Open Journal of Molecular and Integrative Physiology
Vol.1 No.2(2011), Article ID:6951,8 pages DOI:10.4236/ojmip.2011.12002
Physiological and pathophysiological roles of the electrogenic Na+-HCO-3 cotransporter NBCe1
1Department of Internal Medicine, Faculty of Medicine, University of Tokyo, Tokyo, Japan;
2Department of Neurology, University Hospital Gasthuisberg, Belgium;
3Division of Nephrology, Department of Medicine, Tri-Service General Hospital, National Defense Medical Center, Chinses Taipei.
Email: georgeseki-tky@umin.ac.jp
Received 5 July 2011; revised 29 July 2011; accepted 3 August 2011.
Keywords: NBCe1; pRTA; migraine; acidemia; W516X knockin mice
ABSTRACT
The electrogenic Na+-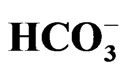 cotransporter NBCe1 encoded by SLC4A4 gene plays essential roles in the regulation of intracellular/extracellular pH. Three NBCe1 variants are thought to mediate distinct physiological roles with different modes of transport stoichiometry. Homozygous inactivating mutations in NBCe1 cause the isolated proximal renal tubular acidosis (pRTA) invariably associated with ocular abnormalities. Functional analyses indicate that more than 50% reduction in NBCe1 activity may be required to induce severe acidemia. Some of the pRTArelated NBCe1 mutations, which show defective membrane expression in mammalian cells, are also associated with migraine. Dysregulation of local pH in brain due to the loss of NBCe1 activity in astrocytes may underlie this association. Two types of NBCe1 deficient animals, NBCe1 knockout and W516X knockin mice, have been reported. Both of them show severe acidemia and early lethality unless they are treated with alkali. In isolated renal proximal tubules from W516X knockin mice, both NBCe1 activity and the rate of bicarbonate absorption are severely reduced, confirming the essential role of NBCe1 in bicarbonate absorption from this nephron segment. In this review, we summarize the recent data about physiological and pathophysiological roles of NBCe1 in health and diseases.
cotransporter NBCe1 encoded by SLC4A4 gene plays essential roles in the regulation of intracellular/extracellular pH. Three NBCe1 variants are thought to mediate distinct physiological roles with different modes of transport stoichiometry. Homozygous inactivating mutations in NBCe1 cause the isolated proximal renal tubular acidosis (pRTA) invariably associated with ocular abnormalities. Functional analyses indicate that more than 50% reduction in NBCe1 activity may be required to induce severe acidemia. Some of the pRTArelated NBCe1 mutations, which show defective membrane expression in mammalian cells, are also associated with migraine. Dysregulation of local pH in brain due to the loss of NBCe1 activity in astrocytes may underlie this association. Two types of NBCe1 deficient animals, NBCe1 knockout and W516X knockin mice, have been reported. Both of them show severe acidemia and early lethality unless they are treated with alkali. In isolated renal proximal tubules from W516X knockin mice, both NBCe1 activity and the rate of bicarbonate absorption are severely reduced, confirming the essential role of NBCe1 in bicarbonate absorption from this nephron segment. In this review, we summarize the recent data about physiological and pathophysiological roles of NBCe1 in health and diseases.
1. INTRODUCTION
The electrogenic Na+-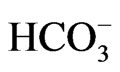 cotransporter NBCe1 is thought to mediate a majority of sodium-coupled bicarbonate absorption from the basolateral membrane of renal proximal tubules [1]. The identification that homozygous mutations in NBCe1 cause proximal renal tubular acidosis (pRTA) has confirmed the essential role of NBCe1 in the regulation of systemic pH [1-3]. NBCe1 also plays important roles in other biological processes such as the maintenance of homeostasis in several ocular tissues, the bicarbonate secretory process in pancreatic ducts and intestinal tracts, and the regulation of synaptic pH in brain [1,4-6]. Consistent with this view, the pRTArelated NBCe1 mutations invariably cause the ocular abnormalities [2,7-12], and some of these NBCe1 mutations are associated with migraine [13]. The recent development of mice deficient for NBCe1 or carrying the W516X mutation further advanced our understanding of the different roles of NBCe1 in a variety of tissues [14, 15].
cotransporter NBCe1 is thought to mediate a majority of sodium-coupled bicarbonate absorption from the basolateral membrane of renal proximal tubules [1]. The identification that homozygous mutations in NBCe1 cause proximal renal tubular acidosis (pRTA) has confirmed the essential role of NBCe1 in the regulation of systemic pH [1-3]. NBCe1 also plays important roles in other biological processes such as the maintenance of homeostasis in several ocular tissues, the bicarbonate secretory process in pancreatic ducts and intestinal tracts, and the regulation of synaptic pH in brain [1,4-6]. Consistent with this view, the pRTArelated NBCe1 mutations invariably cause the ocular abnormalities [2,7-12], and some of these NBCe1 mutations are associated with migraine [13]. The recent development of mice deficient for NBCe1 or carrying the W516X mutation further advanced our understanding of the different roles of NBCe1 in a variety of tissues [14, 15].
In this review, we summarize the recent data about physiological and pathophysiological roles of NBCe1.
2. FUNCTIONAL ROLES OF NBCE1 VARIANTS
NBCe1 encoded by the SLC4A4 gene has three splice variants, which differ at Nor C-terminus, as shown in Figure 1. NBCe1B differs from NBCe1A only at the N-terminus, where the first 41 amino acids of NBCe1A are replaced by the first 85 amino acids of NBCe1B [16]. NBCe1C is identical to NBCe1B except in the C-terminus, where the last 46 amino acids of NBCe1B are replaced by the last 61 amino acids of NBCe1C [17]. The longer N-terminus of NBCe1B and NBCe1C, but not the shorter N-terminus of NBCe1A, has a binding site for inositol 1,4,5-triphosphate (IP3) receptor-binding protein (IRBIT) [18].
These NBCe1 variants show distinct patterns of tissue expression and mediate different biological processes. NBCe1A is predominantly expressed in the basolateral
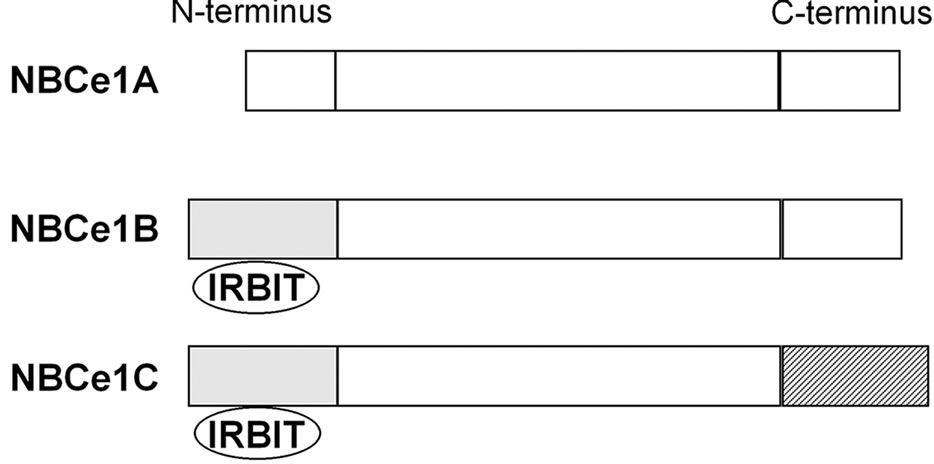
Figure 1. Structures of NBCe1 variants. The longer N-terminus of NBCe1B and NBCe1C has a binding site for IRBIT.
membrane of renal proximal tubules, where it mediates a majority of bicarbonate efflux from tubular cells [3, 19,20]. NBCe1B is expressed in many tissues such as pancreatic ducts, intestinal tracts, ocular tissues, and brain [1,6,21-24]. In these tissues NBCe1B is thought to mediate bicarbonate uptake into cells, which may be essential for several biological processes such as the maintenance of hydration and transparency in cornea or the bicarbonate secretion from pancreas [4,21,25]. NBCe1C seems to be predominantly expressed in brain, but its physiological roles remain speculative [17].
The difference in transport stoichiometry may partially explain the distinct roles of NBCe1 variants. 1Na+ to 3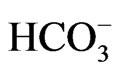 stoichiometry found in the native NBCe1A in renal proximal tubules in vivo is essential for this renal variant to mediate bicarbonate efflux from tubular cells [26,27]. On the other hand, NBCe1B in corneal endothelium or pancreatic ducts is thought to mediate bicarbonate uptake into cells with 1Na+ to 2
stoichiometry found in the native NBCe1A in renal proximal tubules in vivo is essential for this renal variant to mediate bicarbonate efflux from tubular cells [26,27]. On the other hand, NBCe1B in corneal endothelium or pancreatic ducts is thought to mediate bicarbonate uptake into cells with 1Na+ to 2 stoichiometry [4,5,28]. These different modes of transport stoichiometry may not originate from the different structures of NBCe1 variants, but may rather originate from the environment factors such as cell conditions or cell types. For example, NBCe1A is known to function with 1Na+ to 2
stoichiometry [4,5,28]. These different modes of transport stoichiometry may not originate from the different structures of NBCe1 variants, but may rather originate from the environment factors such as cell conditions or cell types. For example, NBCe1A is known to function with 1Na+ to 2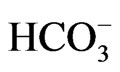 in Xenopus oocytes [29,30], but the increase in cell Ca2+ concentrations can convert its stoichiometry to 1Na+ to 3
in Xenopus oocytes [29,30], but the increase in cell Ca2+ concentrations can convert its stoichiometry to 1Na+ to 3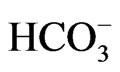 [31]. Similarly, NBCe1A is known to function with 1Na+ to 2
[31]. Similarly, NBCe1A is known to function with 1Na+ to 2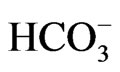 in isolated renal proximal tubules incubated in vitro with the conventional medium [32], but the improvement of incubation conditions can convert its stoichiometry to 1Na+ to 3
in isolated renal proximal tubules incubated in vitro with the conventional medium [32], but the improvement of incubation conditions can convert its stoichiometry to 1Na+ to 3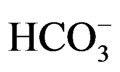 [33]. Conversely, NBCe1B functions with 1Na+ to 2
[33]. Conversely, NBCe1B functions with 1Na+ to 2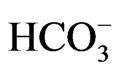 stoichiometry in pancreatic duct cells, but functions with 1Na+ to 3
stoichiometry in pancreatic duct cells, but functions with 1Na+ to 3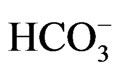 stoichiometry when expressed in renal proximal tubular cells [28].
stoichiometry when expressed in renal proximal tubular cells [28].
3. ROLES OF N-TERMINUS RESIDUES IN THE DISTINCT FUNCTIONS OF NBCE1 VARIANTS
The structural difference in N-terminus may be also partially responsible for the distinct roles of NBCe1 variants. In particular, the longer N-terminus of NBCe1B and NBCe1C may function as an autoinhibitory domain. Indeed, when expressed in Xenopus oocytes, NBCe1B and NBCe1C show very low activities corresponding to 20% - 30% of the NBCe1A activity [34]. However, when co-expressed with IRBIT, NBCe1B shows a dramatically increased activity comparable to that of NBCe1A [18]. The augmentation of NBCe1B activity by IRBIT is also confirmed in pancreatic ducts in vivo [35]. On the other hand, IRBIT does not stimulate NBCe1A, which lacks the binding sites for IRBIT [18].
IRBIT was originally identified as an IP3 receptorsbinding protein that dissociates from the receptors upon the increase in IP3 concentrations [36]. Subsequently, IRBIT was shown to regulate the properties of IP3 receptors by regulating their affinity to IP3 [37,38]. In pancreatic duct cells, IRBIT seems to coordinate epithelial fluid and bicarbonate secretion by stimulating both NBCe1B and cystic fibrosis conductance regulator CFTR through distinct molecular mechanisms [35]. Furthermore, IRBIT can oppose the inhibitory effects of the with-no-lysine (WNK)/Ste20-related proline/alanine-rich kinase (SPAK) pathway by recruiting protein phosphatase 1 in pancreas [39]. Notably, the pancreatic duct cells should change their bicarbonate secretion rates quite widely in response to hormonal stimulation. By contrast, the renal proximal tubular cells mediate bicarbonate absorption at rather constant rates. Thus, the association with IRBIT may represent one of the mechanisms, by which NBCe1B can exert its variant-specific functions in the hormonally regulated secretory epithelia under the control of WNK/SPAK pathway.
The N-terminus residues may also play a role in the responses to inositol 4,5-bisphoshate (PIP2). Thus, in the macropatch from NBCe1A-expressing oocytes, PIP2 can stimulate the NBCe1A activity. PIP2 injection into oocytes also stimulated the whole currents of NBCe1B and NBCe1C. However, PIP2 injection into oocytes failed to enhance the whole currents of NBCe1A [40]. Because NBCe1A with the shorter N-terminus might have a higher affinity for PIP2, NBCe1A could be already maximally activated by endogenous PIP2. On the other hand, the N-terminus of NBCe1B may also play a role in the inhibition by intracellular Mg2+ [41].
4. NBCE1 MUTATIONS IN PRTA
To date, 12 homozygous mutations in SLC4A4 have been reported in pRTA patients as shown in Figure 2.
They are eight missense mutations R298S, S427L, T485S, G486R, R510H, L522P, A799V, and R881C [2, 8,10-12], two nonsense mutations Q29X and W516X [7,15], and two frame shift mutations N721TxfsX29 and S982NfsX4 [9,13]. Topological analysis indicates that
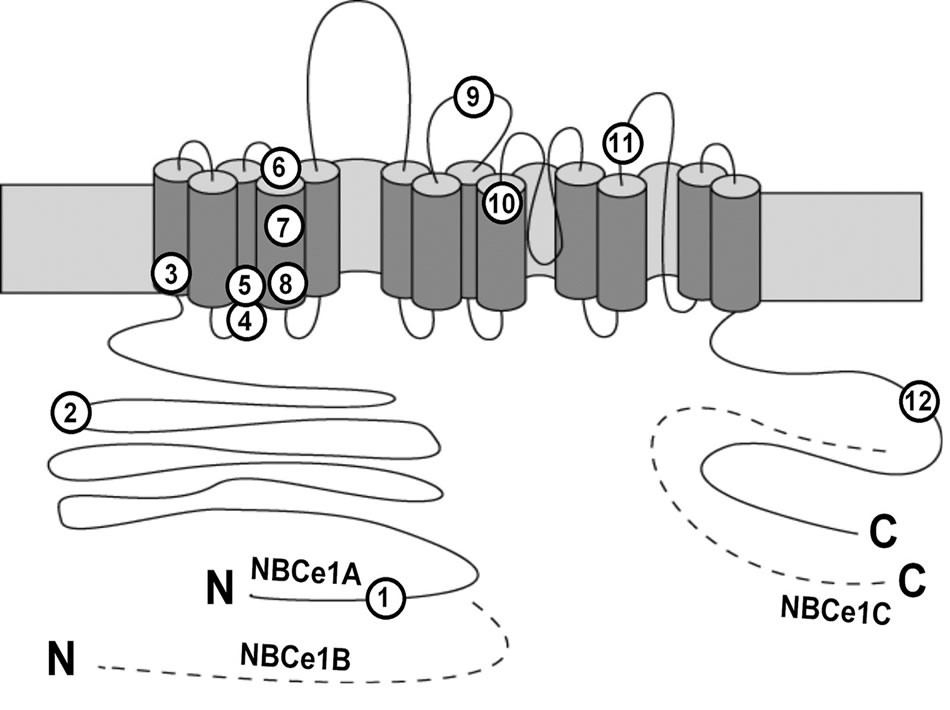
Figure 2. Topology of NBCe1 and pRTA-related mutations. N and C denote N-terminus and C-terminus, respectively. Numbers in circles correspond to Q29X (1), R298S (2), S427L (3), T485S (4), G486R (5), R510H (6), W516X (7), L522P (8), N721TxfsX29 (9), A799V (10), R881C (11), and S982NfsX4 (12).
most of the missense residues causing pRTA lie deep in transmembrane domains, where they perform important structural roles [42]. The pRTA patients carrying homozygous NBCe1 mutations usually presented with the severe acidemia, as defined as the reduction of blood 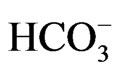 concentration to less than 13 mM, short stature and the ocular abnormalities. The severe acidemia seems to be responsible for short stature, because alkali therapy, if started early in the life, was reported to partially improve the growth rate [43]. Moreover, the two sisters carrying the homozygous S982NfsX4 mutation with a relatively mild pRTA (blood
concentration to less than 13 mM, short stature and the ocular abnormalities. The severe acidemia seems to be responsible for short stature, because alkali therapy, if started early in the life, was reported to partially improve the growth rate [43]. Moreover, the two sisters carrying the homozygous S982NfsX4 mutation with a relatively mild pRTA (blood 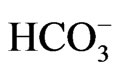 concentration of 15 - 17 mM) showed normal stature [13]. Blood
concentration of 15 - 17 mM) showed normal stature [13]. Blood 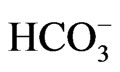 concentration was not reported in one patient carrying L522P mutation [11].
concentration was not reported in one patient carrying L522P mutation [11].
The ocular phenotypes associated with homozygous NBCe1 mutations typically consist of band keratopathy, cataracts, and glaucoma, and these abnormalities sometimes even result in near-total blindness [2,7,12]. Because NBCe1 variants are known to be expressed in several ocular tissues including corneal endothelium, lens epithelium, ciliary epithelium, retina, and trabecular meshwork cells, inactivation of NBCe1 per se is thought to be responsible for the ocular abnormalities [21,25]. For example, the NBCe1-mediated transport in corneal endothelium is indispensable for the maintenance of hydration and transparency in cornea [5,21]. Moreover, the NBCe1 activity seems to be essential also for the lens transparency [21].
Some of the pRTA patients with NBCe1 mutations show enamel abnormalities [7,8,14]. Recent studies indicate that the NBCe1 activity in enamel epithelia cells is required for the normal enamel mineralization [14,44].
5. NBCE1 MUTATIONS AND MIGRAINE
Functional analyses using different expression systems indicate that at least 50% reduction in the NBCe1 activity is required to induce the severe pRTA [2,10,12]. In addition to the functional loss, however, some of the NBCe1 mutants also showed abnormal trafficking in mammalian cells. For example, the T485S and G486R mutants seemed to induce the pure functional impairment with the preserved plasma membrane expression in mammalian cells [10,12]. By contrast, the R510H and R881C mutants seemed to induce both functional impairment and abnormal trafficking [2,45,46]. Because the L522P mutant showed the predominant cytosolic retention in both oocytes and mammalian cells, its function could not be precisely determined [11,12]. The pathophysiological significance of NBCe1 mistargeting remained speculative. Our recent findings, however, have identified a potential association between the NBCe1 mistargeting and the occurrence of migraine [13].
Migraine is a common disease affecting more than 10% of the population [47]. Although genetic factors are believed to play a substantial role, a true Mendelian type of inheritance has been established only in familial hemiplegic migraine (FMH). This is a very rare autosomal dominant subtype of migraine with aura caused by three different genes: CACNA1A encoding the α1 subunit of voltage-gated neuronal Cav2.1 calcium channels [48], ATP1A2 encoding the α2 subunit of Na+/K+ pump [49], and SCN1A encoding the neuronal voltage-gated sodium channel Nav1.1 [50]. Mutations in these transporters are thought to cause migraine by enhancing neuronal excitability [51]. We found two sisters with pRTA, normal stature, severe ocular abnormalities, and hemiplegic migraine. Genetic analysis ruled out pathological mutations in known genes for FMH but identified a novel homozygous 65-base-pair deletion (S982NfsX4) in the C-terminus of NBCe1. The S982NfsX4 mutant, while showing the normal electrogenic activity in Xenopus oocytes, had almost no transport activity due to a predominant endoplasmic reticulum (ER) retention in mammalian cells. By analyzing the other pRTA pedigrees with the distinct NBCe1 mutations, we identified 4 additional homozygous patients with migraine; hemiplegic migraine with episodic ataxia in L522P, migraine with aura in N721TxfsX29, and migraine without aura in R510H and R881C. Evaluation of the corresponding NBCe1B mutants revealed a remarkable coincidence between the lack of plasma membrane expression in C6 glioma cells and the occurrence of migraine. These results strongly suggest that the near total loss of NBCe1B activity in astrocytes can potentially cause migraine through dysregulation of synaptic pH [13].
Cerebral cortical hyperexcitability causing cortical spreading depression (CSD) may be the pathophysiological mechanism underlying migraine aura [51]. In response to neuronal excitation, glial cells depolarize and secrete acid via inward electrogenic Na+-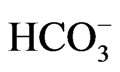 cotransport NBCe1, the process of which is known as depolarization-induced alkalinization DIA [6,52]. Under normal condition, the net extracellular acidosis due to DIA makes surrounding neuronal cells less excitable, because excitatory NMDA receptors are blocked by protons, with a steep sensitivity in the physiological range of extracellular pH [6]. We therefore speculate that absence of DIA due to defective NBCe1 expression in the plasma membrane may cause a positive feedback loop of increased neuronal activity leading to further NMDA-mediated neuronal hyperactivity, causing complete depolarization of a sizable population of brain cells, i.e. CSD.
cotransport NBCe1, the process of which is known as depolarization-induced alkalinization DIA [6,52]. Under normal condition, the net extracellular acidosis due to DIA makes surrounding neuronal cells less excitable, because excitatory NMDA receptors are blocked by protons, with a steep sensitivity in the physiological range of extracellular pH [6]. We therefore speculate that absence of DIA due to defective NBCe1 expression in the plasma membrane may cause a positive feedback loop of increased neuronal activity leading to further NMDA-mediated neuronal hyperactivity, causing complete depolarization of a sizable population of brain cells, i.e. CSD.
Heterozygous mutations in Cl–/ exchanger AE1 cause autosomal dominant distal renal tubular acidosis, through either mistargeting of mutant AE1 to the apical membrane or ER retention of hetero-oligomer complexes consisting of wild-type and mutant proteins [53]. On the other hand, pRTA has never been reported in subjects carrying heterozygous NBCe1 mutations. Because NBCe1 can also form oligomer like AE1 [54], however, some of the phenotypes caused by NBCe1 mutations may be inherited by an autosomal dominant manner. Consistent with this prediction, several family members carrying the heterozygous S982NfsX4 mutation also presented with glaucoma and migraine with or without aura. The co-expression experiments mimicking the heterozygous status indeed identified a dominant negative effect of the mutant through the hetero-oligomer formation with wild-type NBCe1 [13]. While this kind of dominant negative effect may explain the occurrence of ocular and neurological phenotypes by the heterozygous S982NfsX4 mutation, the acid secretory ability of renal distal tubules may be sufficient to compensate for a substantial reduction in bicarbonate absorption from the proximal tubules. Future studies will be required to examine whether heterozygous NBCe1 mutations without pRTA represent a risk factor for common forms of migraine.
exchanger AE1 cause autosomal dominant distal renal tubular acidosis, through either mistargeting of mutant AE1 to the apical membrane or ER retention of hetero-oligomer complexes consisting of wild-type and mutant proteins [53]. On the other hand, pRTA has never been reported in subjects carrying heterozygous NBCe1 mutations. Because NBCe1 can also form oligomer like AE1 [54], however, some of the phenotypes caused by NBCe1 mutations may be inherited by an autosomal dominant manner. Consistent with this prediction, several family members carrying the heterozygous S982NfsX4 mutation also presented with glaucoma and migraine with or without aura. The co-expression experiments mimicking the heterozygous status indeed identified a dominant negative effect of the mutant through the hetero-oligomer formation with wild-type NBCe1 [13]. While this kind of dominant negative effect may explain the occurrence of ocular and neurological phenotypes by the heterozygous S982NfsX4 mutation, the acid secretory ability of renal distal tubules may be sufficient to compensate for a substantial reduction in bicarbonate absorption from the proximal tubules. Future studies will be required to examine whether heterozygous NBCe1 mutations without pRTA represent a risk factor for common forms of migraine.
A recent study using cells cultured from the wild-type and NBCe1 KO mice identified that, in addition to Cl–- dependent mechanism, NBCe1 activity also contributes to the DIA response in hippocampal neurons [55].
6. SINGLE NUCLEOTIDE POLYMORPHISM IN NBCE1
The final adjustments of urinary sodium excretion by distal nephrons are considered to be important for wholebody volume regulation. Indeed, rare heterozygous mutations in sodium handling genes in renal distal tubules such as SCL12A3, SLC12A1, and KCNJ1 have been shown to cause a significant reduction in systolic blood pressure [56]. On the other hand, the sodium handling in renal proximal tubules is also known to affect blood pressure [57-59]. Proximal tubules are responsible for the reabsorption of approximately 60% of the NaCl filtered from glomerulus. This process is accomplished by the coordinated operation of the apical Na+/H+ exchanger isoform 3 (NHE3) and the basolateral NBCe1A [3]. NHE3 knockout (KO) mice show hypotension, even after the genetic rescue of intestinal absorptive defect [58]. Furthermore, abrogation of angiotensin type 1 receptors in proximal tubules alone is shown to be sufficient to reduce blood pressure and to provide substantial protection against hypertension [59].
In this context, we recently performed functional characterization of the four nonsynonymous single nucleotide polymorphisms (SNPs), E122G, S356Y, K558R, and N640I in NBCe1. The detailed analysis revealed that only the K558R variant that lies near the extracellular end of transmembrane 5 has a significantly reduced transport activity corresponding to 41% - 47% of the wild-type activity [60]. K558 may not only represent one of the putative binding sites for 4, 4'-diisothiocyanatostilbene-2, 2'-disulphonic acid DIDS [61], but also play an important role in the NBCe1 transport functions. As already discussed, heterozygous NBCe1 mutations have not been linked to pRTA. Nevertheless, more than 50% reduction in NBCe1 activity in one allele may have a substantial impact on the overall renal sodium handling. It remains to be investigated whether the heterozygous status of K558R variant is associated with the reduction in blood pressure.
7. NBCE1 KO AND W516X KNOCKIN MICE
Two types of NBCe1 deficient mice have been produced. NBCe1 KO mice exhibited severe metabolic acidosis (blood  concentration of 5.3 mM) most likely due to pRTA, growth retardation, hyperaldosteronism, anemia and splenomegaly, abnormal enamel mineralization, intestinal obstruction, and early death before weaning [14]. Abdominal organs appeared pale probably due to anemia and poor perfusion. In NBCe1 KO spleen, the white pulp and the red pulp were severely disrupted. The enhanced expression of Na+-K+-2Cl– cotransporter NKCC1 seemed to compensate for the predicted loss of bicarbonate transport in isolated colon from NBCe1 KO mice. Only when the availability of
concentration of 5.3 mM) most likely due to pRTA, growth retardation, hyperaldosteronism, anemia and splenomegaly, abnormal enamel mineralization, intestinal obstruction, and early death before weaning [14]. Abdominal organs appeared pale probably due to anemia and poor perfusion. In NBCe1 KO spleen, the white pulp and the red pulp were severely disrupted. The enhanced expression of Na+-K+-2Cl– cotransporter NKCC1 seemed to compensate for the predicted loss of bicarbonate transport in isolated colon from NBCe1 KO mice. Only when the availability of 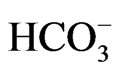 was limited by carbonic anhydrase inhibition with acetazolamide, NBCe1 KO colon exhibited a significant reduction in cAMP-stimulated short circuit current [14].
was limited by carbonic anhydrase inhibition with acetazolamide, NBCe1 KO colon exhibited a significant reduction in cAMP-stimulated short circuit current [14].
We recently identified a homozygous NBCe1 W516X mutation in a Chinese girl with severe pRTA (blood 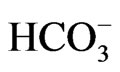 concentration of 10 mM), growth retardation, and the typical ocular abnormalities including band keratopathy, cataracts, and glaucoma. To clarify the pathogenesis of W516X mutation, we have produced mice carrying this mutation by homologous recombination in the embryonic stem cells [15].
concentration of 10 mM), growth retardation, and the typical ocular abnormalities including band keratopathy, cataracts, and glaucoma. To clarify the pathogenesis of W516X mutation, we have produced mice carrying this mutation by homologous recombination in the embryonic stem cells [15].
The expression of NBCe1 mRNA and protein were virtually absent in W516X knockin (KI) mice, consistent with the involvement of nonsense-mediated mRNA decay (NMD) in the defective transcription and translation of this mutation. Like NBCe1 KO mice, homozygous W516X KI mice presented with severe metabolic acidosis (blood  concentration of 3.9 mM) due to pRTA, growth retardation, hyperaldosteronism, anemia and splenomegaly, and early death before weaning. These mice also showed prerenal azotemia, renal failure, and bone dysplasia [15]. Alkali therapy markedly prolonged the survival, and partially improved growth retardation and bone abnormalities. Severe protein catabolism in W516X KI mice as evidenced by a striking increase in E3 ubiquitin ligase was also markedly improved by alkali therapy. In isolated renal proximal tubules form W516X KI mice, the NBCe1 activity as analyzed by cell pH measurement was severely reduced to less than 20% of the wild-type activity. Similarly, the rate of bicarbonate absorption was also markedly reduced to less than 20% of that in wild-type mice. These data directly confirmed the indispensable role of NBCe1 in bicarbonate absorption from renal proximal tubules. The prolonged survival time by alkali therapy uncovered the development of corneal opacities due to corneal edema, indicating that NBCe1 in corneal endothelium is essential for the maintenance of corneal transparency also in mice.
concentration of 3.9 mM) due to pRTA, growth retardation, hyperaldosteronism, anemia and splenomegaly, and early death before weaning. These mice also showed prerenal azotemia, renal failure, and bone dysplasia [15]. Alkali therapy markedly prolonged the survival, and partially improved growth retardation and bone abnormalities. Severe protein catabolism in W516X KI mice as evidenced by a striking increase in E3 ubiquitin ligase was also markedly improved by alkali therapy. In isolated renal proximal tubules form W516X KI mice, the NBCe1 activity as analyzed by cell pH measurement was severely reduced to less than 20% of the wild-type activity. Similarly, the rate of bicarbonate absorption was also markedly reduced to less than 20% of that in wild-type mice. These data directly confirmed the indispensable role of NBCe1 in bicarbonate absorption from renal proximal tubules. The prolonged survival time by alkali therapy uncovered the development of corneal opacities due to corneal edema, indicating that NBCe1 in corneal endothelium is essential for the maintenance of corneal transparency also in mice.
Both NBCe1 KO and W516X KI mice showed a more severe acidemia than human pRTA patients. Surprisingly, the detailed analysis in W516X KI mice revealed that there was no compensatory increase in mRNA of distal acid/base transporters, which could be due to poor renal perfusion and the loss of viable kidney function and mass. Unlike in humans, however, haploinsufficiency of NBCe1 in these genetically engineered mice is associated with mild acidemia [14,15]. Therefore, we cannot exclude a possibility that the compensatory capacity of distal tubular acidification may be less in mice than in humans.
Mutation-specific approaches aiming at the different classes of genetic mutations are now being investigated as new molecular therapies in genetic disorders. For example, PTC124, a novel nonsense suppression agent with apparently less toxicity, has been developed for the treatment of genetic disorders caused by premature terminating codon [62,63]. Thus, W516X KI mice may become a useful model to test the efficiency of PTC as well as other agents aiming at the suppression of NMD.
Unlike NBCe1 KO and W516X KI mice, NHE3 KO mice showed only a mild acidemia [57]. These results suggest that while the basolateral transport system cannot compensate for the lack of NBCe1, the apical transport system may be able to at least partially compensate for the lack of NHE3 in proximal tubules. Indeed, functional analysis using isolated renal proximal tubules from NHE3 KO mice revealed the residual amiloridesensitive NHE activity, which corresponded to approximately 50% of the wild-type activity [64]. This residual NHE activity, also found in NHE2/NHE3 double KO mice, might represent the activity of NHE8, another isoform of NHE recently identified to exist in the apical membrane of renal proximal tubules [64,65]. Until now, mutations in NHE3 or NHE8 have not been identified in human pRTA patients.
8. ACKNOWLEDGEMENTS
This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
REFERENCES
- Romero, M.F. and Boron, W.F. (1999) Electrogenic Na+/
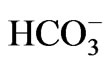 cotransporters: Cloning and physiology. Annual Review of Physiology, 61, 699-723. doi:10.1146/annurev.physiol.61.1.699
cotransporters: Cloning and physiology. Annual Review of Physiology, 61, 699-723. doi:10.1146/annurev.physiol.61.1.699 - Igarashi, T., Inatomi, J., Sekine, T., Cha, S.H., Kanai, Y., et al. (1999) Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nature Genetics, 23, 264-266. doi:10.1038/15440
- Boron, W.F. (2006) Acid-base transport by the renal proximal tubule. Journal of the American Society of Nephrology, 17, 2368-2382. doi:10.1681/ASN.2006060620
- Ishiguro, H., Steward, M.C., Lindsay, A.R. and Case, R.M. (1996) Accumulation of intracellular HCO3− by Na+-HCO3– cotransport in interlobular ducts from guinea-pig pancreas. The Journal of Physiology, 495(Part 1), 169-178.
- Usui, T., Seki, G., Amano, S., Oshika, T., Miyata, K., et al. (1999) Functional and molecular evidence for Na+- HCO3– cotransporter in human corneal endothelial cells. Pflügers Archiv, 438, 458-462. doi:10.1007/s004240051062
- Chesler, M. (2003) Regulation and modulation of pH in the brain. Physiological Reviews, 83, 1183-1221.
- Igarashi, T., Inatomi, J., Sekine, T., Seki, G., Shimadzu, M., et al. (2001) Novel nonsense mutation in the Na+/HCO3– cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. Journal of the American Society of Nephrology, 12, 713-718.
- Dinour, D., Chang, M.H., Satoh, J., Smith, B.L., Angle, N., et al. (2004) A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. The Journal of Biological Chemistry, 279, 52238-52246. doi:10.1074/jbc.M406591200
- Inatomi, J., Horita, S., Braverman, N., Sekine, T., Yamada, H., et al. (2004) Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflügers Archiv, 448, 438-444. doi:10.1007/s00424-004-1278-1
- Horita, S., Yamada, H., Inatomi, J., Moriyama, N., Sekine, T., et al. (2005) Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. Journal of the American Society of Nephrology, 16, 2270-2278. doi:10.1681/ASN.2004080667
- Demirci, F.Y., Chang, M.H., Mah, T.S., Romero, M.F. and Gorin, M.B. (2006) Proximal renal tubular acidosis and ocular pathology: A novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Molecular Vision, 12, 324-330.
- Suzuki, M., Vaisbich, M.H., Yamada, H., Horita, S., Li, Y., et al. (2008) Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflügers Archiv, 455, 583-593. doi:10.1007/s00424-007-0319-y
- Suzuki, M., Van Paesschen, W., Stalmans, I., Horita, S., Yamada, H., et al. (2010) Defective membrane expression of the Na+-HCO3−cotransporter NBCe1 is associated with familial migraine. Proceedings of the National Academy of Sciences of the United States of America, 107, 15963-15968. doi:10.1073/pnas.1008705107
- Gawenis, L.R., Bradford, E.M., Prasad, V., Lorenz, J.N., Simpson, J.E., et al. (2007) Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3- cotransporter. The Journal of Biological Chemistry, 282, 9042-9052. doi:10.1074/jbc.M607041200
- Lo, Y.F., Yang, S.S., Seki, G., Yamada, H., Horita, S., et al. (2011) Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kidney International, 79, 730-741. doi:10.1038/ki.2010.523
- Abuladze, N., Song, M., Pushkin, A., Newman, D., Lee, I., et al. (2000) Structural organization of the human NBC1 gene: KNBC1 is transcribed from an alternative promoter in intron 3. Gene, 251, 109-122. doi:10.1016/S0378-1119(00)00204-3
- Bevensee, M.O., Schmitt, B.M., Choi, I., Romero, M.F. and Boron, W.F. (2000) An electrogenic Na+-HCO3– cotransporter (NBC) with a novel COOH-terminus, cloned from rat brain. American Journal of Physiology—Cell Physiology, 278, C1200-C1211.
- Shirakabe, K., Priori, G., Yamada, H., Ando, H., Horita, S., et al. (2006) IRBIT, an inositol 1,4,5-trisphosphate receptor-binding protein, specifically binds to and activates pancreas-type Na+/HCO3– cotransporter 1 (pNBC1). Proceedings of the National Academy of Sciences of the United States of America, 103, 9542-9547. doi:10.1073/pnas.0602250103
- Schmitt, B.M., Biemesderfer, D., Romero, M.F., Boulpaep, E.L. and Boron, W.F. (1999) Immunolocalization of the electrogenic Na+-HCO3– cotransporter in mammalian and amphibian kidney. American Journal of Physiology, 276, F27-F38.
- Yamada, H., Yamazaki, S., Moriyama, N., Hara, C., Horita, S., et al. (2003) Localization of NBC-1 variants in human kidney and renal cell carcinoma. Biochemical and Biophysical Research Communications, 310, 1213-1218. doi.org/10.1016/j.bbrc.2003.09.147
- Usui, T., Hara, M., Satoh, H., Moriyama, N., Kagaya, H., et al. (2001) Molecular basis of ocular abnormalities associated with proximal renal tubular acidosis. The Journal of Clinical Investigation, 108, 107-115.
- Marino, C.R., Jeanes, V., Boron, W.F. and Schmitt, B.M. (1999) Expression and distribution of the Na+-HCO3– cotransporter in human pancreas. American Journal of Physiology, 277, G487- G494.
- Satoh, H., Moriyama, N., Hara, C., Yamada, H., Horita, S., et al. (2003) Localization of Na+-HCO3- cotransporter (NBC-1) variants in rat and human pancreas. American Journal of Physiology—Cell Physiology, 284, C729- C737.
- Majumdar, D., Maunsbach, A.B., Shacka, J.J., Williams, J.B., Berger, U.V., et al. (2008) Localization of electrogenic Na/bicarbonate cotransporter NBCe1 variants in rat brain. Neuroscience, 155, 818-832. doi:10.1016/j.neuroscience.2008.05.037
- Bok, D., Schibler, M.J., Pushkin, A., Sassani, P., Abuladze, N., et al. (2001) Immunolocalization of electrogenic sodium-bicarbonate cotransporters pNBC1 and kNBC1 in the rat eye. American Journal of Physiology—Renal Physiology, 281, F920-F935.
- Yoshitomi, K., Burckhardt, B.C. and Fromter, E. (1985) Rheogenic sodium-bicarbonate cotransport in the peritubular cell membrane of rat renal proximal tubule. Pflügers Archiv, 405, 360-366. doi:10.1007/BF00595689
- Seki, G., Coppola, S., Yoshitomi, K., Burckhardt, B.C., Samarzija, I., et al. (1996) On the mechanism of bicarbonate exit from renal proximal tubular cells. Kidney International, 49, 1671-1677. doi:10.1038/ki.1996.244
- Gross, E., Hawkins, K., Abuladze, N., Pushkin, A., Cotton, C.U., et al. (2001) The stoichiometry of the electrogenic sodium bicarbonate cotransporter NBC1 is cell-type dependent. The Journal of Physiology, 531, 597-603. doi:10.1111/j.1469-7793.2001.0597h.x
- Heyer, M., Muller-Berger, S., Romero, M.F., Boron, W.F. and Fromter, E. (1999) Stoichiometry of the rat kidney Na+- HCO3– cotransporter expressed in Xenopus laevis oocytes. Pflügers Archiv, 438, 322-329. doi:10.1007/s004240050916
- Sciortino, C.M. and Romero, M.F. (1999) Cation and voltage dependence of rat kidney electrogenic Na+-HCO3– cotransporter, rkNBC, expressed in oocytes. American Journal of Physiology, 277, F611-F623.
- Muller-Berger, S., Ducoudret, O., Diakov, A. and Fromter, E. (2001) The renal Na+HCO3–cotransporter expressed in Xenopus laevis oocytes: change in stoichiometry in response to elevation of cytosolic Ca2+ concentration. Pflügers Archiv, 442, 718-728. doi:10.1007/s004240100592
- Seki, G., Coppola, S. and Fromter, E. (1993) The Na+-HCO3– cotransporter operates with a coupling ratio of 2 HCO3– to 1 Na+ in isolated rabbit renal proximal tubule. Pflügers Archiv, 425, 409-416. doi:10.1007/BF00374866
- Muller-Berger, S., Nesterov, V.V. and Fromter, E. (1997) Partial recovery of in vivo function by improved incubation conditions of isolated renal proximal tubule. II. Change of Na-HCO3 cotransport stoichiometry and of response to acetazolamide. Pflügers Archiv, 434, 383-391.
- McAlear, S.D., Liu, X., Williams, J.B., McNicholas-Bevensee, C.M. and Bevensee, M.O. (2006) Electrogenic Na/HCO3 cotransporter (NBCe1) variants expressed in Xenopus oocytes: functional comparison and roles of the amino and carboxy termini. The Journal of General Physiology, 127, 639-658. doi:10.1085/jgp.200609520
- Yang, D., Shcheynikov, N., Zeng, W., Ohana, E., So, I., et al. (2009) IRBIT coordinates epithelial fluid and HCO3- secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. The Journal of Clinical Investigation, 119, 193-202.
- Ando, H., Mizutani, A., Matsu-ura, T. and Mikoshiba, K. (2003) IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. The Journal of Biological Chemistry, 278, 10602-10612. doi:10.1074/jbc.M210119200
- Devogelaere, B., Nadif Kasri, N., Derua, R., Waelkens, E., Callewaert, G., et al. (2006) Binding of IRBIT to the IP3 receptor: Determinants and functional effects. Biochemical and Biophysical Research Communications, 343, 49-56. doi:10.1016/j.bbrc.2006.02.119
- Ando, H., Mizutani, A., Kiefer, H., Tsuzurugi, D., Michikawa, T., et al. (2006) IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Molecular Cell, 22, 795-806. doi.:10.1016/j.molcel.2006.05.017
- Yang, D., Li, Q., So, I., Huang, C.L., Ando, H., et al. (2011) IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. The Journal of Clinical Investigation, 121, 956-965. doi:10.1172/JCI43475
- Wu, J., McNicholas, C.M. and Bevensee, M.O. (2009) Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates the electrogenic Na/HCO3 cotransporter NBCe1-A expressed in Xenopus oocytes. Proceedings of the National Academy of Sciences of the United States of America, 106, 14150-14155. doi:10.1073/pnas.0906303106
- Yamaguchi, S. and Ishikawa, T. (2008) The electrogenic Na+-HCO3– cotransporter NBCe1-B is regulated by intracellular Mg2+. Biochemical and Biophysical Research Communications, 376, 100-104. doi:10.1016/j.bbrc.2008.08.104
- Zhu, Q., Kao, L., Azimov, R., Newman, D., Liu, W., et al. (2010) Topological location and structural importance of the NBCe1-A residues mutated in proximal renal tubular acidosis. The Journal of Biological Chemistry, 285, 13416-13426. doi:10.1074/jbc.M109.093286
- Shiohara, M., Igarashi, T., Mori, T. and Komiyama, A. (2000) Genetic and long-term data on a patient with permanent isolated proximal renal tubular acidosis. European Journal of Pediatrics, 159, 892-894. doi:10.1007/PL00008363
- Lacruz, R.S., Smith, C.E., Moffatt, P., Chang, E.H., Bromage, T.G., et al. (2011) Requirements for ion and solute transport, and pH regulation, during enamel maturation. Journal of Cellular Physiology, in press. doi:10.1002/jcp.22911
- Li, H.C., Szigligeti, P., Worrell, R.T., Matthews, J.B., Conforti, L., et al. (2005) Missense mutations in Na+:HCO3– cotransporter NBC1 show abnormal trafficking in polarized kidney cells: A basis of proximal renal tubular acidosis. American Journal of Physiology—Renal Physiology, 289, F61-F71. doi:10.1152/ajprenal.00032.2005
- Toye, A.M., Parker, M.D., Daly, C.M., Lu, J., Virkki, L.V., et al. (2006) The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. American Journal of Physiology—Cell Physiology, 291, C788-C801. doi:10.1152/ajpcell.00094.2006
- Lipton, R.B., Scher, A.I., Kolodner, K., Liberman, J., Steiner, T.J., et al. (2002) Migraine in the United States: epidemiology and patterns of health care use. Neurology, 58, 885-894.
- Ophoff, R.A., Terwindt, G.M., Vergouwe, M.N., van Eijk, R., Oefner, P.J., et al. (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell, 87, 543-552. doi:10.1016/S0092-8674(00)81373-2
- De Fusco, M., Marconi, R., Silvestri, L., Atorino, L., Rampoldi, L. et al. (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nature Genetics, 33, 192-196. doi:10.1038/ng1081
- Dichgans, M., Freilinger, T., Eckstein, G., Babini, E., Lorenz-Depiereux, B., et al. (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet, 366, 371-377. doi:10.1016/S0140-6736(05)66786-4
- Goadsby, P.J. (2007) Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends in Molecular Medicine, 13, 39-44. doi:10.1016/j.molmed.2006.11.005
- Brune, T., Fetzer, S., Backus, K.H. and Deitmer, J.W. (1994) Evidence for electrogenic sodium-bicarbonate cotransport in cultured rat cerebellar astrocytes. Pflügers Archiv, 429, 64-71. doi:10.1007/BF02584031
- Alper, S.L. (2002) Genetic diseases of acid-base transporters. Annual Review of Physiology, 64, 899-923. doi:10.1146/annurev.physiol.64.092801.141759
- Kao, L., Sassani, P., Azimov, R., Pushkin, A., Abuladze, N., et al. (2008) Oligomeric structure and minimal functional unit of the electrogenic sodium bicarbonate cotransporter NBCe1-A. The Journal of Biological Chemistry, 283, 26782-26794. doi:10.1074/jbc.M804006200
- Svichar, N., Esquenazi, S., Chen, H.Y. and Chesler, M. (2011) Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization-induced alkalinization. The Journal of Neuroscience, 31, 6997-7004. doi:10.1523/JNEUROSCI.6088-10.2011
- Ji, W., Foo, J.N., O'Roak, B.J., Zhao, H., Larson, M.G., et al. (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nature Genetics, 40, 592-599. doi:10.1038/ng.118
- Schultheis, P.J., Clarke, L,L., Meneton, P., Miller, M.L., Soleimani, M., et al. (1998) Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nature Genetics, 19, 282-285. doi:10.1038/969
- Woo, A.L., Noonan, W.T., Schultheis, P.J., Neumann, J.C., Manning, P.A., et al. (2003) Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. American Journal of Physiology—Renal Physiology, 284, F1190-F1198.
- Gurley, S.B., Riquier-Brison, A.D., Schnermann, J., Sparks, M.A., Allen, A.M., et al. (2011) AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metabolism, 13, 469-475. doi:10.1016/j.cmet.2011.03.001
- Yamazaki, O., Yamada, H., Suzuki, M., Horita, S., Shirai, A., et al. (2011) Functional characterization of nonsynonymous single nucleotide polymorphisms in the electrogenic Na+-HCO3– cotransporter NBCe1A. Pflügers Archiv, 461, 249-259. doi:10.1007/s00424-010-0918-x
- Lu, J. and Boron, W.F. (2007) Reversible and irreversible interactions of DIDS with the human electrogenic Na/HCO3 cotransporter NBCe1-A: role of lysines in the KKMIK motif of TM5. American Journal of Physiology—Cell Physiology, 292, C1787-C1798. doi:10.1152/ajpcell.00267.2006
- Welch, E.M., Barton, E.R., Zhuo, J., Tomizawa, Y., Friesen, W.J., et al. (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature, 447, 87-91. doi:10.1038/nature05756
- Du, M., Liu, X., Welch, E.M., Hirawat, S., Peltz, S.W., et al. (2008) PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proceedings of the National Academy of Sciences of the United States of America, 105, 2064-2069. doi:10.1073/pnas.0711795105
- Choi, J.Y., Shah, M., Lee, M.G., Schultheis, P.J., Shull, G.E., et al. (2000) Novel amiloride-sensitive sodium-dependent proton secretion in the mouse proximal convoluted tubule. The Journal of Clinical Investigation, 105, 1141-1146. doi:10.1172/JCI9260
- Goyal, S., Vanden Heuvel, G. and Aronson, P.S. (2003) Renal expression of novel Na+/H+ exchanger isoform NHE8. American Journal of Physiology—Renal Physiology, 284, F467-F473.