Advances in Biological Chemistry
Vol.3 No.3(2013), Article ID:32386,13 pages DOI:10.4236/abc.2013.33031
Homeostatic cell cycle and the origin of autophagosomal vesicles
![]()
Research Center, University of Medicine and Dentistry of New Jersey, Newark, USA
Email: slomiaam@umdnj.edu
Copyright © 2013 Amalia Slomiany, Bronislaw L. Slomiany. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 23 March 2013; revised 5 May 2013; accepted 14 May 2013
Keywords: Autophagosomes; ER-Transport Vesicles; Cell Organelle Repair; Lysosomal Cell Debridement; Homeostatic Cell Cycle
ABSTRACT
The autophagosomes were identified in the viable cycloheximide (CHX)-treated cells which had an incapacitated translational process and thus disabled synthesis of endoplasmic reticulum (ER)-derived vesicular transporters. They were found devoid of the proteins transported from ER to cell organelles, were unable to fuse with ER, Golgi or mitochondria, and displayed affinity with lysosomes. The analysis of autophagosomes, derived from the CHX cell organelles, revealed that their lipid composition resemble that of the maternal organelle. Thus, the ER-derived autophagosomes were marked with the presence of phosphatidylinositol (PI), Golgi-derived vesicles contained sphingomyelin (SM) and glycosphingolipids (GLL), and the mitochondria-derived autophagosomes contained phosphatidylglycerol (PG) and cardiolipin (CL). The incubation of the vesicles with intact lysosomes afforded their and the lysosome membrane lipids degradation. The analysis of the products derived from incubation of lysosomes and autophagosomes with radiolabeled SM, in the presence and the absence of TritonX100, allowed us to conclude that during autophagosome degradation the lysosomal enzymes are not released to cytosol, and that only lysosomes contain the enzymes degrading membrane lipids. In summary, our findings allowed us to authenticate the vesicles generated in the CHX-treated cells as organelle-specific autophagosomes and to determine that complete cycle of cell restitution and debridement includes intralysosomal degradation of the lysosomal membrane engulfing the autophagosomes vesicles.
1. INTRODUCTION
In theory, an autophagy, the degradation of the dysfunctional portions of cell membrane and its organelles, and its synchronization with biomembranes repair and replacement is crucial to maintain the state of equilibrium in the cell size, specificity and function. To satisfy these homeostatic criteria, the autophagy must be organelle specific, proportional to newly assembled membrane replacement, and be completed by the delivery of the excised dysfunctional portion of the organelles to lysosomes for degradation. Therefore, a vital and the overriding feature in the design of the investigation tracing the intracellular transport is to retain the unmodified membrane lipid and protein make-up, and the setting that closely parallels the in situ environment that would allow tracking the progressive movement of the synthetic cargo transporting vesicles and generation of autophagosomes [1-4]. Knocking out gene technique is the most popular way to probe a protein’s destination and function, but such manipulation can create unnoticeable yet relevant consequence in the transport, by generating central change that destroys destination marker of the transporter [4-9]. Or, in certain situations, the modified by deletion complex may trigger cellular machinery created to eliminate misshapen product, or the product is diverted to inappropriate cellular location [4-10]. Hence, as the consequence of gene knockout the cell architecture and function may be modified or destroyed [9-13]. Just as risky is the approach relying on point mutation technique, which can impact protein folding and its organization in the membrane, and thus disturb the vesicular structure responsible for cellular homeostasis [5-7,10,14]. Moreover, the results of the studies embracing genetic manipulation and point mutation techniques are assessed under the conditions that minimize function of membrane-specific lipids or reduce their involvement to mere hydrophobic background for the protein folding.
These apparent shortfalls in the general strategy that advance solitary contribution of protein in the cellular transport, repair, and debridement, have been addressed by our investigative approach tracing labeled cargo and fusion of the vesicular membranes lipids with the cellular membrane or its organelle [15-20]. This approach allowed us to document the elemental dissimilarities in the composition of the transport vesicles, the dissimilarities of the lipids that are integrated at the transporters conception in the Endoplasmic Reticulum (ER), the differences substantiated further as the vesicles membrane and protein cargo undergo refinement in Golgi, and additional divergences produced until incorporation of the ultimate structure into specific site of the cell is accomplished [15-24]. Cumulatively, these investigations allowed us to conclude that precise, destination-specific vesicular construction is a crucial attribute of the homeostatic repair and restitution [15,17,18,21,25,26]. The validation of the concept that entails crucial role of lipids in ER initiation of biomembrane configuration, and identification of the en bloc membrane inserts in the cellular and organellar membranes, thus cemented the role of specific lipids in the restitution of cells.
To corroborate the deductive logic of our concept of homeostatic criteria, the information on catabolic elimination of the dysfunctional portion of the organelles reflected in the production of the autophagosomes, should endorse organelle-specific elimination of its fragments and reflect surgical resections of the damaged and being replaced membrane segments. Consequently, each such resection, from each site and organelle, should produce unique vesicles that compositionally reflect their derivation site and display affinity for association with lysosomes. Accordingly, every viable cell that is actively involved in the synthesis and secretion, metabolism and energy production and consumption, generates autophagosomes that reflect turnover of the specific organelles and the sites of cellular membrane. This idea of the autophagosomes origin is to some degree supported by numerous reports providing evidence that autophagosomes contain the unique, organelle-specific markers, but again, their membrane origin was not documented and still the subject of intense debate [27-33]. Therefore, in keeping with our advances in organelle specific membrane synthesis and delivery, and the fidelity of cell restitution, we have engaged in the investigation to document that autophagosomal vesicles reflect the organelle-specific membrane resections destined for lysosomal degradation.
2. MATERIALS AND METHODS
2.1. Preparation of Cells for Subcellular Fractionation
For the isolation of cell components, the initial step of fragmentation of the structure is crucial and a method is required that gives minimum breakage. Hence, in the method described herein we used the techniques which allowed us to purify cell organelles with minimum fragmentation of their structure, so that Cell Cytosol (CC) was not admixed with organellar components and that cell membranes were not admixed with intracellular membranes. In further preparation of organelles, we aimed to isolate the undamaged structure so we could use them for incubation with CC and formation of the transport or autophagosomal vesicles.
The cells were prepared from rat gastric mucosa and the liver as described previously [17,18,22,26,34]. The single cells, that were separated from larger debris with the aid of specific cell size nylon mesh, were centrifuged at 50 × g for 2 min, washed twice with the enzyme-free medium, twice with the Minimum Essential Medium (MEM), and counted in hemocytometer. Thus prepared cells were then incubated in MEM for 3 hours with or without radiolabel, then where indicated incubated for 30 min with 100 nM cycloheximide (CHX), and used for preparation of nuclei [22], subcellular organelles, cell cytosol [17,22,23,25,26] and cellular membranes [15,23]. In the experiments dedicated to the vesicles synthesis with translation active CC or RNase or CHX treated CC, the preparations of the organelles were additionally rinsed with phosphate buffered saline (PBS), or 0.5 M NaCl or urea-PBS, in order to remove the associated residual cytosolic proteins that otherwise would remain on their membranes. The synthesis of phospholipids and protein was assessed using radiolabeled [3H] inositol, [3H] arachidonate, [3H] choline, [3H] serine, [3H] palmitate and [32P] ATP [17,18,21-23,26]. The radiolabeled CHX incubated cells’ organelles were incubated with vesiclesdepleted CHX cytosol and the formed products subjected to separation from organelles as described earlier for ER, Golgi and mitochondria-derived biosynthetic transport vesicles [17,18,21-23,25,26]. The vesicles generated in the CHX-treated cells were subjected to fusion experiments with Golgi, ER, mitochondria and lysosomes in the medium containing cold, vesicles-free, CHX-derived CC at concentration of 15 mg protein/ml of incubation mixture enriched with 50 mM ATP, 250 mM CTP, 50 mM GTP, 5 mM creatine phosphate, 8.0 IU/ml creatine kinase, and where indicated 100 nM CHX or 25 mg/ml RNase, 10 mM UDP-Glc and 10 mM palmitoyl CoA [17,18,21- 23,25,26].
2.2. Preparation of the Active Cell Cytosol (CC)
The viable cells (normal, or CHX-treated) were homogenized for 10 sec at 600 rpm in 3 volumes of buffer containing 0.25 M sucrose; 50 mM TRIS-HCl (pH 7.4), 25 mM magnesium acetate and 10 mM each of aprotinin, leupeptin, chemostatin, and 1 mM phenylmethylsulfonylfluoride, were centrifuged at 5000 × g for 15 min. The supernatant, diluted with 2 volumes of homogenization buffer, was re-centrifuged at 10,000 × g for 20 min. The resulting supernatant was then subjected to centrifugation at 100,000 × g for 1 h. Thus obtained soluble fraction was adjusted to 15 - 18 mg protein/ml, admixed with an ATP generating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml creatine phosphokinase, and referred to as transport active cell cytosol (CC) or (CHX CC). Then, the vesicles remaining in the obtained cytosols were removed by centrifugation at 150,000 × g for 1 h, and the spun down material suspended in 55% sucrose, overlaid with 55% - 30% gradient and centrifuged at 150,000 × g for 16 h. The radiolabeled material was recovered from the gradient as reported earlier for isolation of transport vesicles from ER, Golgi and mitochondria [17,18,21-23,25,26,35].
2.3. Preparation of Cellular Organelles and Membranes
The cell membranes and subcellular organelle fractions (mitochondria, ER, Golgi) were recovered from the cold or radiolabeled cells as described earlier [17,18,21-23, 26]. The ER and Golgi organelles sediment, remaining after separation of nuclei, mitochondria, endosomes and lysosomes, and cell cytosol, was suspended in the buffer containing 0.2 M PIPES (pH 6.9), 2.0 M glycerol, 1 mM EGTA and 1.0 mM magnesium acetate and applied on the top of discontinuous gradient of 2.0/1.5/1.3/1.0 M sucrose and centrifuged at 100,000 × g for 16 h. The cell membranes were recovered from 1.0 M sucrose, ER from 1.3 M and 1.5 M sucrose and Golgi from the top of the 2.0 M sucrose. Each sucrose-separated fraction was subjected to further purification. The cell membranes were washed with original PIPES buffer and centrifuged at 3000 rpm for 2 min. To separate apical epithelial membranes, the buffer was adjusted with 0.2% Triton X-100 and the mixture incubated at 4˚C for 5 min [21, 23]. This treatment resulted in breaking up the phospholipids-rich membranes into smaller segments and that allowed us to separate apical membranes containing cholesterol, glycosphingolipids and glycoproteins. The latter membranes were recovered by low speed centrifugation at 3000 rpm for 2 min.
The mitochondria and lysosomes were purified from 10,000 × g spun fraction. From the initial fraction, the fluffy layer of broken mitochondria and ER microsomes that covered the crude mitochondrial pellet was suctioned off, the pellet was gently resuspended in five volumes of medium consisting of 0.07 M sucrose, 0.2 M mannitol, 0.1 mM disodium EDTA, and 1 mM TRIS, pH 7.2, and the suspension was spun for 10 min at low speed (500 × g), the supernatant was recovered and spun for 10 min at 9000 × g [25]. This manipulation was repeated three times. Thus purified preparation of mitochondria contained only occasional broken mitochondria, ER membranes, or lysosomes. The endosomes and lysosomes were recovered from the mitochondrial fraction following treatment that afforded swelling of mitochondria [36-38]. The swollen mitochondria were separated from lysosomes by means of isoosmotic gradient [38], and the lysosomes used for incubation with vesicles recovered from CC, and the organelles subjected to incubation with vesicle-free CHX CC. Mitochondrial fraction recovered from the gradient was used for the preparation of mitochondrial lipids and thin layer chromatography [25].
2.4. Generation and Purification of the Vesicles
ERand Golgi-derived transport vesicles were generated in the presence of radiolabeled precursors according to procedure described previously [17,18,21-23,26], and where indicated, in the presence of Brefeldin A (BFA) [24], and cycloheximide (CHX) [39,40]. The ER or Golgi membranes, mixed with CC, ATP-generating system, UTP, CTP GTP, fatty acyl CoA and water soluble cold or radiolabeled lipids precursors, were incubated for 30 min at 37˚C, centrifuged over 0.3 M sucrose and treated with stripping buffer at 2˚C for 15 min followed by centrifugation at 10,000 × g for 10 min to separate transport vesicles from ER or Golgi membranes. The separated from maternal membranes transport vesicles were recovered from the supernatant resulting from centrifugation of the supernatant mixture at 150,000 × g for 1 h. The crude fraction of the transport vesicles was suspended in 55% sucrose, overlaid with 55% - 30% gradient and centrifuged at 150,000 × g for 16 h. The purified transport vesicles were recovered from the gradients as reported earlier [17,18,21-23,26].
2.5. Incubation of Transport Vesicles with ER, Golgi, Mitochondria and Lysosomes
One volume of radiolabeled vesicles (1.3 - 1.5 mg protein/ml) was suspended in one volume of CC or CHX CC (15 mg protein/ml), and added to one volume of cold cell organelles (5 mg protein/ml). The reaction was allowed to proceed from 0 - 30 min at 4˚C (control) and at 37˚C in the presence of ATP regenerating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml of creatine phosphokinase, or in the ATP depleting system containing 5 mM glucose and 500 units/ml hexokinase. After incubation, the respective organelles were recovered by centrifugation through three volumes of 0.5 M sucrose at 3000 rpm for 5 min. The vesicles recovered from the supernatant after incubation with Golgi were purified on 55% - 30% sucrose gradient and used in fusion experiments with cold mitochondria, and with lysosomes. One volume of the recovered vesicles (0.9 - 1.1 mg/ml) was resuspended in one volume of CC (15 mg/ml) and added to one volume of purified mitochondria, or lysosomes (5 mg/ml). Each reaction was allowed to proceed for up to 30 min under same conditions as described for the ER vesicles fusion with Golgi. In the experiments estimating en bloc fusion of transport vesicles with Golgi and/or mitochondria or lysosomes, the associated but not fused vesicles were released from the membrane by subjecting the membrane fraction to treatment with 2 M urea at 4˚C or 0.5 M NaCl, and then the recovered organelles were centrifuged through 0.5 M sucrose, washed and subjected to radiolabeled lipid analysis.
3. RESULTS
In the investigations presented herein we concentrated on the process of elimination of the superfluous and unwanted elements of the organelles that in viable cells are released to cytosol in form of autophagosomes and require removal by means of catabolic degradation. In order to reduce contribution of the vesicles from the synthetic arm of the transport/restitution cycle, the cells were subjected to the translation/elongation inhibition treatment with CHX [39-41], and the cytosolic vesicles produced by autophagocytic activity characterized. Also, the subcellular fractions representing ER, Golgi, and mitochondria were subjected to the incubation with the transport active CHX CC to produce organelle specific vesicles which then were tested for their ability to associate and fuse with cell organelles and their lipid composition characterized. The vesicles recovered from active CC of the CHX treated cells are shown in Figure 1.
As illustrated above, the CHX-treated cell cytosol contained serine labeled vesicles, and displayed similar
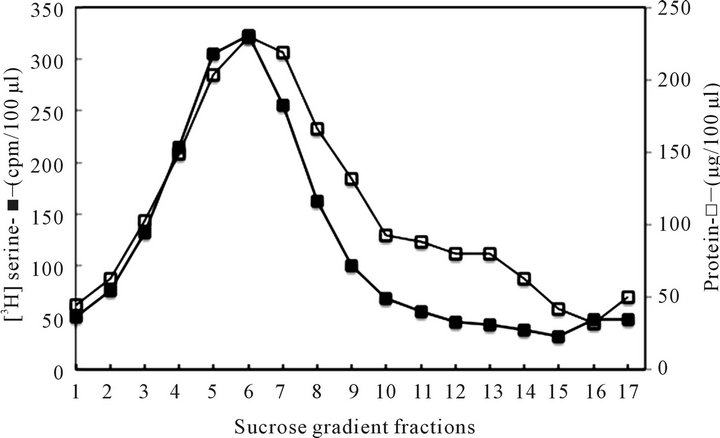
Figure 1. The vesicles isolated from CHX treated cells cytosol. Prior to subcellular fractionation, the purified hepatocytes were subjected to 24 h labeling with serine and/or palmitate and then treated with CHX for 1 h to inhibit translation/elongation and the transport/restitution processes that produce ER-derived transport vesicles. By eliminating the contribution of the synthetic arm of the cycle, the products of the phagosomal activity were uncovered.
sedimentation pattern as the cargo carrying transport vesicles produced in ER during active translation/translocation and vesicles formation activity. The cytosol derived from the CHX treated cells was used for vesicles isolation according to procedures described in Materials and Methods. As apparent, the sedimentation profile of autophagosomes was found to be similar to that of the synthetic transport vesicles characterized in our previous investigations [15,18-20,23,26]. Therefore, to document that these structures are not the remains of the synthetic arm of the cycle, but are created in the process of intracellular catabolic elimination of the organelle fragments, first we determined their cargo content, and their ability to fuse with the organelles that are restored by ER-derived transporters (Golgi, mitochondria), and then use them in experiments determining their reactivity with lysosomes. As demonstrated in Figure 2, the serine and palmitate labeled CHX-treated hepatocytes’ cytosol contained protein and lipid-labeled vesicles, which were cargo-free, were not fusing with Golgi, and the majority associated with lysosomes.
In the preliminary experiment with not separated ERGolgi fraction (not shown in Figure 3), the traces of the vesicular material, that during incubation was not re-
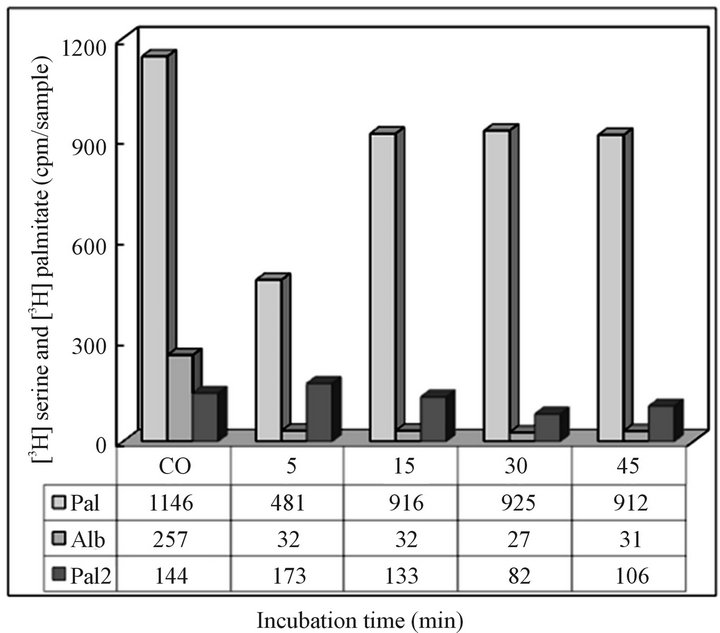
Figure 2. Recovery of radiolabeled CHX cytosol-derived vesicles following 5 - 45 minutes incubation with Golgi (Pa l) and with lysosomes (Pal 2). The corresponding samples of the vesicles used in reaction with organelles were also subjected to PAGE protein separation and estimation of the radiolabeled albumin. The results from the experiment are presented under (Alb). As shown, the vesicles remaining in CHX cytosol contained only traces of the albumin transporting vesicles and were not able to react with Golgi, instead readily associated with lysosomes. Due to small amount of labeled lipids, the origin and the destination of the remaining in cytosol fraction (Pal 2) was not investigated further. The CO column represents the radiolabel recovered from cytosol immediately after combining fractions for incubation with Golgi and/or lysosomes.
tained with lysosomes or fused with Golgi was not changed or displayed the property of the synthetic vesicles at the stage of retrograde transport. Fusion was not detected neither with ER-Golgi, nor Golgi or mitochondrial membranes, and the retained material was easily separated by the treatment with 0.5 M NaCl (Figure 3).
Based on the results presented in Figures 2 and 3 we could afford the conclusion that the CHX treatment-derived cytosol contained still other than autophagosomes, or cargo transporting vesicles, or the postulated retrograde transporters between Golgi and ER [42] since they were not fusing with ER, Golgi or mitochondria, or display affinity for lysosomes. However, as shown in Figure 4, the overwhelming majority of the material was retained with lysosomes. Within 5 min of incubation, the association was nearly complete, and only minimal changes were observed when incubation was continued for up to 45 min. The ability of the CHX cells vesicles to associate with lysosomes sets them apart from synthetic transport vesicles which as demonstrated in earlier investigations displayed capacity to fuse with Golgi, mitochondria, endosomes and specific sites of cell membrane, could not be separated by treatment with 2 M urea or 0.5 M NaCl.
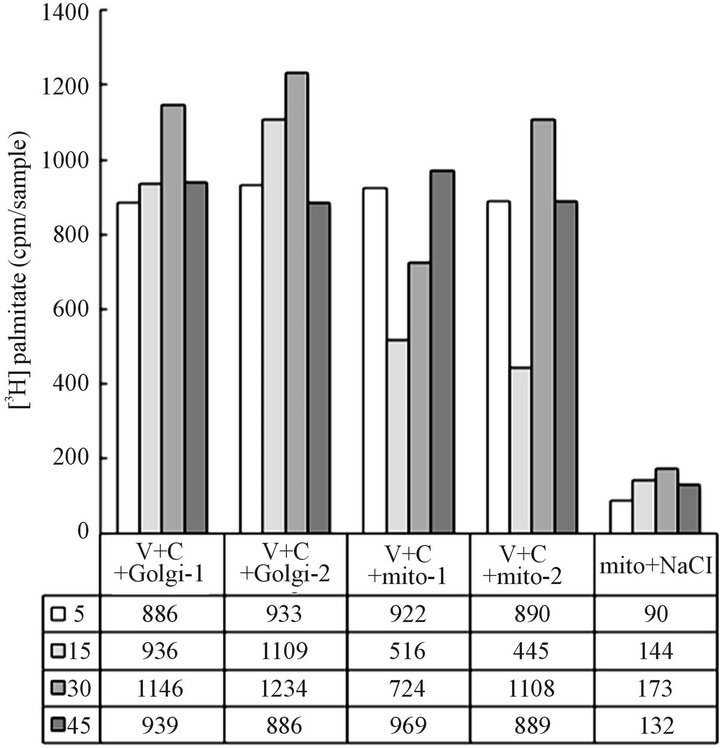
Figure 3. Recovery of CHX cytosol vesicles from the transport-specific reaction with Golgi (V + C + Golgi-1, 2) and with mitochondria (V + C + mito-1, 2). The data under columns (mito + NaCl) depict the results of stripping of the recovered mitochondria with 0.5 M NaCl. It appears that the vesicles which have not reacted with lysosomes (Figure 2) were not the synthetic mitochondrial transporters either. The possibility remains that the fraction of the vesicles not reacting with Golgi, mitochondria or lysosomes may be the transport product destined to endosomes. However, without specific lipid markers, we could not identify the vesicles destined to endosomes.
Hence, the experiments depicted in Figures 1-4, demonstrate that the vesicular material isolated from cytosol of the CHX-treated cells represents cellular vesicles destined for lysosomes, and hence represents the cell autophagosomes awaiting digestion. The latter is proven by the fact that the recovered mixture of the cytosolic vesicles (Figure 4) reacts indiscriminately with lysosomes, the attribute that is shared by all catabolism-destined cellular autophagosomes.
In order to investigate the concept that the autophagosomes are not identical in their lipid composition but reflect compositional similarities with the maternal organelle undergoing repair and restitution, we concentrated our further efforts on isolation of the vesicles which were discharged from CHX treated cells’ organelles when subjected to incubation in active CHX CC. The purified ER, Golgi, and mitochondria were incubated in the CHX-derived vesicle-free CC and then the discharged vesicular material was isolated from the cytosol. As presented in Figures 5 and 6, the incubation of the organelles in the CHX vesicle free cytosol produced material that separated from organelle main fraction in similar manner as the synthetic ER-derived transport vesicles highlighted in our earlier publications [15,17,19, 25]. However, upon their further characterization we found critical differences in their features. As already presented in Figures 2 and 3, for the mixture of vesicles
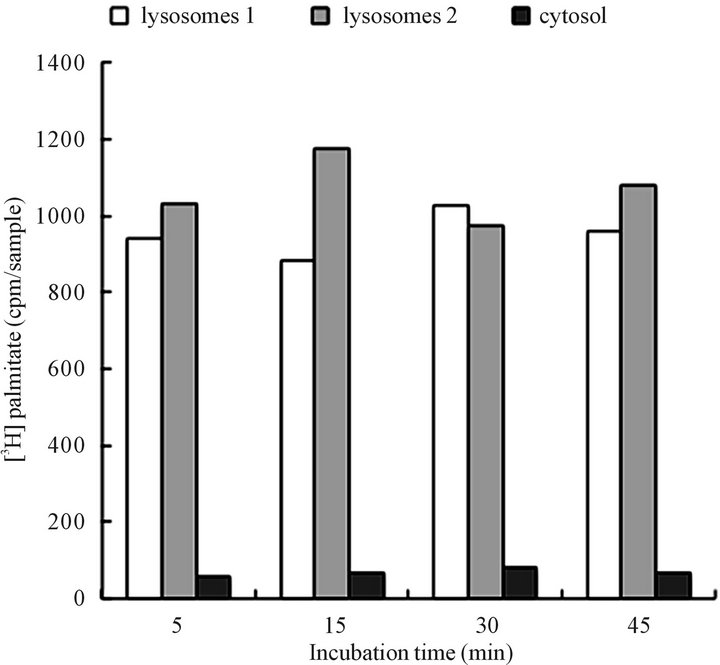
Figure 4. Association of CHX cytosol-derived vesicles with lysosomes. The recovery of radiolabeled tracer from lysosomes (cpm in lysosomes 1, 2) and in cytosol following 5 - 45 minutes incubation with label-free lysosomes and cytosol. The results demonstrate recovery of the palmitate labeled lipids recovered from the intact lysosomes, and from the cytosol following centrifugation of the individual samples. On the average, each incubation sample contained from 1200 cpm - 1400 cpm. The detailed description of the incubation is presented under Materials and Methods.
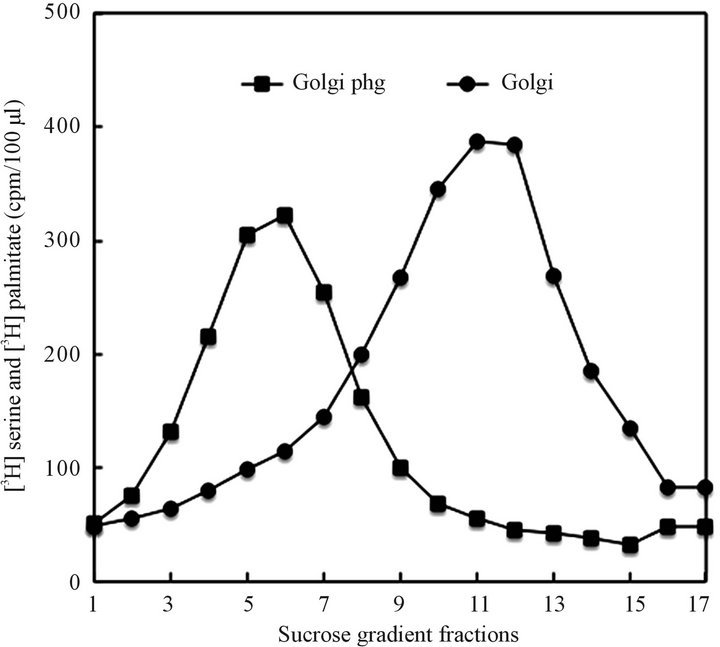
Figure 5. Golgi derived autophagosomal vesicles (Golgi phg) generated upon incubation of the CHX-derived Golgi organelle in CHX cytosol and separation of the vesicles released from Golgi on the sucrose gradient shown in Figure 1, and described in detail in Materials and Methods. As shown in Figure 2 for the mixture of vesicles recovered from the CHX cytosol, the CHX-organelle-released vesicles, in contrast to synthetic transport vesicles, were cargo-free and readily associated with lysosomes. In contrast, the Golgi membranes were not associating with lysosomes.
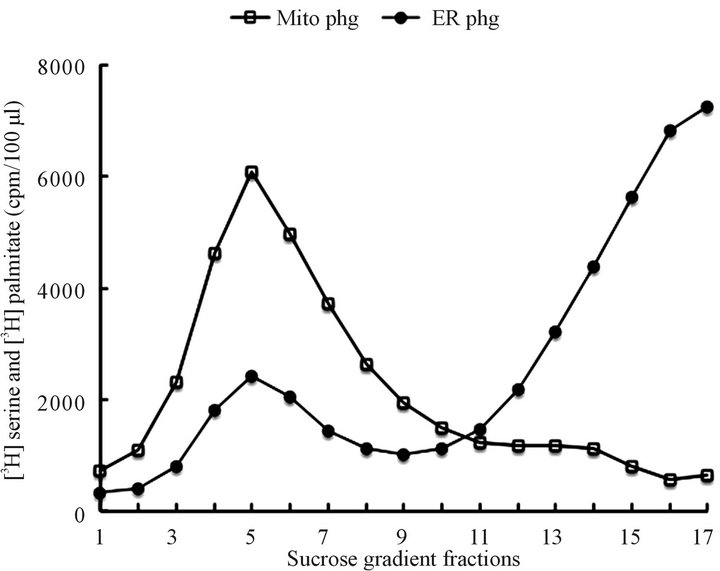
Figure 6. Mitochondriaand ER-derived autophagosomal vesicles generated upon incubation of the CHX cells derived organelles in CHX vesicle-free cytosol. The released vesicles were separated and purified as described in Materials and Methods and illustrated here in Figures 1 and 5. The mitochondrial incubates, prior to sucrose gradient centrifugation, were subjected to 12,000 rpm centrifugation to separate the maternal mitochondria from cytosol-contained mitochondrial autophagosomes (mitoPhg). The ER incubates were recovered from cytosol as described in Materials and Methods and the ER autophagosomes (ERPhg) were separated from the ER on the sucrose gradient depicted above. As apparent the maternal ER fraction consisted of heterogeneous population of the microsomal vesicles.
derived from CHX cytosol, the vesicles derived from the organelles of CHX treated cells would not fuse with Golgi, the Golgi-derived vesicles have not displayed affinity for apical or basolateral membranes, and the mitochondria-derived material would not associate with either Golgi or plasma membranes.
As verified by thin layer chromatography each organelle-derived vesicles consisted of lipids specific for the organelle from which they were derived (Figure 7), and displayed the ability to associate with and be degraded by the lysosomes (Figure 8). As identified by thin-layer chromatography, the Golgi derived vesicles from gastric epithelial cells, in addition to PE, PC, and PI, contained SM and GSL (Figure 7), whereas mitochondria-derived material contained PC, PE, phosphatidylglycerol (PG) cardiolipin (CL), lysoPG, dilysoCL but not GSL, SM or Cer (Figure 9). Moreover, as established for CHX CC derived mixture of vesicles featured in Figures 1-4, the ERand Golgi-derived material did not have the synthetic cargo specific for gastric epithelial cells (mucin) or hepatocytes-specific cargo (albumin). The latter finding
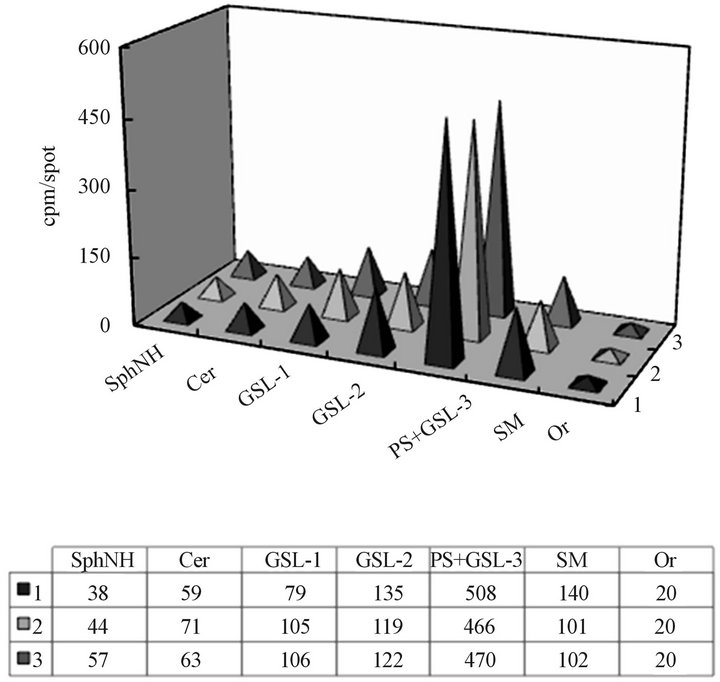
Figure 7. The composition and the serine labeling of CHX Golgi lipids before (1) and after (2) incubation with CHX cytosol, and the vesicles recovered from the cytosol (3). The purified CHX derived Golgi, and the material recovered from fractions 5-7, and 10-12 (Figure 5) corresponding to the cytosol recovered vesicles (Golgi phg) and Golgi (Golgi), respectively were subjected to lipid extraction and thin layer chromatography. The equal amount of radiolabeled lipid extract (2000 cpm/spot together with cold internal standard of Golgi-derived lipids) was applied and subjected to separation in the solvent system consisting of chloroform/methanol/water (65:35:8, v/v/v). Following iodine visualization, the areas corresponding to sphingosine (SphNH), ceramide (Cer), glycosphingolipds 1-3 (GSL 1-3), phosphatidylserine (PS), sphingomyelin (SM) and origin (Or) were scraped and the radiolabel quantitated.
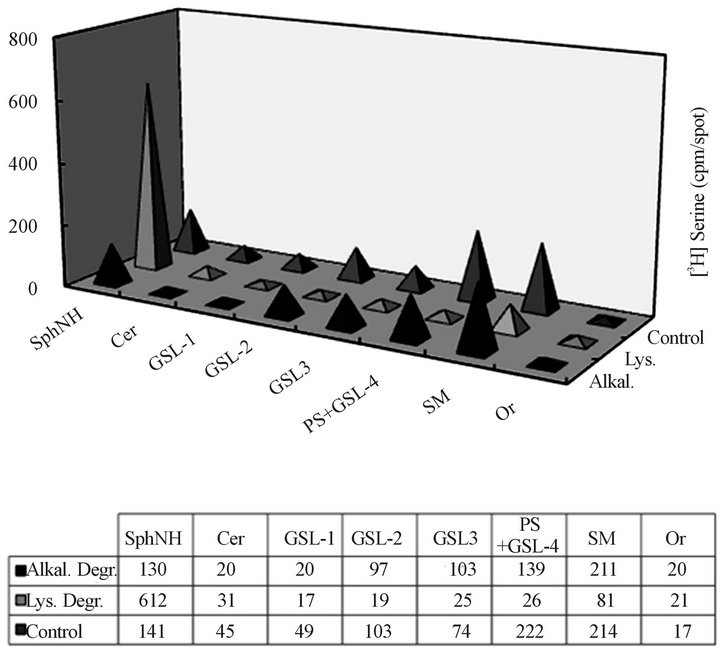
Figure 8. Lysosomal and alkaline degradation products of CHX Golgi-derived autophagosomal vesicles derived from serine labeled cells. The control (untreated), the lysosomal incubation extracts (Lys.) and the products of alkaline degradation (Alkal.) were subjected to TLC in solvent system consisting of chloroform/methanol/water (65:35:8, v/v/v) and radioactivity counting. As reflected in the results, the products of the incubation with lysosomes afforded labeled sphingosine (SphNH), while alkaline degradation impacted PS only. Thus, we concluded that incubation with lysosomes produces terminal degradation of the membrane lipids contained in the Golgiderived autophagosomes.
alone was indicative that the vesicles are representing cellular process that does not contribute to cargo transport and could not represent empty transporters either, since they were lacking ability to associate with cargoproducing organelle (ER).
In summary, this portion of our study has demonstrated that the cells treated with CHX and thus with arrested translational ER activity, continue to release vesicular material whose lipid composition reflects maternal organelle membrane from which they were isolated (Figures 8 and 9). Importantly, the common feature of all those organelle-produced and released to cytosol vesicles is their affinity to lysosomes.
The retention of Golgi-derived autophagosomes with lysosomes was not a static association, and as shown by lipid analysis, the incubation resulted in autophagosome degradation manifested in the production of radiolabeled sphingosine, the degradation product derived from SM and GLL (Figure 10).
To demonstrate phagosomal vesicles lipid digestion we used the radiolabeled vesicles from individual organelle preparations and analyzed products after incubation with cold lysosomes. As shown, (Figure 10), upon incubation, the lipids of Golgi autophagosomal vesicles yielded labeled shingosine (SphNH2). To reveal whether
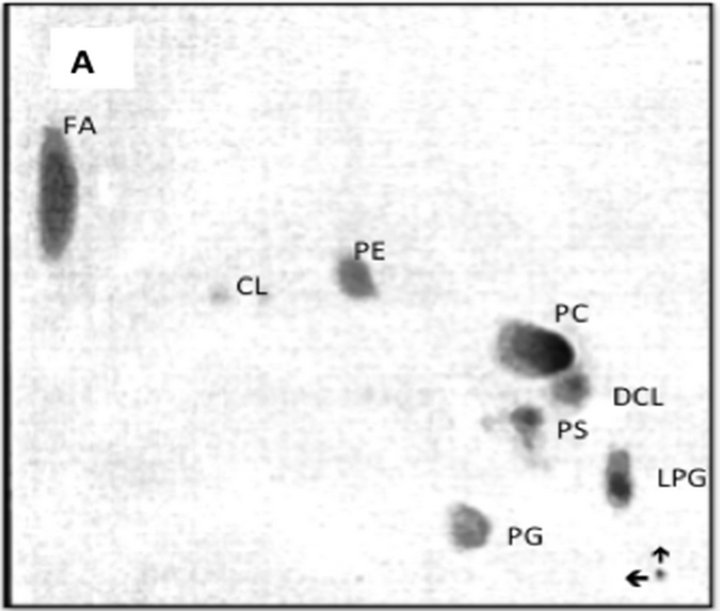
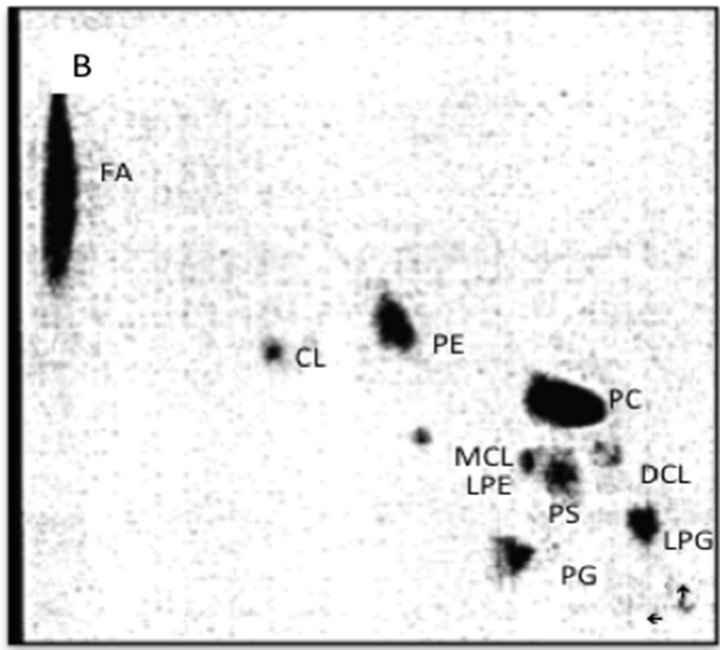
Figure 9. TLC analysis of the lipid composition of the mitochondrial autophagosomes (A) and mitochondria (B) following incubation of the purified organelles in the CHX cytosol. The mitochondria-released autophagosomal vesicles were recovered from the cytosol as shown in Figure 6. The recovered material following extraction with chloroform/ methanol (2:1, v/v) was applied to thin layer plate and subjected to two-dimensional chromatography in three solvent system. First solvent system consisting of petroleum ether/ethyl ether (7:3, v/v) afforded movement of neutral lipids to the top of the plate. Second system consisting of chloroform/methanol/28% ammonia (65:35:6, v/v/v) was applied in the same direction. The third solvent system consisting of chloroform/acetone/methanol/acetic acid/ water, (30:40:10:10:5, by vol.) in second dimension. The separated lipids were visualized using iodine vapors and identified by compering their migration with external standards and their reactivity with ninhydrin (PE, LPE, PS), orcinol (GSL), and phosphate detecting sprays (SM, phosphoglycerides). Abbreviations: FA, fatty acids, CL, cardiolipin, PE, phosphatidylethanolamine, MCL, mono-lyso-cardiolipin, LPE, lysophosphatidylethanolamine, PS, phosphatidylcholine, DCL, di-lyso-cardiolipin, PG, phosphatidylglycerol, LPG, lysophosphatidylglycerol.
catabolism-dedicated vesicles degradation proceeds with or without effect on lysosomal membrane lipid compo-
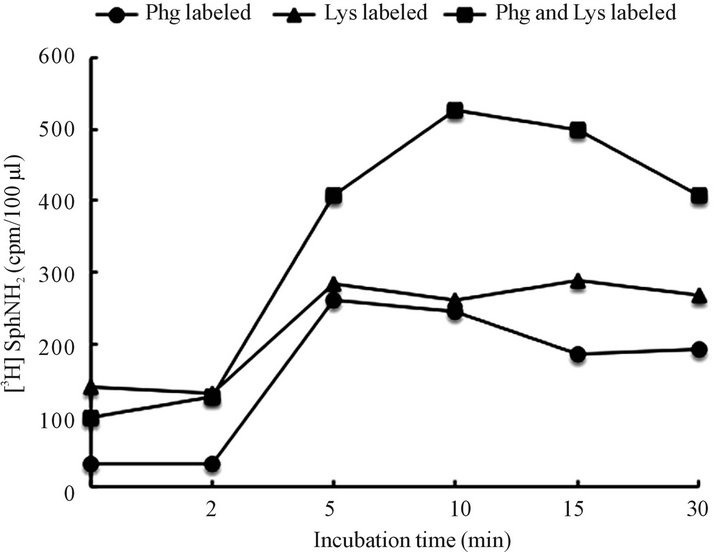
Figure 10. Identification of shingolipid-derived radiolabeled products of Golgi-derived autophagosomes, labeled lysosomes or both labeled organelles following their incubation in CHX cytosol. The incubated samples of the organelles ware subjected to lipid extraction and radiolabeled tracer quantitation. As evident both organelles were releasing free sphingosine (SphNH2) which initially was incorporated in the Golgi derived autophagosomes (shown in Figures 5 and 7) or SM that was found in lysosomes. The fact that radiolabel release was potentiated when both membranes were labeled suggest further that both organelles undergo degradation.
nents, we performed incubation experiments using preparations of labeled lysosomes with cold autophagosomes. Interestingly, within 5 min - 10 min of incubation, the [3H] serine-labeled SphNH2 were released from autophagosomes and from lysosomes.
The issue, which resulted from the demonstration that reaction between autophagosomes and lysosomes releases labeled lipid precursors from both organelles, was whether the degradation was intralysosomal, and if so, why the lysosome membrane-derived SM was digested (Figure 10).
Thus, we faced two alternatives; either autophagosomes had capacity to digest lysosomal membranes or that autophagosomes engulfed by lysosomal membrane were internalized and then digested. The endorsement of the concept of lysosome self degradation was obtained when the sphingomyelinase (SMase) activity was determined by subjecting radiolabeled SM to lysosomal and autophagosomal digestion in the presence and absence of TritonX100. As shown in Figure 11, the SMase activity was only demonstrated in the lysosomes preparation treated with TritonX100. Neither in the intact lysosomes or autophagosomes, nor TritonX100 disrupted autophagosomal vesicles, the SMase activity was discerned.
In our vision of the event, the digestion of autophagosome and lysosomal membrane is only feasible through invagination onto itself of the portions of lysosome engulfing autophagosome and the exposure of its own membrane to hydrolytic milieu. Also, since the re-
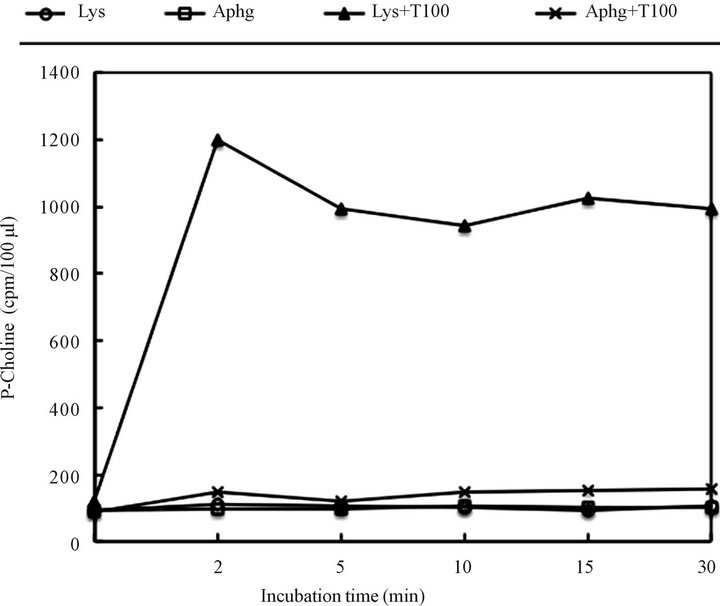
Figure 11. Sphingomyelinase (SMase) activity in the intact and Triton X100 treated lysosomes and autophagosomes. Aliquots of purified organelles (30 - 50 μg protein) were incubated for the indicated time at 37˚C in a buffer (50 μl final volume) containing 20 mM HEPES, 1 mM MgCl2 (pH 7.4) and 4 μl [N-methyl14C] SM (44,000 cpm/tube) in the presence and absence of 0.2% Triton X100. After indicated time of incubation, the tubes ware placed in melting ice-water bath and phosphorylcholine (P-choline) was extracted by adding 250 μl of water and 800 μl chloroform and centrifugation for 3 min at 2700 rpm. Then, 200 μl of water (upper phase) was removed into scintillation vials and counted. To establish P-choline release, a 50 μl of water phase was subjected to thin layer chromatography in the solvent system consisting of methanol/0.9% NaCl/28% NH4OH (50:50:1, v/v/v). The area corresponding to P-choline standard was then scraped and counted.
covered cytosol was not displaying hydrolytic activity toward added substrate, we deduced that the process between autophagosome and lysosome is internalized and contained within the intact lysosome. Otherwise, partial, or complete opening of the lysosomal membranes would show SM degradation in the absence of the detergent. The obtained results let us to speculation that lysosomes engulf the phagosomal vesicles, reseal, and consequently, the organelle-released phagocytic vesicle and the engulfing lysosomal membrane are subjected to intra-lysosomal catabolic dismantling. Hence, the final process of cellular debridement is contained within the lysosome and, as evident in the release of basic lipid metabolites derived from autophagosome and lysosome, is linked with the lysosome self recycling, but without release of the enzymes to cytosol. And thus, the cycle of cell restitution is complete, and as long as cellular cues faci-litate the balanced synthetic and catabolic responses, the cell composition and function remain in homeostatic equilibrium.
4. DISCUSSION
Our investigations of cellular vesicular transport provide evidence that supports the concept of cell restitution as the highly controlled specific process during which portions of cell membrane and its organelles are rebuilt and catabolized with utmost precision and fidelity. The meticulous task of restitution is achieved by ER site-specific, synchronized with lipid synthesis, assembly of the exact replacements of the membranes with their intercalated proteins that form vesicles and thus transport specific cargo, and refurbish the specific organelle [15-23,25,26]. In several and significant to the topic details, the concept outlined above, driven by the results of the investigation on biomembrane synthesis and catabolic turnover, diverges from the view emphasizing role of structural details of the individual proteins that were identified in Golgi transporters [43,44]. The initial findings captured in Golgi and created with components derived from variety of cells, organs and their cytosolic fractions [43-49] grew into theory that epitomized several proteins and generalized Golgi-derived transporters as universal structures in cellular transport. The adherence to this simplified scheme of protein-induced transport created incongruous course of research that grew to represent fundamental phenomena of cell cycle rebuilding and catabolism [50- 53]. An a priori assumption that the proteins find their configuration in premade fictitious lipid mixtures, when in fact the protein under best of circumstances associates with artificial membrane imitation, generated interpretations and answers that do not translate or represent the real cell structures [10,31]. In continuation, the assumption that vesicles characteristics and the intricate positioning of the proteins within the vesicular membrane or its surrounding is protein structure-dependent moved the field into exclusive investigations of the elements that determine protein placement based on the in vitro structural visualization [1,10,29,32,42,54,55]. These monochromatic and one-dimensional views of the real cell membranes ignore the indispensible role of lipids in biomembrane genesis and create doubts whether voluminous collection of the information that arises from the artificial systems find true answers or reflect the nonexistent cellular phenomena and details that grew out of methodological restrictions.
Our scrutiny of the process has found that the ER-localized events set the cellular transport pathway and establish synthesis of the cell membranes with organellespecific proteins within organelle-specific lipids, and hence are not lipid-identical, or lipid-universal [15-18,21, 23-26]. Cogently, these structures must be restored, time and again, with utmost fidelity, the fidelity which cannot be achieved by depositing fully assembled protein in simulated imitation of lipid mixture [8,31,47,48,50,51]. For this reason, it is difficult to reconcile the information on intracellular transport that emanates from the studies offering countless details and explanations of the events viewed from narrowly drown protein involvement and to coalesce it into the comprehensive system operating in the living cells.
In contrast to above described mainstream theoretical framework, our investigations are not restricted by protein methodologies and remain focused on the events determined in systems closely mimicking the intracellular in vivo cycle that involves translation, translocation and membrane biogenesis [15-18,21,23-26]. Thus approached investigation of cell membranes biogenesis generated data that support the idea of inherent, cotranslational integration of proteins within concomitantly assembled membrane lipids that takes place at the specific sites of ER [22]. We detected the codependence between protein cohort translation and specific membrane lipids synthesis and rigorous transfers of the signal that is initiated in nuclear release of mRNAs geared to produce and deliver organelle-specific membranes [15,25,26]. By uncovering that membrane structure is configured in amalgamation with translation of the appropriate cytosolic mRNAs, the harmony which ascertains synthesis of appropriate spectrum of transport vesicles that are destined and precisely fitting Golgi, apical epithelial membrane, basolateral membrane, endosomes and mitochondria became clear and obvious. The fact that from the inception, the ER-assembled vesicles carry specific proteins and membrane lipids which represent hallmarks of their destination site combined with the en bloc incorporation of the vesicular membrane into organelle and cell membrane portrays highly organized meticulous system that affords homeostatic restitution of the cell without compromising the fidelity of the renewal [15,24-26]. Certainly, our investigative results and the evolved concept depart drastically from the idea that biomembrane biogenesis is controlled by the protein complexes associateing with vesicles during intracellular transport, but explains their affinity to differently structured vesicles [47-51].
As shown in this communication, the restitution of cell components, is not concluded with the delivery of newly assembled membrane fragments, but proceeds into the clearance process of the reproduced elements. The course of debridement is well recognized as cell catabolism observed in assembly of autophagosomes, but the numerous studies on the subject continue to pose the questions relating to phagosome biogenesis, origin, composition and overall function in cell renewal and survival [2,27-33, 56-59]. Thus far it has been determined that phagosomes carry characteristic proteins of ER, Golgi, mitochondria, but together they lack uniformity that is regarded as paramount feature of the individual cellular structures. Even sophisticated technologies used in the investigations of numerous cell pathologies, evoked by neuroand myodegenerative disorders, or cellular storage disorders associated with lysosomal dysfunction [5,7,28,33,60-62], have not so far uncovered the triggers of the debridement process which is mandated by cell re-embodiment.
In our concept, affirmed be the results of the investigations, the autophagosomes dissimilarity is genuine feature of their cellular structures and the specific organellar markers indicate their derivation. They reflect release of the material from various cellular organelles undergoing metabolic restitution. Therefore, just as organelles, the composition of the autophagosomes can not be identical and uniform. The data supporting this notion were obtained from our studies of the CHX-treated cells cytosol and their organelles. Our efforts concentrated on seeking the detached, catabolism-destined products, released to the cytosol from the overgrown or damaged and nonfunctional structures eliminated from the organelles. This fact was ascertained by demonstrating that the vesicles remaining in CHX treated cells were not ER-derived synthetic products since they were not carrying secretory cargo and failed to fuse with Golgi and mitochondria, and importantly to our concept, displayed affinity for lysosomes. Together, the results of the studies presented herein demonstrate that autophagosomes originate from all organelles of the cell and are dissimilar in quantity and structure, and reflect the composition of the maternal sites [15,18,20-26].
The persistent, yet unanswered issues that is raised in the investigations underscoring the singular role of proteins in cellular transport is the notion of retrograde transport and return of transporters to ER [1,2,4,5,14, 45,48,50,63]. As we deduced in earlier study, the process is not plausible, since ER lipid composition would be continually modified on the account of lipid composition of the returning vesicles [15,17,21,23,25,26]. Moreover, as we observed in the preparation of the autophagosomes, the material recovered from cytosol and organelle incubates has minimal affinity for association with ER, Golgi or mitochondria, while majority associates with lysosomes. Hence, in our experimental model of the restitution cycle, with arrested translational processes in ER and paused synthesis of ER-transport vesicles and hence arrested synthetic arm of the renewal cycle, the material remaining in the cytosol consists of excised portions of the membranes delegated for degradation. And, in our understanding of the restitution cycle, the CHX treated cells cytosol-derived vesicles reflected mixture of autophagosomes released from all cell membrane and organelles.
The validation of the concept that these vesicles are not cargo free retrograde transport vesicles but autophagosomes produced by every organelle, and as such their lipid composition differ from the vesicles recovered from CHX cytosol, was gained in the study of individual organelles subjected to incubation in CHX derived and vesicle-free cytosol. As predicted from extending the concept of ER-specific assembly of the membranes, the autophagosomes derived from isolated organelles displayed characteristic lipid composition of their maternal source. Thus, the phagosomes derived from ER consisted of PC, PE, and PI, the Golgi derived vesicles contained sphingolipids, while mitochondria-derived structures contained PG and CL. Moreover, as demonstrated in the results, the incubation of these vesicles with ER, Golgi or mitochondria would not afford membrane fusion, instead, their affinity for lysosomes. Indisputably, the differences in lipid composition, missing ability to fuse with ER, Golgi or mitochondria, and the communal affinity for lysosomes, support our contention that cytosol recovered vesicles and the organelle-derived products represent material destined for lysosomes and that lipid composition of the organelle-produced material reflect organelle membrane composition, and is destined for degradation in lysosomes. Thus, as assumed by others, these vesicles could not represent the retrograde transport to Golgi, ER or mitochondria or other than lysosomes cellular structures [45,47,48,50,52,53,63-66].
The subsequent question that required investigation was whether the affinity of these autophagosomal vesicles to lysosomes reflects process of cell debridement through intralysosomal degradation. Indeed, as shown in the results, the incubation of cold lysosomes with radiolabeled autosomal vesicles led to degradation of their membranes lipids, and the lipid extracts contained radiolabeled lipid precursors (sphingosine) or radiolabeled fatty acids tracer (palmitate). The release of radiolabeled lipid precursors was also observed when the radiolabeled lysosomes were used instead. In our interpretation, these results demonstrate the mode of lysosomal degradation, the process that must involve the engulfment of autophagosomal vesicle and invagination into lysosome together with the engulfing lysosomal membrane. As such, both internalized membranes can be and are degraded. As demonstrated the release of radiolabeled lipid precursors was evident when either component of the incubation mixture contained radiolabeled membrane lipids and that made plausible that engulfment and invagination determine autophagosomal degradation and lysosomal turnover. This results-driven conclusion, fits well with overall concept of membranes synthesis, organelle restitution and cell debridement through lysosomal engulfment of phagosomes, and the observation of cellular structures containing double lipid membrane in the lysosomes [2,3,27,28,61,67]. It appears that in the final stage of the cycle, the degradative process in lysosomes accomplishes the task of autophagosomes removal and catabolism of the engulfing portion of lysosomal membrane. Lysosomes, just as other cellular organelles enlarge during endosomal membranes fusion and delivery of newly assembled lysosomal enzymes necessary for continuous catabolic degradation, and thus the renewal dictates their own catabolic resizing.
Thus, the concept advanced by the results of our investigations on membranes with the specific emphasis on lipid synthesis, aligned with translational activity in ER, explains the cellular phenomena that are responsible for organized replacement of cell elements without loosing their function and authenticity [20,22-26]. In comparison, the concepts based on theory that is neglecting the role of celland organelle-specific lipids, promulgates interpretation of the process that cannot fulfill the requirements of the exact reproduction of the cell and its organelles and their highly specialized functions.
In the envisioned process, the cell restoration comes to a full cycle; starting with synthesis of precisely designed cellular fragments, their delivery to specific sites, excision of the superfluous dysfunctional portions of the cell and their degradation in lysosomes. The experimental approach based on tracking of organelle-specific lipids provided evidence that firmly supports our original hypothesis that the vesicles remaining in CHX treated cells’ cytosol and the vesicles produced during incubation of CHX-derived organelles with CHX-cytosol produced organelle-specific vesicles, have characteristic features of autophagosomes because they do not fuse with cell organelles active in the synthetic cell cycle, display affinity for and are degraded during the course of the incubation with intact lysosomes.
In final conclusion, our results on the syntheticcatabolic cycle of the cell demonstrate the presence of a very rigid principle determining every cell’s homeostatic reproduction. Each initiation of replacement elements that provide specifically crafted for the site and the organelle segment of membrane is followed by the excision of its dysfunctional impaired portion and its release to cytosol for the catabolic degradation along with portion of the lysosome. It is quite possible that disconcerted protein and lipid synthesis in ER membrane contribute to ER stress pathway or apoptosis [63], the processes that are employed to eliminate cell products or entire cells which may induce change in the cell function or structure. In the study presented here, we could not determine whether the phagosome affinity to lysosomes, the common trait of all catabolism destined vesicles, is engendered by unrecognized as yet phagosomal lipid markers such as substantial increase in presence of lysophospholipids observed in mitochondrial vesicles (Figure 9(A)), or other similarities that control the final destination of the cellular fragments that must be removed from the cell milieu and converted in lysosomes into reusable catabolites [5,6,7,11,13]. Certainly, they must be different from the markers of the transport vesicles recognizing and rebuilding Golgi, endosomes, mitochondria and plasma membranes, since they are unable to associate or fuse with them. Conversely, transport vesicles carrying cargo and replacing intracellular organelles are not digested by contact with lysosomes. Thus challenge still awaits whether lipids alone or the presence of the specific lipid metabolites and cytosolic proteins [5] determine association of autophagosomes with lysosomes.
REFERENCES
- Toulmay, A. and Prinz, W.A. (2011) Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Current Opinions in Cell Biology, 23, 458-463. doi:10.1016/j.ceb.2011.04.006
- Orsi, A., Polson, H.E. and Tooze, S.A. (2010) Membrane trafficking events that partake in autophagy. Current Opinions in Cell Biology, 22, 150-156. doi:10.1016/j.ceb.2009.11.013
- Saftig, P. and Klumperman, J. (2009) Lysosome biogenesis and lysosomal proteins: Trafficking meets function. National Review of the Molecular Cell Biology, 10, 623- 635. doi:10.1038/nrm2745
- Mellman, I. and Nelson, W.J. (2008) Coordinated protein sorting, targeting and distribution in polarized cells. Nature Reviews/Molecular Cell Biology, 9, 833-855.
- Walter, P. and Ron, D. (2011) The unfolded protein response: From stress pathway to homeostatic regulation. Science, 334, 1081-1086. doi:10.1126/science.1209038
- Smith, M.H., Ploegh, H.L. and Weissman, J.S. (2011) Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science, 334, 1086-1074. doi:10.1126/science.1209235
- Keiser, M., Alfalah, M., Propsting, M.J., Castelletti, D. and Naim, H.Y. (2006) Altered folding, turnover, and polarized sorting act in concert to define a novel pathomechanism of congenital sucrose-isomaltase deficiency. Journal of Biological Chemistry, 281, 14393-14399. doi:10.1074/jbc.M513631200
- Bilder, D., Schober, M. and Perrimon, N. (2003) Integrated activity of PDZ complexes regulates epithelial polarity. Nature Cell Biology, 5, 53-58. doi:10.1038/ncb897
- Sharma, N., Low, S.H., Misra, S., Pallavi, B. and Weimbs, T. (2006) Apical targeting of synthaxin 3 is essential for epithelial cell polarity. Journal of Cell Biology, 173, 937- 948. doi:10.1083/jcb.200603132
- Lukacs, G.L. and Verkman, A.S. (2012) CFTR; folding, misfolding and correcting the ΔF508 conformational defect. Trends in Molecular Medicine, 18, 81-91. doi:10.1016/j.molmed.2011.10.003
- Ron, E., Shenkman, M., Groisman, B., Izenshtein, Y.B., Leitman, J. and Lederkremer, G.Z. (2011) Bypyss of glycan-dependent glycoprotein delivery to ERAD by upregulated EDEM 1. Molecular Biology of the Cell, 22, 3945-3954. doi:10.1091/mbc.E10-12-0944
- Rodriguez-Boulan, E. and Musch, A. (2005) Protein sorting in Golgi complex: Shifting paradigms. Biochimica Biophysica Acta, 1744, 455-464. doi:10.1016/j.bbamcr.2005.04.007
- De Matteis, M.A. and Luini, A. (2008) Exiting the Golgi complex. Nature Reviews of Molecular Cell Biology, 9, 273-284. doi:10.1038/nrm2378
- Folch, H. (2008) Regulation of membrane trafficking in polarized epithelial cells. Current Opinions in Cell Biology, 20, 208-213.
- Slomiany, A. and Slomiany, B.L. (2010) Cell membranes composition is defined in ER and their restitution proceeds by en bloc fusion of ER generated transport vesicles. Health, 2, 1444-1454. doi:10.4236/health.2010.212214
- Slomiany, A. and Slomiany, B.L. (2003) Lipidomic processes in homeostatic and LPS-modified cell renewal cycle. Role of phosphatidylinositol 3-kinase pathway in biomembrane synthesis and restitution of apical epithelial membrane. Journal Physiology and Pharmacology, 54, 533-551.
- Slomiany, A., Grabska, M., Piotrowski, E. and Slomiany, B.L. (1994) Intracellular processes associated with vesicular transport from endoplasmic reticulum to Golgi and exocytosis. Archives of Biochemistry and Biophysics, 310, 247-255. doi:10.1006/abbi.1994.1164
- Slomiany, A., Grzelinska, E., Grabska, M. and Slomiany, B.L. (1992) Intracelular processes associated with glycolprotein transport and processing. Archives of Biochemistry and Biophysics, 298, 167-175. doi:10.1016/0003-9861(92)90108-9
- Slomiany, A., Grzelinska, E., Yamaki, K. and Slomiany, B.L. (1992) Function of intracellular phospholipase A2 in vectorial transport of apoproteins from ER to Golgi. International Journal of Biochemistry, 24, 1397-1406.
- Slomiany, A., Grzelinska, E., Kasinathan, C., et al. (1992) Biogenesis of endoplasmic reticulum transport vesicles transferring gastric apomucin from ER to Golgi. Experimental Cell Research, 201, 1669-1682. doi:10.1016/0014-4827(92)90280-L
- Slomiany, A. and Slomiany, B.L. (2011) Transformations of phosphatidylinositol phosphates in the outer and inner nuclear membrane are linked to synthesis and restitution of cellular membranes. Health, 3, 187-199. doi:10.4236/health.2011.34035
- Slomiany, A., Grabska, M. and Slomiany, B.L. (2006) Homeostatic restitution of cell membranes. Nuclear membrane lipid biogenesis and transport of protein from cytosol to intranuclear spaces. International Journal of Biological Sciences, 2, 216-226. doi:10.7150/ijbs.2.216
- Slomiany, A., Nowak, P., Piotrowski, E. and Slomiany, B.L. (1998) Effect of ethanol on intracellular vesicular transport from Golgi to the apical membrane. Role of phosphatidylinositol 3-kinase and phospholipase A2 in Golgi transport vesicles association and fusion with the apical membrane. Alcohol, Clinical and Experimental Research, 22, 167-175. doi:10.1111/j.1530-0277.1998.tb03634.x
- Slomiany, A., Grabska, M., Slomiany, B.A., et al. (1993) Intracellular transport, organelle biogenesis and establishment of Golgi identity: Impact of Brefeldin A on the activity of lipid synthesizing enzymes. International Journal of Biochemistry, 25, 891-901. doi:10.1016/0020-711X(93)90245-A
- Slomiany, A. and Slomiany, B.L. (2012) Phosphatidylglycerol-containing ERtransport vesicles built and restore outer mitochondrial membrane and deliver nuclear DNA translation products to generate cardiolipin in the inner mitochondrial membrane. Advances in Biological Chemistry, 2, 132-145.
- Slomiany, A., Sano, S., Grabska, M., Yamaki, K. and Slomiany, B.L. (2004) Gastric mucosal cell homeostatic physiome. Critical role of ER-initiated membrane restitution in the fidelity of cell function renewal. Journal of Physiology and Pharmacology, 55, 837-860.
- Mari, M., Tooze, S.E. and Reggiori, F. (2011) The puzzling origin of the autophagosomal membrane. F1000 Biology Reports, 3, 25-34.
- Maiuri, M.C., Zalckvar, E., Kimchi, A. and Kroemer, G. (2007) Self-eating and self-killing: Cross talk between autophagy and apoptosis. National Reviews in Molecular Cell Biology, 8, 741-752. doi:10.1038/nrm2239
- Yoshimori, T. and Noda, T. (2008) Toward unraveling membrane biogenesis in mammalian autophagy. Current Opinions in Cell Biology, 20, 401-407. doi:10.1016/j.ceb.2008.03.010
- Tooze, S.E. and Yoshimuri, T. (2010) The origin of the autophagosomal membrane. National Cell Biology, 12, 831-835. doi:10.1038/ncb0910-831
- Brodsky, J.L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell, 151, 1163-1167. doi:10.1016/j.cell.2012.11.012
- Ragusa, M.J., Stanley, R.E. and Hurley, J.H. (2012) Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell, 151, 1501-1512. doi:10.1016/j.cell.2012.11.028
- Wong, E. and Cuervo, A.M. (2010) Autophagy gone awry in neurodegenerative diseases. National Neurosciences, 13, 805-811.
- Croze, E.M. and Morre, D.J. (1984) Isolation of plasma membrane, Golgi apparatus, and endoplasmic reticulum from single homogenates of mouse liver. Journal of Cellular Physiology, 119, 46-57. doi:10.1002/jcp.1041190109
- Parsons, D.F., Willims, G.R. and Chance, B. (1966) Characteristics of isolated and purified preparations of the outer and inner membranes of mitochondria. Annals New York Academy of Sciences, 137, 643-666. doi:10.1111/j.1749-6632.1966.tb50188.x
- Tjelle, T.E., Brech, A., Juvet, L.K., Griffiths, G. and Berg, T. (1996) Isolation and characterization of early endosomes, late endosomes, late endosomes and terminal lysosomes: Their role in protein degradation. Journal of Cell Sciences, 109, 2905-2914.
- Rome, L.H. and Crain, L.R. (1981) Degradation of mucopolysaccharide in intact isolated lysosomes. Journal of Biological Chemistry, 256, 10763-10768.
- Kawashima, A., Sato, A., Kawashima, M., Nitta, K.,Yamura, W., Sugino, N., Nihei, H. and Natori, Y. (1998) A simple procedure for the isolation of rat kidney lysosomes. Kidney International, 54, 275-278. doi:10.1046/j.1523-1755.1998.00958.x
- Schneider-Poetsch, T., Ju, J., Eyler, D.E., Dang, Y., Bhat, S., Merrick, W.C., Green, R., Shen, B. and Liu, J.O. (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nature Chemistry and Biology, 6, 209-217.
- Obrig, T.G., Culp, W.J., McKeehan, W.L. and Hardesty, B. (1971) The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. Journal of Biological Chemistry, 246, 174-181.
- Kapp, L.D. and Lorsch, J.R. (2004) The molecular mechanics of eukaryotic translation. Annual Reviews of Biochemistry, 73, 657-704. doi:10.1146/annurev.biochem.73.030403.080419
- Lee, M.C., Miller, E.A., Goldberg, J., Orci, L. and Schekman, R. (2004) Bi-directional protein transport between the ER and Golgi. Annual Reviews of Cell Development and Biology, 20, 87-123. doi:10.1146/annurev.cellbio.20.010403.105307
- Rothman, J.E. (1994) Mechanisms of intracellular protein transport. Nature, 372, 55-63. doi:10.1038/372055a0
- Beck, R., Prinz, S., Distellkoter-Bechart, P., et al. (2011) Coatomer and dimeric ADP ribosylation factor 1 promote distinct steps in membrane scission. Journal of Cell Biology, 194, 765-777.
- Idone, V., Tam, C. and Andrews, N.W. (2008) Two-way traffic on the road to plasma membrane repair. Trends in Cellular Biology, 18, 552-559. doi:10.1016/j.tcb.2008.09.001
- Sarrl, E., Sicart, A., Lazaro-Dieguez, F. and Egea, G. (2011) Phospholipid synthesis participates in the regulation of diacylglycerol required for membrane trafficking at Golgi complex. The Journal of Biological Chemistry, 286, 28632-28643. doi:10.1074/jbc.M111.267534
- Sudhof, T.C. and Rothman, J.E. (2009) Membrane fusion: Grappling with SNARE and SM proteins. Science, 323, 474-477. doi:10.1126/science.1161748
- Glick, B.S. and Nakano, A. (2009) Membrane traffic within the Golgi apparatus. Annual Review of Cell Development Biology, 25, 113-132. doi:10.1146/annurev.cellbio.24.110707.175421
- Kawano, M., Kumagai, K., Nishijima, M. and Handa, K. (2006) Efficient trafficking of ceramide from endoplasmic reticulum to the Golgi apparatus requires a VAMPassociated protein interacting FFAT motif of CERT. The Journal of Biological Chemistry, 281, 30279-30288. doi:10.1074/jbc.M605032200
- van Meer, G., Halter, D., Sprong, H., Somerharju, P. and Egmond, M.R. (2006) ABC lipid transporters: Extruders, flippases, or flopless activators? FEBS Letters, 580, 1171- 1177. doi:10.1016/j.febslet.2005.12.019
- Mellman, I. and Nelson, W.J. (2009) Coordinated protein sorting, targeting and distribution in polarized cells. National Review of the Molecular Cell Biology, 9, 833-845.
- Neupert, H. and Herrmann, J.M. (2007) Translocation of proteins into mitochondria. Annual Review of Biochemistry, 76, 723-749. doi:10.1146/annurev.biochem.76.052705.163409
- Lemmon, M.A. (2008) Membrane recognition by phospholipids binding proteins. National Review of the Molecular Cell Biology, 9, 99-111.
- Reggiori, V. and Tooze, S.H. (2012) Autophagy regulation through Atg9 traffic. Journal of Cell Biology, 198, 151-153. doi:10.1083/jcb.201206119
- Whitelegge, J. (2011) Up close with membrane lipidprotein complexes. Science, 334, 320-321. doi:10.1126/science.1214084
- Hayashi-Nishino, M., Fujita, M., Noda, T., Yamaguchi, A., Yoshimori, T. and Yamamoto, A. (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. National Cell Biology, 5, 1180-1185.
- Hailey, D.W., Rambold, A.S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P.K. and Lippincott-Schwartz, J. (2010) Mitochondria supply membranes for autophagosomal biogenesis during starvation. Cell, 141, 656-667. doi:10.1016/j.cell.2010.04.009
- Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C. and Rubinsztein, D.C. (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. National Cell Biology, 12, 747-757.
- van der Vaart, A. and Reggiori, F., (2010) The Golgi complex as a source for yeast autophagosomal membranes. Autophagy, 6, 800-801. doi:10.4161/auto.6.6.12575
- [61] Marion, G., Madeo, F. and Kroemer, G. (2011) Autophagy for tissue homeostasis and neuroprotection. Current Opinions in Cell Biology, 23, 198-206. doi:10.1016/j.ceb.2010.10.001
- [62] Cay, Q., Lu, L., Tian, J.H., Zhu, Y.B., Qiao, H. and Sheng, Z.H. (2010) Snapin regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron, 68, 73-86.
- [63] Platt, F.M., Boland, B. and van der Spoel, A.C. (2012) Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. Journal of Cell Biology, 199, 723- 734. doi:10.1083/jcb.201208152
- [64] Kale, J., Liu, Q., Leber, B. and Andrews, D.W. (2012) Shedding light on apoptosis at subcellular membranes. Cell, 151, 1179-1184. doi:10.1016/j.cell.2012.11.013
- [65] Osman, C., Voelker, D.R. and Langer, T. (2011) Making heads or tails of phospholipids in mitochondria. Journal of Cell Biology, 192, 7-16.
- [66] Gohil, V.M. and Greenberg, M.L. (2009) Mitochondrial membrane biogenesis: Phospholipids and proteins go hand in hand. Journal of Cell Biology, 184, 469-472.
- [67] Klausner, R.D., Donaldson, J.G. and Lippincott-Schwartz, J. (1992) Brefeldin A: Insights into the control of membrane traffic and organelle structure. Journal of Cell Biology, 116, 1071-1080. doi:10.1083/jcb.116.5.1071