and HPLC-UV analysis of polyisoprenoids. These analyses were performed as already reported [3].
3. RESULTS
The systemic efficacy of simvastatin was verified by checking rat plasma cholesterol and triglyceride content. As expected, both the metabolic parameters were significantly decreased following statin treatment (Figure 1).
HMGR chronic inhibition did not statistically modify animal weight (data not shown).
The effects exerted by simvastatin in rat cardiac ventricles were investigated assessing the levels of HMGR main end-products namely cholesterol and CoQ9. Ven-
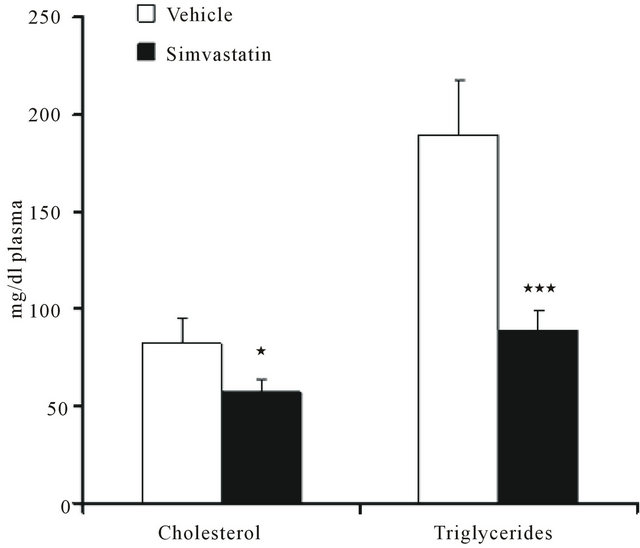
Figure 1. Plasma cholesterol and triglyceride content in rats treated with vehicle (DMSO) or simvastatin for 21 days. Data are expressed as mean ± SD of 7 different animals. *P < 0.05, ***P < 0.001 as determined by Student’s t test.
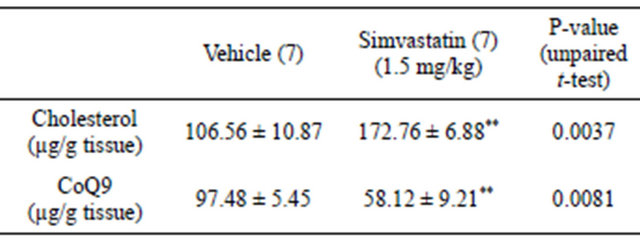
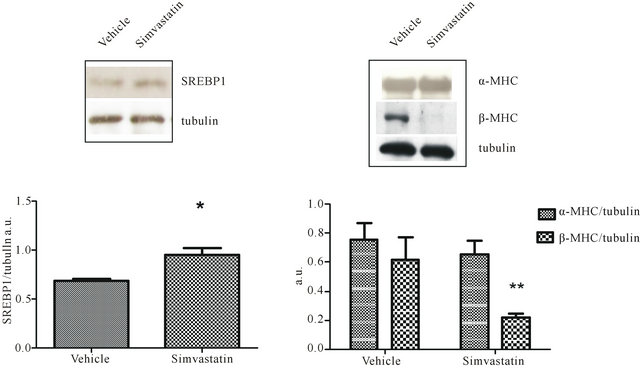
tricle cholesterol content was analyzed to ascertain whether simvastatin was effective even in the heart; CoQ9 amount was checked due to its implication in cardiomyocite energy supply. Tissue content of cholesterol was increased in cardiac ventricles of simvastatin-treated animals when compared to control rats (vehicle). On the contrary, the amount of ventricular CoQ9 was significantly reduced (Table 1). As further proof of simvastatin efficacy within cardiac ventricles, the protein levels of HMGR transcription factor SREBP-1 were assessed. As expected, the amount of transcriptionally active SREBP- 1 fragment was significantly increased following simvastatin treatment (Figure 2(a)).
Moreover, as we previously described that simvastatin was able to induce a shift of MHC isoforms in skeletal muscle [3] ventricular content of β-MHC was evaluated in order to investigate if simvastatin treatment could cause modification of MHC protein profile even in cardiac muscle. Intriguingly, as shown in Figure 2(b), tissue amount of β-MHC was strikingly reduced, on the contrary non-statistically significant differences were detected in ventricular α-MHC content of simvastatintreated animals when compared to controls.
4. DISCUSSION
Our work highlights the strict connection between HMGR main end-products and cardiac physiology showing that the inhibition of the enzyme, which catalyzes the key and rate limiting step of cholesterol biosynthetic pathway, was able to reduce the amount of CoQ9 and β-MHC in rat ventricle. The enzyme activity was chronically inhibited by means of simvastatin, among the lipophilic statins generally taken up much more widely into a
Table 1. Effect of simvastatin treatment on HMGR end-products of the mevalonate pathway. Number of ventricles contributing to each value are shown in brackets. **P < 0.01.
broad range of tissues compared with hydrophilic ones [13] .
The lipid lowering property of simvastatin was displayed by the reduction of both plasma cholesterol and triglyceride content. The efficacy of simvastatin on cardiac muscle was supported by the reduced amount of ventricular CoQ9 which guarantees the energy supply to the heart being involved in mitochondrial electron transport chain. On the contrary, tissue cholesterol levels increased. This result is not surprising if it is considered that statin treatment induces a compensatory up-regulation of the transcriptionally active fragment of SREBP- 1 responsible for the expression of those genes involved in cholesterol uptake, among others LDLR.
Given that fibers having distinct MHC isoforms exhibit different metabolisms and since an alteration in the energy metabolic system can affect cardiac gene expression [14], we investigated whether the observed reduction of ventricular CoQ9 content, following statin treatment, could cause putative modifications in the phenoltype of ventricular myocytes. Very interestingly, our hypothesis was confirmed by a strong reduction of β- MHC protein expression without any detectable modi fication in α-MHC content. These results seem apparently to contradict our previous data obtained from Extensor digitorum longus muscle of simvastatin treated rats, where the statin induced a shift of fast MHC isoforms towards a slower phenotype [3]. Actually, this discrepancy might be ascribed to the reduction of CoQ9 (among the effectors of fiber oxidative metabolism) content which occurs in rat heart but was not observed in rat skeletal muscle [3].
Moreover, MHC isoform pattern can be modulated by transcriptional repressors BHLHB2 and BHLHB3, both able to inhibit β-MHC expression more consistently than that of α isoform [4]. It is worth noting that both repressors are target genes of SREBP-1 which, as previously shown, increases following statin treatment.
The hypothetical mechanism underlying the modification of fiber phenotype in cardiac ventricle of rats under statin treatment could depend on the homeostatic response of the tissue to HMGR inhibition: the rise of SREBP-1 active fragment can lead to a putative increase
 (a) (b)
(a) (b)
Figure 2. SREBP1, α-MHC, and β-MHC in simvastatin treated cardiac ventricle. Panel a illustrates a typical Western blot (top) and the densitometric analysis (bottom) of SREBP1 detected in total lysates prepared from cardiac ventricle of rats undergone simvastatin treatment and those receiving vehicle. Panel b shows a representative Western blot (top) and the densitometric analysis (bottom) α-MHC and β-MHC detected in total lysates prepared from cardiac ventricle of rats undergone simvastatin treatment and those receiving vehicle. Protein levels were normalised to tubulin content. Data are mean values ± SD of seven different animals per group expressed as arbitrary units. *P < 0.05, **P < 0.001 as determined by Student’s t test.
of BHLHB2 and BHLHB3 involved in β-MHC reduction. This event, together with the statin-induced decrease of CoQ9 content could cause a disproportion in fiber phenotype inducing a shift towards a glycolytic metabolism.
Since the different cardiac MHC isoforms exhibit distinct functional characteristics and are distributed regionally within the heart to match the mechanical and functional demands of the different areas, Krenz and co-workers assess that even relatively minor changes in overall MHC isoform content could have significant functional consequences [15].
In addition, we previously demonstrated that HMGR inhibition by simvastatin delays skeletal muscle regeneration reducing myoblast fusion index and fast and slow MHC expression [16]. Thus, we cannot exclude that after myocardial infarction, when statins are always prescribed to patients, these drugs could further interfere with the already inefficient regeneration of cardiac tissue after injury.
Our results seem to highlight a paradox: drugs such as statins, prescribed to prevent cardiovascular diseases, could cause cardiac metabolic and structural modifications whose functional implications might be dangerous. In the light of our observations and of conflicting data present in literature, it is worthwhile investigating to what extent it is advantageous to prevent cardiovascular diseases through statin administration running the risk of undergoing myocardial function changes.
5. ACKNOWLEDGEMENTS
This work was supported by a grant from the University of Roma Tre (CLAR) to VP.
REFERENCES
- Goldstein, J.L. and Brown, M.S. (1990) Regulation of the mevalonate pathway. Nature, 343, 425-430. doi:10.1038/343425a0
- Draeger, A., Monastyrskaya, K., Mohaupt, M., Hoppeler, H., Savolainen, H., Allemann, C. and Babiychuk, E.B. (2006) Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. Journal of Pathology, 210, 94-102. doi:10.1002/path.2018
- Trapani, L., Melli, L., Segatto, M., Trezza, V., Campolongo, P., Jozwiak, A., Swiezewska, E., Pucillo, L.P., Moreno, S., Fanelli, F., Linari, M. and Pallottini, V. (2011) Effects of myosin heavy chain (MHC) plasticity induced by HMGCoA-reductase inhibition on skeletal muscle functions. FASEB Journal, 25, 4037-4047. doi:10.1096/fj.11-184218
- Lecomte, V., Meugnier, E., Euthine, V., Durand, C., Freyssenet, D., Nemoz, G., Rome, S., Vidal, H. and Lefai, E. (2009) A new role for sterol regulatory element binding protein 1 transcription factors in the regulation of muscle mass and muscle cell differentiation. Molecular and Cellular Biology, 30, 1182-1198. doi:10.1128/MCB.00690-09
- Heynekamp, J.J., Hunsaker, L.A., Vander Jagt, T.A., Royer, R.E., Deck, L.M. and Vander Jagt, D.L. (2008) Isocoumarin-based inhibitors of pancreatic cholesterol esterase. Bioorganic & Medicinal Chemistry, 16, 5285- 5294. doi:10.1016/j.bmc.2008.03.016
- Trapani, L. and Pallottini, V. (2010) Age-related hypercholesterolemia and HMG-CoA reductase dysregulation: Sex does matter (a gender perspective). Current Gerontology and Geriatrics Research, 2010, 420139. doi:10.1155/2010/420139
- Golomb, B.A. and Evans, M.A. (2008) Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. American Journal of Cardiovascular Drugs, 8, 373-418. doi:10.2165/0129784-200808060-00004
- Pisarenko, O.I., Studneva, I.M., Lankin, V.Z., Konovalova, G.G., Tikhaze, A.K., Kaminnaya, V.I. and Belenkov, Y.N. (2001) Inhibitor of beta-hydroxy-beta-methylglutaryl coenzyme A reductase decreases energy supply to the myocardium in rats. Bulletin of Experimental Biology and Medicine, 132, 956-958. doi:10.1023/A:1013611110134
- Nawarskas, J.J. (2005) HMG-CoA reductase inhibitors and coenzyme Q10. Cardiology in Review, 13, 76-79.
- Morkin, E. (2000) Control of cardiac myosin heavy chain gene expression. Microscopy Research and Technique, 50, 522-531. doi:10.1002/1097-0029(20000915)50:6<522::AID-JEMT9>3.0.CO;2-U
- Hoh, J.F., McGrath, P.A. and Hale, P.T. (1978) Electrophoretic analysis of multiple forms of rat cardiac myosin: Effects of hypophysectomy and thyroxine replacement. Journal of Molecular and Cellular Cardiology, 10, 1053- 1076. doi:10.1016/0022-2828(78)90401-7
- Swynghedauw, B. (1986) Developmental and functional adaptation of contractile proteins in cardiac and skeletal muscles. Physiological Review, 66, 710-771.
- Izumo, N., Fujita, T., Nakamuta, H. and Koida, M. (2001) Lipophilic statins can be osteogenic by promoting osteoblastic calcification in a Cbfa1- and BMP-2-independent manner. Methods and Findings in Experimental and Clinical Pharmacology, 23, 389-394. doi:10.1358/mf.2001.23.7.662123
- Kakinuma, Y., Miyauchi, T., Suzuki, T., Yuki, K., Murakoshi, N., Goto, K. and Yamaguchi, I. (2002) Enhancement of glycolysis in cardiomyocytes elevates endothelin-1 expression through the transcriptional factor hypoxia-inducible factor-1 alpha. Clinical Science (London), 103, 210S-214S.
- Krenz, M., Sadayappan, S., Osinska, H.E., Henry, J.A., Beck, S., Warshaw, D.M. and Robbins, J. (2007) Distribution and structure-function relationship of myosin heavy chain isoforms in the adult mouse heart. Journal of Biological Chemistry, 282, 24057-24064. doi:10.1074/jbc.M704574200
- Trapani, L., Segatto, M., La Rosa, P., Fanelli, F., Moreno, S., Marino, M. and Pallottini, V. (2012) 3-hydroxy 3- methylglutaryl coenzyme a reductase inhibition impairs muscle regeneration. Journal of Cellular Biochemistry, 113, 2057-2063. doi:10.1002/jcb.24077