Open Journal of Internal Medicine
Vol.2 No.3(2012), Article ID:22969,28 pages DOI:10.4236/ojim.2012.23024
Recent advances of miRNA involvement in hepatocellular carcinoma and cholangiocarcinoma
![]()
1USC Research Center for Liver Diseases, Keck School of Medicine USC, Los Angeles, USA
2College of Health Science, Ewha Womans University, Seoul, Korea
3Ministry of Education, Second Affiliated Hospital, Chongqing Medical University, Chongqing, China
4Xiangya Hospital of Central South University, Central South University, Changsha, China
5School of Medicine, Complutense University, Madrid, Spain
Email: *Hheping@usc.edu
Received 26 June 2012; revised 25 July 2012; accepted 3 August 2012
Keywords: MicroRNAs; Hepatitis C virus; Hepatitis B Virus; Hepatocellular Carcinoma; Cholangiocarcinoma
ABSTRACT
MicroRNAs (miRNAs), which are a class of highly evolutionarily conserved non-coding RNAs, modulate gene expression and are regulated by specific genes. Several studies have shown that the expression of miRNAs is deregulated in Hepatitis C virus (HCV) & Hepatitis B virus (HBV) infection, liver cancer progression, tumor invasion and metastasis. There are a number of high-quality review articles relative to the general role of miRNA alterations in carcinogenesis and specific reviews dealing with the miRNA changes in hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA). Since primary liver cancer is predominantly comprised of HCC and intrahepatic cholangiocarcinoma (ICC), in the present review we specifically focus on recent advances of miRNAs related to tumorigenesis, invasion and metastasis of primary liver cancer, with special emphasis on their relationships to their target genes. HCV & HBV are major causes of liver disease, including acute and chronic hepatitis, liver cirrhosis, and HCC, while HCV infection is a risk factor for ICC. We also discuss the miRNA alterations involved in HCV & HBV infection. We briefly describe advances in molecular signaling of miRNAs in liver cancers and present insights into new therapeutic clues that target liver cancer.
1. INTRODUCTION
MicroRNAs (miRNAs) are about 19 - 25 nucleotides long, non-coding RNAs that are important regulators of gene expression [1-3]. Up to now, a large number of miRNAs have been discovered in animals and plants. Figure 1 shows miRNA biogenesis and possible mechanisms of the miRNA-mediated post-transcriptional repression. MiRNAs are transcribed by an RNA polymerase II (Pol II) to generate primary transcripts (primiRNAs). The initiation step (cropping) is mediated by Drosha, class 2 RNase III. The cytoplasmic RNase III Dicer participates in the second processing step (dicing) to produce miRNA duplexes. The duplex is separated and one strand is selected as the mature miRNA, whereas the other strand is degraded. The mature miRNA is incorporated into ribonucleoprotein complexes which function in RNA interference-mediated gene silencing.
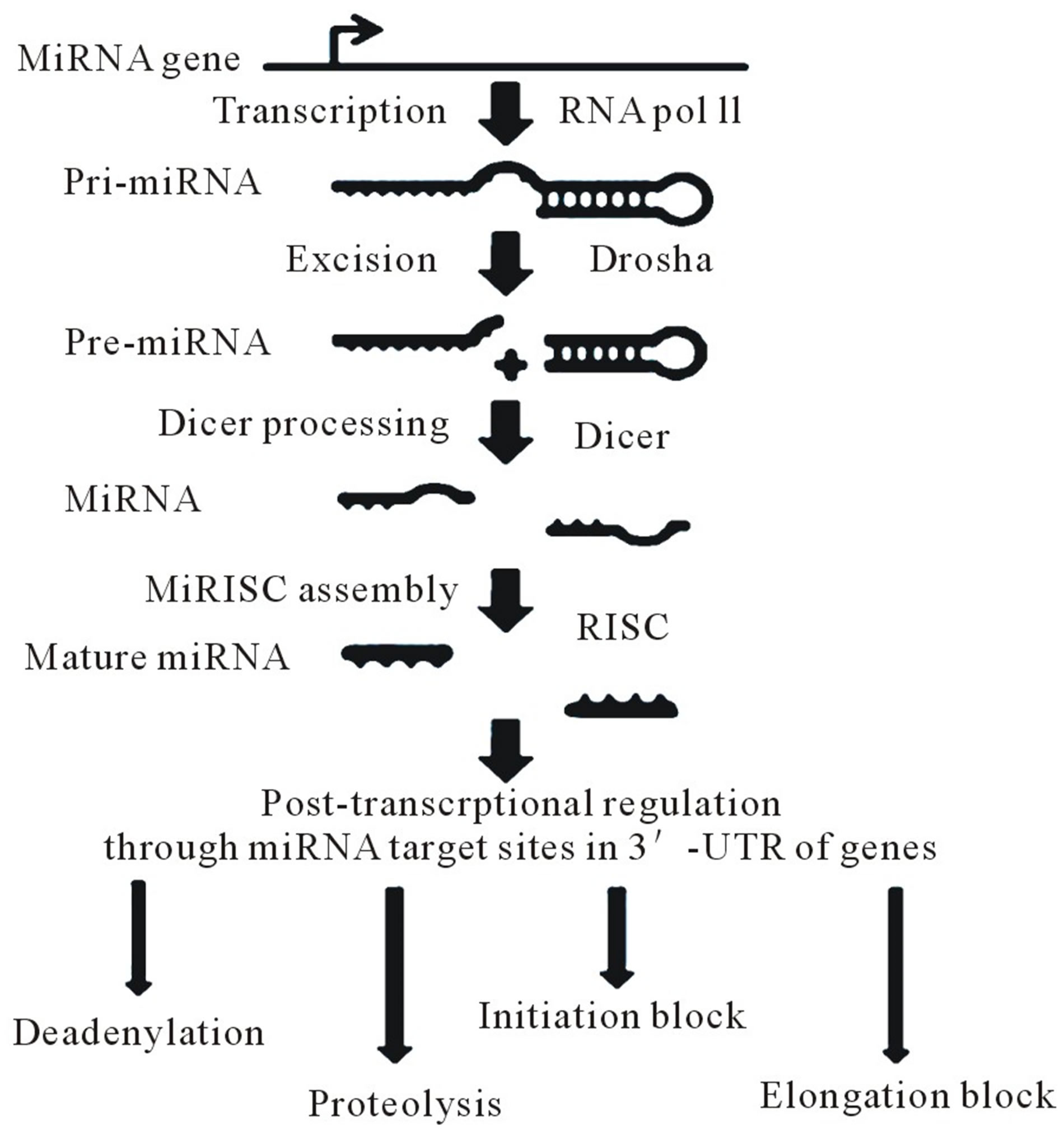
Figure 1. miRNA biogenesis and possible mechanisms of the miRNAs-mediated post transcriptional repression.
The complete complementarity between the miRNA seed sequence (nucleotides 2-8 at the 5’ end of the miRNA) and the targeted RNA are essential for target recognition. miRNA target sites are located at the 3’-UTR region of each gene. Even though a shorter seed pairing may be compensated by extensive matching between the target and the 3’ regions of the miRNAs [4], a single mismatch block can impair binding. miRNAs are widely thought to have two functions in post-transcriptional regulation, mRNA degradation and translation inhibition [5]. In addition to promoting mRNA degradation via deadenylation, miRNA can interfere at different steps of translation, including the initiation step [6], translational elongation [7], and proteolysis of nascent translated products [8] (Figure 1). The 3’-UTRs of mRNAs play crucial roles in post-transcriptional regulation of gene expression including mRNA localization [9], stability [10], and translation [11]. Apart from having miRNA binding sites, 3’-UTRs can also harbor motifs that interact with specific RNA-binding proteins. These motifs are normally short-sequence elements whose activity can be influenced by their secondary structure. Interestingly it has been shown that miRNAs are involved in the development, invasion and metastasis of hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA).
1.1. miRNAs and Epigenetics
Epigenetics is the study of heritable changes in gene expression caused by mechanisms other than changes in the underlying DNA sequence. Gene epigenetic changes usually include DNA methylation and histone modifications. DNA methylation involves the addition of a methyl group to the 5 position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring, and this is a reversible process. It is catalyzed by three major DNA methyltransferases: DNMT1 (maintenance DNMT), which preserves the methylation patterns throughout each cell division [12], and DNMT3a and DNMT- 3b (de novo DNMTs), which transfer a methyl group to previously unmethylated genomic regions [13]. CpG islands are frequently located at the promoter region of a gene and their hypermethylation usually prevents gene expression.
Methylation contributing to epigenetic inheritance can occur through either DNA methylation or protein methylation. Acetylation is a reaction that introduces an acetyl functional group into a chemical compound. Glycosylation is the enzymatic process that attaches glycans to proteins, lipids, or other organic molecules. SUMOylation (SUMO: small ubiquitin like modifier) is a posttranslational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptionnal regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle [14]. Ubiquitination is an enzymatic, protein posttranslational modification process in which the carboxylic acid of the terminal glycine from the di-glycine motif in the activated ubiquitin forms an amide bond to the epsilon amine of the lysine in the modified protein. Histones, which are highly alkaline proteins in the eukaryotic cell nuclei, package and order the DNA into structural units and are the main protein components of chromatin. They can undergo post-translational modifications (such as methylation, acetylation, phosphorylation, glycosylation, SUMOylation and ubiquitination) which can determine whether chromatin is in the accessible and early replicating form (called euchromatin), or in the inaccessible and late replicating form (called heterochromatin) [15]. For example, while acetylation of histones H3 and H4 and methylation of lysine 4 of histone H3 (H3K4me) are found in euchromatin, di or trimethylation of lysine 9 of histone 3 (H3K9me) is a characteristic mark of heterochromatin, which is highly conserved in fungi, plants, and animals. These histone modifications are catalyzed by several histone deacetylases (HDACs) and histone methyltransferases. Finally, the polycomb repressive complex 2 (PRC2) can mediate epigenetic gene silencing by trimethylating histone H3 lysine 27 (H3K27me3), a mark of heterochromatin.
Some miRNAs target, either directly or indirectly, effectors of the epigenetic machinery. miR-29a, miR-29b, and miR-29c directly targets DNMT3a and DNMT3b [16]. Members of the miR-290 cluster directly target retinoblastoma-like 2 (RBL2), an inhibitor of DNMT3 genes [17]. The miR-148 family targets the human DNMT3b protein coding region [18]. Interestingly, miR- 148a promoter is frequently hypermethylated in different tumors [19,20]. miR-148a, miR-152, and miR-301 can all directly target DNMT1 [21]. Meanwhile, some miRNAs are also involved in regulating the expression of HDACs and PRC1 genes. HDAC4 is a direct target of both miR-1 and miR-140 [22,23], while miR-449a targets the 3’-UTR region of HDAC1 [24]. miR-101 directly targets the enhancer of zeste homolog 2 (EZH2), mainly acting as a gene scilencer [25,26].
1.2. miRNA Target Prediction
miRNAs have a profound effect on the modulation of gene expression. More than 17,000 microRNAs have been identified, however, these numbers are rapidly increasing with improvements in miRNA detection and prediction approaches. Several computational methods have been developed to predict miRNA targets in animals [27-45]. The functional consequence of miRNA dysregulation lies in the mRNA targets whose expression is altered. Discovery of targets becomes a critical issue regarding miRNA biology. Each miRNA can target several or more mRNAs, so to identify the targets of all of these small RNAs would be an overwhelming challenge by current technology alone. Combination use of computational target prediction tools (for example with TargetScan, PicTar, miRanda, miR-Base/MicroCosm, Targets Diana-microT, RNAhybrid and RNA22), helps researchers to save time and money. Different tools are designed for specific purposes depending on research needs. Most of the tools offer online access and these are shown in Table 1.
1.3. miRNA Involvement in the Embryonic, Adult and Regenerative Liver
miRNA expression is highly dynamic in the embryonic, adult and regenerative liver. The disexpression of miRNAs perturbs tissue homeostasis, cell apoptosis and proliferation. Before describing miRNA involvement in HCC and CCA, we briefly introduce miRNA involvement in the embryonic, adult and regenerative liver. In order to enhance our understanding of liver development and regeneration and to explore the role of miRNAs during these processes, we have summarized miRNA expression in Table 2. Certain miRNAs are abundantly expressed in human embryonic livers such as let-7c, miR-92, miR-106b, miR-122, miR-124, miR-125a-5p, miR-125b, miR-140-3p, miR-199a-3p, miR-199a-5p, miR-205, miR-206, miR-451 and miR-483-3p [46]. The three most abundantly expressed miRNAs are miR-122, miR-192, and miR-199a/b-3p [47]. In addition, let-7a, miR-483-3p are also abundantly expressed in human

Table 1. microRNA target prediction tools.

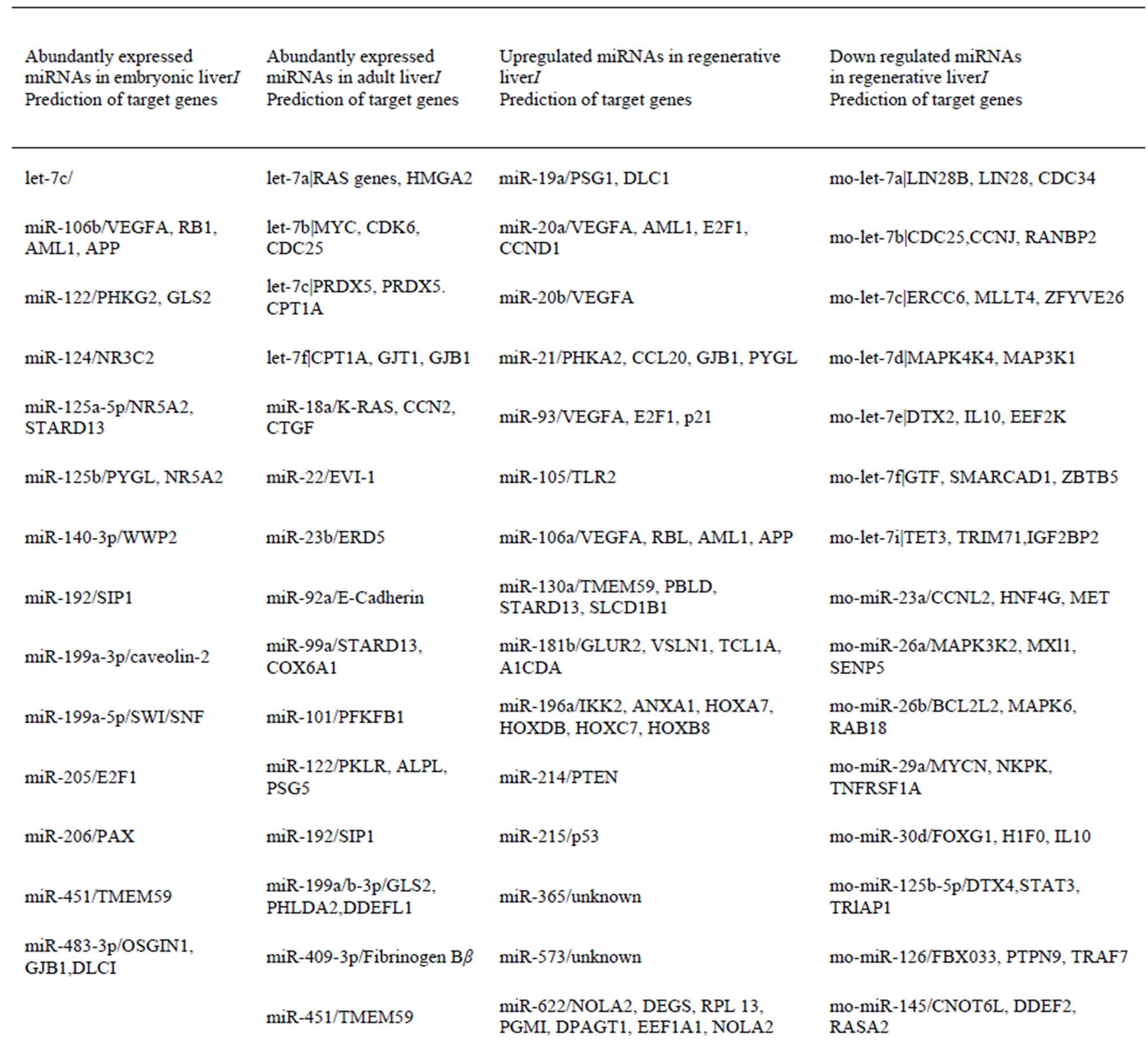
Table 2. miRNA expression patterns and predicted target genes in embryonic, adult and regenerative liver tissues [53-70].
adult liver tissue.
Since miR-92, miR-106b, miR-124, miR-125a-5p, miR-125b, miR-140-3p, miR-205 and miR-206 are highly-expressed in embryonic liver compared to the adult liver, these miRNAs may regulate embryonic progression such as the cell cycle. In humans, two different miR-92 loci, miR-92A and miR-92B, are encoded in the polycistronic miR-17-92 and miR-106A-363 clusters, respectively [48] and miR-92 was considered to function as an oncogene [49]. miR-92 reveals a novel and essential role in early vertebrate development [50]. Let-7a, let-b, let-7c and let-7f were all more highly expressed in the adult liver compared to the embryonic and regenerative liver. These let-7 family members have been shown to target proliferation-promoting genes, such as high mobility group AT-hook protein 2 (HMGA2) [51] and insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1) [52]. The let family may mediate cell cycle arrest in the adult liver. Downregulation of the let-7 family may be associated with increase in cell proliferation in the embryonic and regenerative liver. The targets of detailed miRNAs are summarized in Table 2. Since targets of the let-7 family have the role of cross-action, we put their targets together in let-7 targets. It is necessary to focus on cell-type expression patterns of miRNAs in the embryonic, adult and regerative liver.
2. MICRORNA INVOLVEMENT IN HEPATOCELLULAR CARCINOMA
2.1. General Background
miRNAs regulate various physiological and pathological processes by modulating the expression of their target mRNAs, which play important roles in diverse cellular processes including differentiation, proliferation, growth, migration and survival. Given the critical role that these small regulatory RNAs play in biology, it is speculated that the alterations of miRNA expression patterns can have pathological consequences. More than 50% of miRNA genes are located at fragile sites of chromosomes or in cancer-associated genomic regions, indicating that they are cancer-related and could be used as new diagnostic and prognostic cancer markers as well as potential molecular targets. HCC is a highly aggressive tumor that currently ranks as the fifth most prevalent cancer worldwide. Despite great advances in the disease treatment, relapse and metastasis are frequently observed, and the 5-year survival rate remains low. The progression of HCC involves the deregulation of genes that are critical to cellular processes such as cell cycle control, cell growth, apoptosis, cell migration, invasion and metastasis. The genes and proteins underlying the development and progression of HCC have been extensively investigated in the past decades and miRNAs have only recently been found to be frequently deregulated in HCC. This deregulation is related to HCC progression. and specific miRNAs were found to be associated with the metastasis, recurrence, and prognosis of HCC. We will focus on the most recent knowledge of miRNA expression patterns, their interaction and the predicted target genes in HCC as well as their clinical potentials.
2.2. Expression Patterns
The expression profiles of microRNAs are associated with the initiation and progression of human tumors. Comparable studies on miRNA profiling in HCC and adjacent non tumor liver tissue have revealed characteristic miRNA signatures for HCC and suggest the significance of miRNA involvement in tumorigenesis. Murakami et al. profiled miRNA gene expression in 24 HCCs and 22 non-HCC tissues and uncovered miR-18, precursor miR-18, miR-125, miR-195, miR-199a, miR- 200a, and miR-224 as miRNAs that were differentially expressed between HCC and the adjacent Non-HCC samples [71]. Five other groups also identified miRNA signatures that differentiated tumor from adjacent non tumor liver tissues [72]. The results from these groups differ, suggesting heterogeneous expression of these miRNAs and reflecting the heterogeneous nature of HCC. Furthermore different miRNA signatures are associated with unique clinicopathologic features of tumor and distinct HCC outcomes. Table 3 shows clinicopathologically associated miRNA signatures in HCC [52, 71,72,79,104,226,247,254-259].
Among the identified miRNAs, some are involved in HCC carcinogenesis by promoting the stemness of cancer stem cells (CSC) and by controlling cell proliferation and apoptosis, while others are associated with HCC progression by controlling cell migration and invasion. Downregulation of miR-15b, miR-122a, miR-124, miR- 199a, miR-199b, and miR-203 regulates multiple genes during HCC progression [73,74]. In contrast, miR-141 [75], miR-92 [76], miR-222 [77], miR-221 [78], miR-224 [79] and miR-199a-3p [80] are upregulated and could promote cell cycle progression, reduce cell death and favor angiogenesis and invasion. Therefore, pathogenesis or progression of HCC is closely related to differentially expressed miRNAs. Discovery of the expression profiles of miRNAs in HCC will help to better understand the regulation of signaling networks in the process of HCC carcinogenesis. Since HCC is a heterogeneous tumor that develops via activation of multiple pathways and molecular events, identification of HCC molecular subtypes by miRNA signature is promising for design of specific treatment strategies for each subtype. By using a ligation-mediated amplification method and unsupervised clustering analysis, Toffanin et al.

Table 3. miRNA signature in HCC with different clinicalpathological characteristics.
identified 3 main clusters of HCCs: the wingless-type WNT (MMTV) integration site, interferon-related, and proliferation subclasses [81]. Among the proliferation classes, miR-517a is an oncogenic miRNA that promotes tumor progression and is suggested to be a therapeutic target for patients with HCC.
2.3. Interaction of miRNAs and Their Predicted Target Genes
An important feature of miRNAs is that a single miRNA can regulate multiple target mRNAs. For instance, miR- 122 has 32 validated cellular mRNA targets. This property enables miRNAs to exert wide control on a network of genes that play a critical role in cell division, cell death, and DNA repair. Alterations in these genes results in unrestrained cell proliferation that predisposes to liver cancer. Functional and target association studies on dysregulated miRNAs in HCC have enabled us to gain a more comprehensive understanding of their roles in HCC carcinogenesis.
2.3.1. Interaction of miRNAs in Gene Networks
miRNAs control cell cycle progression by interacting with key cell cycle regulators. Different miRNAs could interact with certain cell cycle regulators such as cyclin/
cyclin-dependent kinase (CDK) complexes, and CDK inhibitors of the CDK interacting protein/kinase inhibitory protein (Cip/Kip) family. On the other hand, certain miRNAs simultaneously affect multiple pro-oncogenic pathways, including receptor tyrosine kinase pathway, apoptosis and transforming growth factor (TGF)-β signaling pathways. Therefore, miRNAs seem to contribute to normal cell functions.
Functionally, miRNAs could be categorized as tumor suppressive or oncogenic. In HCC, there can be aberrant expression of miRNAs, overexpression of oncogenic miRNAs or loss of expression of tumor suppressive miRNAs. These changes cause defects in cell cycle control, inactivation of tumor suppressors and activation of oncogenic signaling pathways, which offer liver cancer cells a growth advantage, resulting in the development and progression of HCC. For example, miR-26a was found to negatively target G1/S cyclins (cyclin D2 and cyclin E2), resulting in reduced expression of these cyclins and induction of cell growth arrest [82]. Liver specific miR-122 directly targets cyclin G1 expression thus inhibiting cell cycle progression [83], miR-195 inactivated cyclin D1, CDK4/6, and E2F transcription factor 3 (E2F3) in G1/S transition [84]. CDK6 was also shown to be negatively targeted by miR-124, which was silenced through CpG methylation in HCC and induced cell cycle arrest at the G1/S checkpoint [73]. miR-199a-3p was identified to target mechanistic target of rapamycin (serine/threonine kinase) (mTOR), which can block the G1/S transition and influence HCC cells (HepG2, Huh-7 and SNU475) to doxorubicin sensitivity [80]. Let-7g inhibits the proliferation of HCC cells by downregulation of c-Myc and upregulation of p16INK4A [85]. All of the above mentioned miRNAs have tumor supperssive functions and are down regulated in HCC, resulting in loss of cell cycle control. miR-221 and miR-222 downregulate CDK inhibitors, p27 and p57 [86,87]. miR- 106b and miR-93 which target another CDK inhibitor p21 [88], have been found to be overexpressed in HCC, stimulating HCC proliferation. In addition, the protein phosphatase 6 catalytic subunit (PPP6C), a negative cell cycle regulator, was recently identified as a direct target of miR-373. miR-373 is up-regulated in HCC tissues and promotes the proliferation of HCC cell lines HepG2 and QGY-7703 by regulating the G1/S transition [89].
miR-122 is a major miRNA in the liver cell and has a suppressive function in cell cycle control. However, it was also reported to negatively target antiapoptotic Bcl-2 family protein Bcl-w, which subsequently activates caspase-3 and reduces cell viability [90]. By regulation of a desintegrin and metalloprotease (ADAM) 17, miR-122 inhibits intrahepatic metastasis of HCC by angiogenesis suppression. Therefore, frequently decreased expression of miR-122 in HCC promotes cancer cell growth and metastasis. miR-221 is another example of the multifunctional effects that miRNAs have in tumorigenesis. Besides the modulation of CDK inhibitor in cell cycle regulation, miR-221 inhibits apoptosis by targeting proapoptotic protein Bcl2 modifying factor (BMF). The miR-221-222 cluster prevents tumor necrosis factorrelated apoptosis in HCC cells. miR-222 inhibited the regulatory subunit B of the protein phosphatase 2A (PP2A) which could inactivate Akt by dephosphorylation. The cluster of miR-221 and miR-222 activates PI3K/Akt pathway by repressing phosphatase and tensin homolog (PTEN), a negative regulator of the pathway. PTEN is also a target of miR-221. The PI3K/Akt pathway could be supressed by miR-125b through reducetion of Akt phosphorylation. In addition, DNA-damageinducible transcript 4 (DDIT4), a modulator of the mTOR pathway was found to be targeted by miR-221 and miR-222 [91]. miR-221 and miR-222 also target TIMP, an inhibitor of matrix metalloproteinase which plays a role in promoting cell invasion and metastasis. The consequence of frequent overexpression of miR- 221 and miR-222 in HCC leads to promotion of liver cancer cell growth, invasion, migration and resistance of apoptosis.
An in-depth analysis of miRNA expression profiles (miRNomes) was done looking at human livers that are normal, that have hepatitis, or that have HCC. Through this analysis, miR-199a/b-3p was identified to target the tumor-promoting p21 protein (Cdc42/Rac)-activated kinase 4 (PAK4) to suppress HCC growth through inhibition of PAK4/Raf/MEK/ERK pathway [54].
Taken together, deregulated miRNAs play dynamic roles in HCC tumorigenesis and progression through enormous gene networks and interactions in a cell. Knowledge on miRNA interactions in gene networks will help to identify miRNAs as therapeutic targets. There are a number of high-quality reviews on miRNA involvement in HCC. In order to better to understand the roles of miRNA involvement in HCC, we have listed the miRNAs and their targets that are mentioned in this review. Table 4 summarizes the most recently published miRNA function and their validated targets mentioned above.
2.3.2. Predicted Target Genes
Using the algorithms from TargetScan, PicTar, and miRanda, Gramantieri identified cyclin G1 as a predicted target for miR-122a and this has been successfully validated by experiments [83]. Using function association analysis, miRNA functions can be predicted in association with disease, pathway, gene oncology, and transcription regulation. miR-520b and miR-525-5p were enriched in human embryonic stem cells (hESCs) by using targetscan and the predicted targets of miR-520b and miR-525-5p were ras-related protein Rab-22A (RAB22A), polycomb group ring Figure 5 (PCGF5), left right determination factor 1 (LEFTY1), insulin-like growth factor 1 receptor (IGF1R), toll-like receptor 7 (TLR7), and homeobox D10 (HOXD10). A functional association study indicated that miR-520b and miR- 525-5p may have important roles for maintaining the stemness or pluripotency of hESCs by regulating the target genes [54]. When facing the colossal task of identifying miRNA targets, bioinformative approches have proven to be an effective and efficient aid that has been widely used in miRNA research.
2.4. Metastasis
Identification of a metastatic mechanism is important to improve the long term survival of patients with HCC due to its high rate of intrahepatic metastasis. However, the mechanism of miRNAs in HCC metastasis is poorly understood. Hurst and colleagues gave the term “metastamir” to describe the set of miRNAs that participate in the metastatic cascade [110]. In many cases, these miRNAs are specific for metastatic development and are not involved in tumorigenesis. Some miRNAs, such as miR-122 and let-7g, function as suppressors of metastasis. miR-122 significantly decreases the tumor volume and suppresses metastasis by reducing blood vessel formation [95]. Recently, miR-142-3p was shown to suppress migration and invasion of a HCC cell by directly and negatively regulating ras-related C3 botulinum toxin substrate 1 (RAC1), which is a regulator of cell migration and invasion [106].
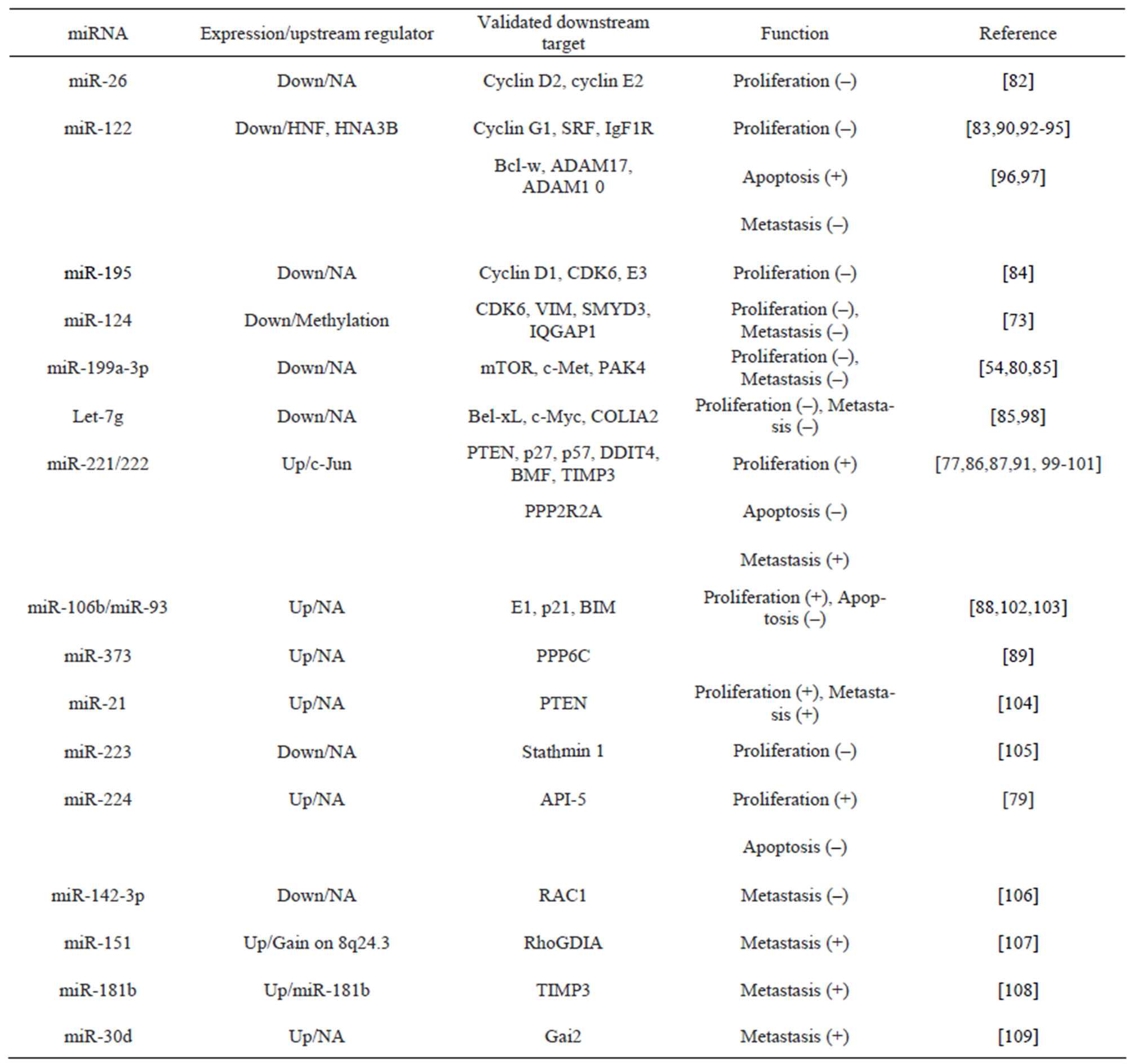
Table 4. Recent advances of miRNA involvement in HCC.
Other miRNAs, such as miR-21, miR-9, miR-143, miR-221, miR-222, miR-181b, miR-30d and miR-151 function as pro-metastatic miRNAs. Meng et al. first reported miR-21-mediated HCC cell invasion by direct targeting of PTEN which facilitates cell migration and invasion [104]. miR-221, miR-222, and miR-181b are further identified as pro-metastatic miRNAs through metalloproteinase inhibitor 3 (TIMP3) [111]. In addition, miR-30d has been shown to be involved in HCC invasion and metastasis by repressing the direct target Galphai2 (G-ai2) [109]. miR-151 increases HCC cell migration and invasion by directly targeting RhoGDIA, resulting in the activation of RAC1, Cdc42 and Rho GTPases [107].
Regulation of the metastatic pathway by miRNAs varies in different types of tumors. For example, overexpression of members of the miR-200 family of miRNAs could act to inhibit metastasis by confining the tumor to the primary location or to promote metastasis by facilitating colonization of a second anatomical site [112]. Nevertheless, elucidating the role of miRNAs in HCC metastasis will provide the potential for development of novel therapeutic agents to fight against tumor metastasis and to improve long term survival of patients.
3. MIRNA INVOLVEMENT IN CHOLANGIOCARCINOMA
3.1. General Background
CCA results from the malignant transformation of bile duct epithelium in the biliary tree including small bile ducts and bile ductules (intrahepatic cholangiocarcinomas; ICC), to large bile ducts at the hilum of the liver or outside the liver (extrahepatic cholangiocarcinomas; ECC). Thus, CCA can be classified as either intraor extrahepatic based on its site of origin along the biliary tree [113]. ICC includes the periductal-infiltrating type, intraductal growth type and mass-forming type. The periductal-infiltrating type grows mainly longitudinally along the bile duct. The intraductal growth type grows toward the lumen of the bile duct. The mass-forming type is found in the liver parenchyma [113]. Risk factors for CCA include primary sclerosing cholangitis (PBC), hepatobiliary fluke, hepatolithiasis, Caroli disease, choledochal cyst, bile duct adenoma, biliary papillomatosis, chronic viral hepatitis, chronic heavy alcohol intake, chronic non-alcoholic liver disease, obesity, and old age [113]. Even though identification of miRNAs in CCA is lagging behind HCC, there have been great improvements in understanting miRNA involvement in CCA [114-120].
3.2. Identification of miRNA Involvement in Cholangiocarcinoma
Masyuk et al. analyzed the expression of 76 miRNAs that were present in both normal and cystic cholangiocytes and 91% of miRNAs in cystic cholangiocytes were found to be downregulated [121]. With the use of miRNA profiling for chronic cholestasis-mediated CCA in the mouse, we found aberrant expression patterns of 40 miRNAs occurring in mouse liver tissue of CCA as shown in Table 5. The aberrant miRNA expression in CCA involves the multiple important factors including: 1) WNT/β-catenin; 2) regulators of G1/S transition (p21, p53, cyclin D1, and p16INK4A); 3) switch from Mnt to c-Myc [113]; 4) MT1-MMP; 5) Notch; 6) hedgedog; and 7) tumor growth factor-beta (TGF-β).
3.2.1. WNT/β-Catenin and MiR-21/MiR-200
Wnt signaling is involved in many aspects of embryonic development, survival, proliferation, and change in cell fate. Dysregulated WNT signaling results from both genetic and epigenetic changes and is associated with a range of diseases, such as colon cancer, HCC and CCA [122-126]. One third of all HCCs have aberrant WNT signaling by mutational and non-mutational events (e.g. cross-talk with other signaling pathways such as TGCC cells). Decreased membrane expression of β-catenin is well correlated with that of E-cadherin [127]. Downregulation of the membrane expression of β-catenin is associated with the progression, invasion and metastasis of ICC cells [128].
miR-21 was one of the first mammalian miRNAs identified and is one of the first to be described as an oncogeneic miRNA as most of the targets of miR-21 are tumor suppressors. miR-21 is associated with a wide variety of cancers including HCC and CCA. WNT1 can be repressed by miR-21. Inhibition of miR-21 or exogenous addition of WNT1 can inhibit human dendritic cell differentiation, suggesting that miR-21 has a regulatory role in dendritic cell differentiation [129]. miR-21 is upregulated in CCA and effectively blocks the expression of the tumor suppressor gene PTEN [117], RECK, TGFβRII, Tap63, p12/CDK2AP1 and PDCD4. It mediates tumorgenesis [104,130-137], suggesting that miR- 21 plays an important role in CCA progression.
The miR-200 family contains miR-200a, miR-200b, miR-200c, miR141 and miR-429. Acumulating data shows that miR-200 is involved in cancer metastasis [138]. miR-200a targets the mRNA of the E-cadherin repressors ZEB1 and ZEB2, leading to an increase in the total E-cadherin available for binding to β-catenin. It also induces formation of the cell-cell adhesion complex. Reduction of miRNA-200a upregulates cytoplasmic and nuclear β-catenin levels, resulting in epithelial to mesenchymal transition (EMT) [139-151]. miR-200a is downregulated in most meningiomas, the most common neoplasm of the nervous system, and regulates expression of β-catenin and activation of Wnt/β-catenin signaling via direct targeting of the 3’UTR of β-catenin mRNA [151]. miR-200b is upregulated in CCA [117]. The target gene is the protein tyrosine phosphatase, nonreceptor type 12 (PTPN12), and the dysregulation of this may contribute to tumorigenesis and tumor cell survival [117].
3.2.2. Regulators of G1/S Transition
The G1/S transition is a stage in the cell cycle at the boundary betweenn the G1 phase, where new organelles are being synthesized, and the S phase, where DNA is replicated. The G1/S transition is disrupted during the development of malignant neoplasms and is characterized by uncontrolled cell proliferation [152,153]. p16- INK4A, p21, p53, cyclin D1 and the retinoblastoma (Rb) gene product are all important as regulators of G1/S transition. G1/S-specific cyclin D1 functions as a regulator of cyclin-dependent kinase (CDK) and forms a complex with CDK4 and CDK6, which phosphorylates tumor suppressor Rb. Phosphorylated Rb allows cells to enter the S phase [154]. Both p16INK4A and p21 function as CDK checkpoints by binding to cyclin D1/CDK complexes [155]. miRNA involvement in the regulation of the G1/S transition is currently a hot topic.
p53 is a tumor suppressor protein. p21 is known as a cyclin-dependent kinase inhibitor or CDK-interacting protein 1. p53 can induce p21 expression under physiological conditions [156,157]. Interestingly, these cell-cycle inhibitors or promoters are abnormally expressed not only in advanced malignant tumors, but also in non-
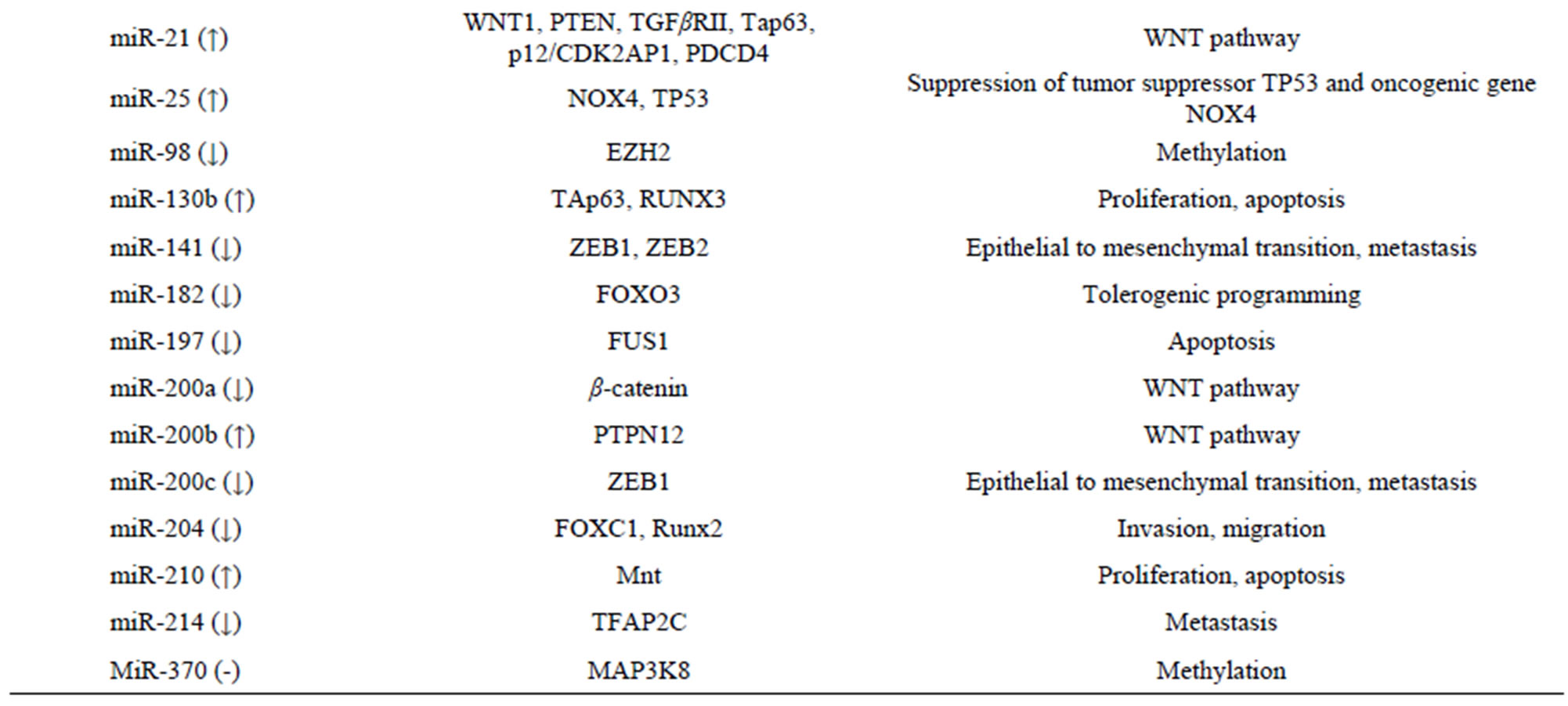
Table 5. miRNA differentially expression in liver tissues of CCA compared with normal liver tiisues by miRNAs profiling.
invasive premalignant lesions during ICC progression [158-160].
Functionally, miR-34a was found to affect tumor cell apoptosis, senescence, proliferation and invasion [161- 163]. A few studies reported that miR-34a was a transcriptional target of p53 [164-166]. It was shown that miR-34a is downregulated [113] and p53 upregulated in CCA progression. A large number of human malignant neoplasms have the p53 mutation. A mutant p53 cannot bind DNA in an effective way and the p21 protein is not available to suppress cell division.
miR-15a is inversely correlated with p53 in lymphocyte leukemia [167]. Our data showed that miR-15a was decreased in chronic cholestasis-mediated CCA (Table 5, Figure 2). Increased apoptosis and proliferation seem to contradict the conventional idea that apoptosis is reduced during carcinogenesis. However, miR-15a downregulation and p53 upregulation, which promotes apoptosis are thought to provide the selective pressures needed for the cells to override apoptosis during the multi stage process of tumorigenesis [168,169]. This leads to the final malignant cholangiocyte population that retains highly proliferative but reduced apoptotic potential [113].
3.2.3. Hypermethylation and miR-98/miR-370
The p16INK4A gene displays its tumor-suppressive function by binding and inactivating cyclin-dependent kinases such as death associated protein 6, SERTA D1, E4F1, cyclin-dependent kinase 4, p53, cyclin-dependent kinase 6, RPL11, PPPIR9B and Mdm2. Frequent p16INK4A inactivation is an early and frequent event of intraductal papillary neoplasm of the liver arising in hepatolithiasis. p16INK4A expression is shown to be decreased in progression through early phase neoplasm to invasive CCA [158]. p16INK4A hypermethylation is associated with this downregulation of p16INK4A. Histone-lysine N-methytransferase (EZH2) encodes a numbers of the polycomb-group (PcG) family. PcG members produce multimeric protein complexes, which are associated with a transcriptional repressive state of genes. EZH2 acts mainly as a gene siliencer by the addition of three methyl groups to lysine 27 of histone 3, leading to chromatin condensation. Abnormal expression of EZH2 is involved in tumorigenesis, including malignant transformation. Aberrant expression of EZH2 is regarded as a potential marker of advanced or aggressive cancer with a poor prognosis. EZH2 is involved in multi-step cholangiocarcinogenesis with respect to the tumor suppressor gene p16INK4A. EZH2 expression increases gradually during ICC, suggesting that hypermethylation of the p16INK4A promoter was related to the aberrant expression of EZH2. The knockdown of EZH2 in cultured ICC cell lines HuCTT-1 and TFK-1 decreased p16INK4A methylation and decreased the binding of

Figure 2. Morphologic features and miR-15a/miR-98 expression of cholangiocarcinoma (CCA) in a mouse model of cholestasis-associated cholangiocarcinoma [113]. H & E staining was done to assess liver morphology at week 28. A week 28 liver showing atypical cell proliferation in the reactive ductules and papillary proliferation of the atypical biliary epithelium and loss of cell polarity in atypical cells (original magnification, X200). Northern blot analysis of miR-15a and miR-98 in the treated groups (CCA) and control groups (Con) at week 28. Oligonucleotide probes for miR-15a and miR-98 for the microRNA Northern were from Exiqon, Inc (Woburn, MA). Densitometric measurements using Quantity One Software (Bio-Rad, Hercules, CA) were done to assess for changes in expression. Densitometric values were derived from at least 3 mice for each time point. P < 0.01 vs control.
EZH2 to the p16INK4A promoter, suggesting that direct binding of EZH2 is involved in the regulation of the p16INK4A gene [170]. It therefore seems conceivable that overexpression of EZH2 may induce hypermethylation of the p16INK4A promoter, followed by decreased expression of p16INK4A in the multi-step cholangiocarcinogenesis, particularly in cases of hepatolithiasis [171].
Using a combinatorial approach of miRNA profiling, bioinformatics, and biochemical tools, and Northern blot, we demonstrated that miR-98 is downregulated in CCA (Table 5, Figure 2) and could negatively regulate EZH2. Further identification of miR-98 targets will determine whether miR-98 is an important target of EZH2 in CCA. miR-370, which targets mitogen-activated protein kinase 8 (MAP3K8), is under tight epigenetic regulation by hypermethylation. MAP3K8 is upregulated in cholangiocarcinoma cell lines as well as in tumor cell xenografts in vivo [118].
3.2.4. MT1-MMP and Let-7
Membrane type 1 metalloprotease (MT1-MMP) is a transmembrane metalloprotease. MT1-MMP plays a major role in the extracellular matrix remodeling and is directly linked to tumorigenesis and metastasis. MT1- MMP also regulates cell and extracellular matrix interaction by processing cell adhesion molecules and eventually promotes cell migration as well. MT1-MMP was expressed in invasive ICC. Interestingly, decreased membrane expression of E-cadherin and β-catenin in the ICC was associated with the aberrant expression of MT1-MMP, suggesting that disruption of the membranous distribution of β-catenin and E-cadherin may result in conditions favorable for the invasion and metastasis of carcinoma cells which express MT1-MMP.
The let-7 miRNA family was regarded as the most representative type of tumor suppressor miRNA. Let-7 expression increases with differentiation and is present in high levels in mature tissue where it inhibits cell proliferation by monitoring DNA replication, mitosis and cytokinesis. Let-7a was decreased in development of CCA [113]. Let-7 downregulation is associated with upregulation of MT1-MMP in pancreatic ductal adenocarcinoma [172].
3.2.5. Notch/c-Myc and MiR-34a
The evolutionarily conserved Notch pathway plays a pivotal role in cellular differentiation, proliferation, fate decision (e.g. EMT), apoptosis, and cell adhesion. In the liver, it is involved in development by coordinating biliary epithelial cell differentiationn and morphogenesis [173]. Disrupted Notch signaling is recognized in a growing number of cancers including liver cancer [174]. The Notch signaling pathway could represent a potential target in CCA treatment. Notch-1 and Notch-2 are upregulated by cholangiocytes in primary sclerosing cholangitis and CCA [175,176]. miR-34a expression can inhibit Notch-1 and Notch-2 protein expression in glioma cells [177].
c-Myc is a transcription factor that binds to E-box sequences as part of a heterodimeric complex with another, Max, to activate transcription. Mnt is an antagonist of c-Myc and represses transcription. Thus, the relative levels of Myc-Max and Mnt-Max heterocomplexes dictate whether target genes are activated or repressed. It is known that Notch targets and upregulates c-Myc. Also, the c-Myc target gene cyclin D1 is a CCA molecular biomarker, and aberrant expression of cyclin D1 contributes to dedifferentiation and cell proliferation in CCA [113]. We found that the up-regulation of miR-210 via hypoxia-inducible factor (HIF)-2α was involved in downregulation of Mnt in CCA. Activation of the miR-34a-cMyc and HIF-2α-miR-210-Mnt pathways in chronic cholestasis-mediated CCA causes c-Myc to bind the E-box element of cyclin D1, instead of Mnt, resulting in cyclin D1 up-regulation [113].
3.2.6. Hedgehog and miR-17-92 Cluster/miR-214
Hedgehog signaling is crucial for many cellular processes involved in cell and stem cell biology. Hedgehog signaling pathways including Sonic, India and Desert have been identified in mammals [178]. Binding of Hedgehog ligands to pass membrane Patched (PTCH) receptors activates transcription factors (Gli-1, Gli-2, Gli-3) [179]. Gli accumulates in the nucleus and induces transcription of genes related to cyclins, β-catenin and growth factors. Like Gli-1, Gli-2 also acts as a transcriptional activator, while Gli-3 plays a repressor role in the absence or inhibition of Hedgehog signaling [180]. Hedgehog signals upregulate cyclin D1 and cyclin D2 for cell cycle acceleration, forkhead box A2 (FOXA2), forkhead box C2 (FOXC2), forkhead box E1 (FOXE1), forkhead box F1 (FOXF1), forkhead box L1 (FOXL1), forkhead box P3 (FOXP3), POU class 3 homeobox 1 (POU3F1), runt-related transcription factor 2 (RUNX2), sex determining region Y (SRY)-box 13 (SOX13), and T-box 2 (TBX2) for cell fate determination. Hedgehog signals also upregulate jagged 2 (JAG2), and inhibin beta C chain (INHBC) to regulate Notch and Activin signaling cascades, respectively. In addition, Hedgehog signals upregulate secreted frizzled-related protein 1 (SFRP1) at least in mesenchymal cells without its promoter CpG hypermethylation to inhibit canonical WNT signaling cascade in epithelial cells. Activation of this pathway is observed in a variety of human cancers, including basal cell carcinomas (BCCs), medulloblastomas, leukemia, gastrointestinal, lung, ovarian, breast, prostate, and heaptobiliary malignancies [181,182]. Sonic is the predominant isoform in the liver. Up to 60% of human HCCs express Sonic [183]. The Hedgedog-Sonic pathway regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans [184]. Biliary fibrosis is accompanied by activation of the Hedgehog pathway in rodents and humans [185-187]. Diverse types of liver injury stimulate cholangiocytes to generate hedgedog ligands, and cholangiocyte-derived hedgedog ligands interact with receptors on cholangiocytes and neighboring cells to modulate virtually every aspect of the ductular reaction to injury. Excessive hedgedog signaling promotes dysfunctional repair and results in chronic hepatic inflammation, fibrogenesis, and carcinogenesis [188].
The miR-17-92 cluster encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92-1) in the human genome. Sufu mRNA, encoding a negative regulator of Hedgehog signaling, is targeted by miR-214. Zebrafish miR-214 binds to the 3’-UTR of Sufu to downregulate Sufu [189]. Since Sufu is implicated in the nuclear trafficking of Gli activator and repressor, miR- 214-induced downregulation of Sufu results in maximal activation of Gli in the presence of Hedgehog and complete repression in the absence of Hedgehog. Interstingly, miR-214 induces cell survival and cisplatin resistance by targeting PTEN in ovarian cancer, to downregulate PTEN for the activation of PI3K-AKT signaling cascade [190]. This can also induce EMT. Effects of miR-17-92 and miR-214 on the Hedhehog-GLI signaling cascade in HCC and ICC remain to be elucidated.
3.2.7 TGF-β and MiR-23b
Members of the transforming growth factor-beta (TGF-β)
family are under intensive investigation in liver cancer due to their importance in both HCC and ICC [191, 192]. A reduction of TGF-β receptors was found in up to 70% of HCC [193,194] and up to 40% of HCC has increased TGF-β expression. TGF-β levels in serum and urine are increased in HCC patients [195,196] and high TGF-β levels have been correlated with advanced clinical stages of HCC [197,198]. TGF-β signaling has also been shown to induce an EMT process in tumor cells. EMT leads to enhanced migration and invasion. Loss of embryonic liver fodrin (ELF), a TGF-β adaptor and signaling molecule, in the liver leads to cancer formation by deregulated hepatocyte proliferation and stimulation of angiogenesis. TGF-β1 and its receptor mRNA were confirmed to be expressed at elevated levels in an animal model of ICC [199,200]. TGF-β1 has also been found to be elevated in human ICC [201,202]. Expression of miR- 29b, which targets Mcl-1, was suppressed in the cholangiocarcinoma cell line, KMCH, as well as in 33% of human cholangiocarcinoma samples [118].
4. MIRNA INVOLVEMENT IN HCV & HBV INFECTION
4.1. General Background
HCV is a small-enveloped positive-stranded RNA virus belonging to the Flaviviridae family, and HBV is a hepatotropic DNA virus from the Hepadnaviridae family [203,204]. According to the World Health Organization (WHO), approximately 170 million people are infected with HCV and 350 million people are infected with HBV worldwide [205]. HCV & HBV are major causes of liver disease, including acute and chronic hepatitis, liver cirrhosis, and HCC. In addition, HCV infection is a risk factor for intrahepatic CCA.
As we described previously, miRNAs can affect a series of fundamental cellular processes through posttranscriptional regulation of target mRNA expression. These processes include cell development, signal transduction, cell proliferation, differentiation, apoptosis and tumorigenesis [206,207]. Recent studies focused on miRNAs have been increasing, especially those studying the relationship between miRNA and infection or cancer [208-210]. miRNA deregulation has been linked to the pathophysiology of human diseases, including those resulting from viral infections. Accumulated evidence demonstrates that host-cellular miRNAs can modulate the expression of various viral genes, thereby playing a crucial role in the host-virus interaction [211-214]. On the other hand, mammalian viruses tend to interfere with or subvert the cellular miRNA to benefit their replication, which differs from plant and insect organisms using RNA interference (RNAi) pathways as a major antiviral pathway [215,216]. Furthermore, viruses can also encode viral miRNAs to alter host physiology or to influence its replication. Over 200 viral miRNAs have now been identified [213,217]. However, no viral miRNAs have yet been identified in HCV-infected cells, whereas HBVencoded miRNA target none of host cellular gene [213, 218]. Herein, we discuss our current knowledge of virally influenced miRNAs of cellular origin and the relationship between HCV & HBV infections and cellular miRNAs.
4.2. Expression Patterns in Elation to miRNAs of HCV & HBV Infection
Recent data indicates that HCV & HBV might have evolved a strategy to prevent infected cells from undergoing apoptosis and to evade the innate and adaptive immune responses of their hosts through modulating the expression of cellular miRNAs. Although both HCV and HBV are hepatitis viruses, there are different clinical features between the gene expression profiles of liver tissue infected with chronic hepatitis C versus chronic hepatitis B [219]. Thus, the miRNA expression patterns following HCV and HBV infections will be described separately.
Braconi et al. [220] profiled miRNA expression in the human hepatoma cell line, Hep-394, (HepG2 transfected with full-length HCV genome) and the control cell line by a custom microarray, and then validated the expression of selected miRNAs by real-time PCR. They found that 10 miRNAs were downregulated >2-fold and 23 miRNAs were upregulated >2-fold in Hep-394 cells, including miR-193b predicted to target Mcl-1, an antiapoptotic protein. Furthermore, when comparing HCV+ and HCV-human liver biopsy samples from 29 liver transplantation patients, multiple miRNAs were differentially expressed. Among them, miR-122, a liver-specific miRNA, and miR-320, a cell-cycle associated miRNA, were downregulated in the HCV+ samples. miR-16 induced cell cycle arrest and was upregulated among HCV+ samples [221]. In different HCV genotypes and replicon models, there was altered expression of anticorrelated mRNAs. These HCV-associated anticorrelated miRNAs and mRNA pairs were related to metabolism, cell growth, cytokine pathways, immunization and so on [222,223].
To clarify the interactions between HBV and the host cellular miRNAs, the expression profiles of cellular miRNAs of HepG2.2.15 (a stable HBV-expressing cell line) and its parent cell line, HepG2, were compared using miRNA microarray. Eighteen miRNAs were differentially expressed between the two cell lines. Among them, eleven were over-expressed and seven were under-expressed in HepG2.2.15 cells. Finally, Northern blot analysis confirmed that the expression of miR-181a, miR-181b, miR-200b and miR-146a were upregulated whereas miR-15a was downregulated [224]. Another recent study examined circulating miRNAs in HBV-infected patients who were divided into three groups: those who were chronic asymptomatic carriers (ASC), those with chronic hepatitis B (CHB) and those with HBVassociated acute-on-chronic liver failure (ACLF), compared with healthy controls (HC). The levels of most miRNAs were over-expressed in HBV-infected patients, including miR-122, miR-16, miR-223, miR-19b, miR- 20a, miR-92a, miR-106a, let-7b and miR-194. Strikingly, these miRNAs levels in CHB and ACLF were higher than in HC and ASC, and the expression of miR-122 and miR-194 was associated with HBeAg production in patients with CHB. These miRNAs were considered to be associated with the progression of symptom severity [225].
Also, the differences in miRNA expression pattern in liver tissues obtained from HCV or HBV infected patients were described. The pathway analysis of predicted target genes by selected miRNAs reflected that 13 miRNAs, under-expressed in the HCV group, regulate genes related to immune response, antigen presentation, cell cycle, proteasome, and lipid metabolism. On the other hand, six miRNAs, downregulated in the HBV group, activate pathways related to cell death, DNA damage and recombination, and transcription signalling. This result demonstrates the different gene expression profiles between HCV and HBV infected individuals, and suggests different pathways between those who have HCC infected with HCV versus HBV [226].
4.3. Interaction of miRNAs in Relation to HCV & HBV Infection and Their Targeted Genes
These studies prove that HCV and HBV expression lead to altered expression of host cellar miRNAs. Some of these miRNAs may exert a profound regulatory effect on both host and virus genes.
miR-122, a liver-specific miRNA, is an abundant miRNA in the liver and is comprised of 70% of the miRNAs in the liver [227]. Sequestration of endogenous miR-122 demonstrates that miR-122 positively mediates HCV in hepatoma cells [228]. Two miR-122 binding sites in HCV in the 5’-UTR were predicted. Mutations in the HCV 5’-UTR and compensatory mutations in the miR-122 seed sequence exhibited that the two sites are both occupied by the miR-122 and function cooperatively to regulate HCV replication [228,229]. miRNAs are known to play a negative role in gene expression by incompletely or completely complementary with target mRNA triggering mRNA cleavage or translational repression [230]. miRNAs positively regulate HCV replication. Thus, it is imperative to know whether a distinct functional complex exists between miR-122 and the viral RNA versus its normal cellular target mRNAs. Machlin et al. proposed a miR-122-HCV complex model in which two miR-122 molecules form an oligomeric complex by binding to the two target sites in the 5’-NTR of HCV RNA with 3’ overhanging nucleotides, respectively. This model argues that the miR-122-HCV RNA complex is an unconventional microRNA-target mRNA complex [231]. Furthermore, Ago2, a component of the RNA-induced silencing complex (RISC), was also necessary for ef- ficient miR-122 enhancement of HCV RNA accumulation. So, we could speculate that Ago 2 may play an important role in this unconventional RISC [232].
Three mechanisms may contribute to the increase of HCV abundance by miR-122. First, a direct enhancement of viral RNA replication by miR-122 is dependent upon its interaction with both seed sites in the 5’-UTR, but binding to the 5’-S1 site is more important for efficient replication than adjacent S2 site [228,229]. Second, miR- 122 positively regulates HCV translation and is dependent upon direct interactions with both sites equally, and, at least in part, by enhancing the association of ribosomes with the viral RNA to stimulate HCV translation [233-235]. Third, miR-122 indirectly modulates HCV abundance through regulation of host gene expression, such as Heme oxygenase-1 (HO-1), which is an antioxidant defense and key cytoprotective enzyme repressed by BTB and CNC homology 1 (BACH1, a heme-binding transcription factor) [236]. On the contrary, another study confirmed that miR-122 could negatively regulate HBV replication. The over-expression of miR- 122 inhibited HBV expression, whereas increased expression of HBV resulted in the depletion of endogenous miR-122 by miR-122 inhibitor. They also found that the downregulation of HO-1 by miR-122 plays a negative role in the miR-122-mediated inhibition of viral expression [225,237]. The opposite effect of miR-122 on HCV and HBV is consistent with the opposite expression of miR-122 in HCVand HBV-infected patients.
Unlike miR-122, miR-199a directly suppresses both HCV and HBV replication by targeting a sequence in domain II of the internal ribosome entry site (IRES) region in the HCV 5’-UTR and S protein coding region in the HBV, respectively [238,239]. There are also some other miRNAs regulating virus expression. For example, miR-196 significantly downregulates HCV expression in both HCV replicon cell lines, Con1 and JFH1, cell culture systems through upregulation of HO-1 [240,241]. miR-141 facilitates efficient HCV replication by miR-141-mediated suppression of DLC-1 [242]. miR-1 was able to enhance the HBV core promoter transcription activity and miR-125a-5p was able to downregulate the expression of HBV S gene [243, 244]. Wu et al. used miRanda to identify the highly probable targets of miRNAs in the HBV genome. They found that miR-7, miR-196b, miR-433, and miR-511 all target the polymerase or HBV S gene, that miR-205 targets the HBV X gene, and that miR-345 targets the preC gene of HBV. These miRNAs have the potential to affect the expression of HBV genes and may in the future be used to develop therapeutic approaches to inhibit HBV [245].
4.4. Implication
At present, HCV & HBV infections are still outstanding public health problems because of high morbidity, and they are two major causes of liver cirrhosis and HCC, which have high mortality [246]. There are still many questions regarding the biological behavior of liver diseases associated with the hepatitis virus. With these diseases, the aberrant expression pattern of miRNAs may provide some ideas to help us understand the pathogenic mechanisms. Practical implications of miRNAs as biomarkers, novel drug targets and therapeutic tools for diagnosis, treatment, and prognosis of liver diseases are also discussed. In particular, there are distinct miRNA expression profiles for HCC and intrahepatic CCA related to infection [247]. A recent study indicated that treatment of chronic HCV infection leads to an efficient antiviral effect in chimpanzees by anti-miR-122 with a locked nucleic acid (LNA) antisense oligonucleotide [248].
In addition, regulation of miRNA was found to be one mechanism for the antiviral effects of interferon (IFN) system [249]. The study by Pedersen et al. showed that IFNβ could alter the expression of cellular miRNAs derived from IFNβ-stimulated cells, and eight of these IFNβ-induced miRNAs have predicted targets within HCV RNA [250]. The altered miRNA expression was also found in HCV infected patients treated with IFNα [251]. Furthermore, the different miRNA expression in CHC patients before combination of IFN and ribavirin therapy was associated with their therapeutic outcome [252]. Both findings strongly support the notion that mammalian organisms use cellular miRNAs to combat viral infections through the interferon system. However, there is a differing viewpoint that it may be unlikely that humans have evolved IFN-induced miRNAs to inhibit HCV. It is thought that it is more likely that HCV uses these miRNAs to avoid immune function and to establish persistent infection by suppressing its own replication [253]. Thus, the role of cellular miRNAs in the antiviral action in mammalian organisms is complicated and would be worth future investigation.
5. PROSPECT
The significance of HCCand CCA-associated miRNAs not only provide new insights into the molecular basis of HCC and CCA but may also have value in clinical diagnosis, prognosis, recurrence, and assessment of individual response to therapy. There is also potential for development of novel therapeutic reagents against HCC and CCA. Since HCC and CCA are pathologically and genetically heterogeneous, it has been a challenge to identify molecular classes of HCC & CCA and design treatment strategies for each specific subtype. Profiling of miRNA expression signatures in HCC and CCA subtypes or even individual HCC and CCA patients will be critical to the above clinical applications.
Since the expression signatures between HCC & CCA and the adjacent non-tumor liver tissue are significantly different, it would be promising to develop HCC and CCA biomarkers for diagnosis. To achieve this goal, it is essential to validate these different signatures by large scale clinical follow up investigations before they can actually be used for diagnosis. miR-222 and miR-223 have been validated to be biomarkers for HCC [105]. Biomarkers of miRNAs for CCA still need to be identified.
Because HCC and CCA tissues secrete various tumor-related proteins and miRNAs into the blood, measurement of serum levels of these miRNAs may serve as biomarkers for early diagnosis. Compared with conventional protein serum markers like AFP, serum levels of miR-16 had the highest sensitivity for HCC, followed by miR-199a [260]. Serum miR-500 levels have been shown to be commonly elevated in HCC patients and values returned to normal after surgical treatment [261]. These results are encouraging for clinical diagnosis. With identification of more miRNAs in CCA, we may find biomarkers for CCA stages, different clinic biopathologic variables and a novel class for diagnosis.
The prognosis of HCC and CCA after surgery is far beyond satisfaction because of the high frequency of recurrence and metastasis. miRNA signatures have been investigated in relation to HCC and CCA prognosis. Serum miR-221 levels have been shown to be correlated with tumor size and tumor stage, indicating that upregulation of serum miR-221 levels in HCC can provide prognostic information in HCC patients [262]. By using a large, clinically well-defined cohort, a unique 20- miRNA signature was identified that can predict venous metastases in primary HCC tissues as well as overall survival in an independent cohort of HCC cases, including early stage HCC [259]. Another study demonstrated a set of 19-miRNAs correlated with HCC disease outcome [255]. However, miRNA profiling, specific miRNA identification, and specific target genes are premature in CCA. Identification of aberrant miRNA signatures of HCC & CCA and their corresponding targets has started to benefit the development of miRNA based therapies for HCC and CCA. Administration of miR-26a in a mouse model resulted in inhibition of HCC cell proliferation and induction of tumor apoptosis without toxicity [82]. It has been shown that osteopontin (OPN) is highly overexpressed in HCC with metastasis. Sun et al. developed artificial miRNAs for targeting OPN, and showed that artifical miRNAs significantly blocked OPN expression in a HCC cell line, leading to decreased lung metastasis through inhibition of matrix metalloproteinase (MMP)-2 [263]. Given that the biochemical structure between miRNAs and siRNAs are similar, delivery reagents developed for siRNAs could also be useful for miRNAs. Since miRNAs could target a large number of transcripts, caution must be taken in the application of miRNAbased therapies. From the work described above, it is clear that there are large gaps in our knowledge concerning both miRNAs and CCA. Taken together with the increasing incidence of CCA worldwide, the lack of effective treatment options is concerning [176]. Increased efforts are needed to identify miRNAs and their targets of this complicated and resistant CCA.
The prospects of miRNA based clinical applications are promising. miRNA signatures of HCC and CCA and corresponding targets not only provide new insights into the molecular origin of liver cancer, but also serve potentially as highly sensitive new biomarkers for diagnosis, prognosis and recurrence for HCC and CCA. There is still much to be learned before these clinical applications and therapies can be put to use.
6. ACKNOWLEDGEMENTS
The authors thank Dr. Shelly Lu at the University of Southern California for great help and support. Studies were supported by Pilot/feasibility grant from the USC Research Center for Liver Diseases (P30DK48522), NIH Grants (DK45334, DK51719 and AT1576) and Basic Science Ressearch Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1- A1012261).
REFERENCES
- A. Ichimura, Y. Ruike, K. Terasawa and G. Tsujimoto, “miRNAs and Regulation of Cell Signaling,” FEBS Journal, Vol. 278, No. 10, 2011, pp. 1610-1618. Hdoi:10.1111/j.1742-4658.2011.08087.x
- S. Djuranovic, A. Nahvi and R. Green, “A Parsimonious Model for Gene Regulation by miRNAs,” Science, Vol. 331, No. 6017, 2011, pp. 550-553. Hdoi:10.1126/science.1191138
- A. Aigner, “MicroRNAs (miRNAs) in Cancer Invasion and Metastasis: Therapeutic Approaches Based on Metastasis-Related miRNAs,” Journal of Molecular Medicine, Vol. 89, No. 5, 2011, pp. 445-457. Hdoi:10.1007/s00109-010-0716-0
- J. Brennecke, A. Stark, R. B. Russell and S. M. Cohen, “Principles of microRNA-Target Recognition,” PLOS Biology, Vol. 3, No. 3, 2005, p. e85. Hdoi:10.1371/journal.pbio.0030085
- W. Filipowicz, S. N. Bhattacharyya and N. Sonenberg, “Mechanisms of Post-Transcriptional Regulation by microRNAs: Are the Answers in Sight?” Nature Reviews Genetics, Vol. 9, No. 2, 2008, pp. 102-114. Hdoi:10.1038/nrg2290
- R. S. Pillai, “MicroRNA function: multiple mechanisms for a tiny RNA?” RNA Society, Vol. 11, No. 12, 2005, pp. 1753-1761. Hdoi:10.1261/rna.2248605
- T. Nissan and R. Parker, “Computational Analysis of miRNA-Mediated Repression of Translation: Implications for Models of Translation Initiation Inhibition,” RNA Society, Vol. 14, No. 8, 2008, pp. 1480-1491. Hdoi:10.1261/rna.1072808
- R. S. Pillai, S. N. Bhattacharyya and W. Filipowicz, “Repression of Protein Synthesis by miRNAs: How Many Mechanisms?” Trends in Cell Biology, Vol. 17, No. 3, 2007, pp. 118-126. Hdoi:10.1016/j.tcb.2006.12.007
- T. Du and P. D. Zamore, “Beginning to Understand microRNA Function,” Cell Research, Vol. 17, No. 8, 2007, pp. 661-663. Hdoi:10.1038/cr.2007.67
- A. Bashirullah, R. L. Cooperstock and H. D. Lipshitz, “Spatial and Temporal Control of RNA Stability,” Proceedings of the National Academy of Sciences, Vol. 98, No. 13, 2001, pp. 7025-7028. Hdoi:10.1073/pnas.111145698
- S. Kuersten and E. B. Goodwin, “The Power of the 3’ UTR: Translational Control and Development,” Nature Reviews Genetics, Vol. 4, No. 8, 2003, pp. 626-637. Hdoi:10.1038/nrg1125
- E. Li, T. H. Bestor and R. Jaenisch, “Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality,” Cell, Vol. 69, No. 6, 1992, pp. 915-926. Hdoi:10.1016/0092-8674(92)90611-F
- M. Okano, D. W. Bell, D. A. Haber and E. Li, “DNA Methyltransferases Dnmt3a and Dnmt3b are Essential for de novo Methylation and Mammalian Development,” Cell, Vol. 99, No. 3, 1999, pp. 247-257. Hdoi:10.1016/S0092-8674(00)81656-6
- R. T. Hay, “SUMO: A History of Modification,” Molecular Cell, Vol. 18, No. 1, 2005, pp. 1-12. Hdoi:10.1016/j.molcel.2005.03.012
- M. Fabbri and G. A. Calin, “Epigenetics and miRNAs in Human Cancer,” Advance in Genetics, Vol. 70, 2010, pp. 87-99. Hdoi:10.1016/B978-0-12-380866-0.60004-6
- M. Fabbri, R. Garzon, A. Cimmino, Z. Liu, N. Zanesi, E. Callegari, S. Liu, H. Alder, S. Costinean, C. FernandezCymering, et al., “MicroRNA-29 Family Reverts Aberrant Methylation in Lung Cancer by Targeting DNA Methyltransferases 3A and 3B,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 104, No. 40, 2007, pp. 15805-15810. Hdoi:10.1073/pnas.0707628104
- L. Sinkkonen, T. Hugenschmidt, P. Berninger, D. Gaidatzis, F. Mohn, C. G. Artus-Revel, M. Zavolan, P. Svoboda and W. Filipowicz, “MicroRNAs Control de novo DNA Methylation through Regulation of Transcriptional Repressors in Mouse Embryonic Stem Cells,” Nature Structural & Molecular Biology, Vol. 15, No. 3, 2008, pp. 259-267. Hdoi:10.1038/nsmb.1391
- A. M. Duursma, M. Kedde, M. Schrier, C. le Sage, R. Agami, “miR-148 Targets Human DNMT3b Protein Coding Region,” RNA Society, Vol. 14, No. 5, 2008, pp. 872- 877. Hdoi:10.1261/rna.972008
- U. Lehmann, B. Hasemeier, M. Christgen, M. Muller, D. Romermann, F. Langer and H. Kreipe, “Epigenetic Inactivation of microRNA Gene Hsa-mir-9-1 in Human Breast Cancer,” The Journal of Pathology, Vol. 214, No. 1, 2008, pp.17-24. Hdoi:10.1002/path.2251
- A. Lujambio, G. A. Calin, A. Villanueva, S. Ropero, M. Sanchez-Cespedes, D. Blanco, L. M. Montuenga, S. Rossi, M. S. Nicoloso, W. J. Faller, et al., “A microRNA DNA Methylation Signature for Human Cancer Metastasis,” Proceedings of the National Academy of Sciences USA, Vol. 105, No. 36, 2008, pp. 13556-13561. Hdoi:10.1073/pnas.0803055105
- C. Braconi, N. Huang and T. Patel, “MicroRNA-Dependent Regulation of DNA Methyltransferase-1 and Tumor Suppressor Gene Expression by Interleukin-6 in Human Malignant Cholangiocytes,” Hepatology, Vol. 51, No. 3, 2010, pp. 881-890.
- J. F. Chen, E. M. Mandel, J. M. Thomson, Q. Wu, T. E. Callis, S. M. Hammond, F. L. Conlon and D. Z. Wang, “The Role of microRNA-1 and microRNA-133 in Skeletal Muscle Proliferation and Differentiation,” Nature Genetics, Vol. 38, No. 2, 2006, pp. 228-233. Hdoi:10.1038/ng1725
- L. Tuddenham, G. Wheeler, S. Ntounia-Fousara, J. Waters, M. K. Hajihosseini, I. Clark and T. Dalmay, “The Cartilage Specific microRNA-140 Targets Histone Deacetylase 4 in Mouse Cells,” FEBS Letters, Vol. 580, No. 17, 2006, pp. 4214-4217. Hdoi:10.1016/j.febslet.2006.06.080
- E. J. Noonan, R. F. Place, D. Pookot, S. Basak, J. M. Whitson, H. Hirata, C. Giardina and R. Dahiya, “miR- 449a Targets HDAC-1 and Induces Growth Arrest in Prostate Cancer,” Oncogene, Vol. 28, No. 14, 2009, pp. 1714-1724. Hdoi:10.1038/onc.2009.19
- J. M. Friedman, G. Liang, C. C. Liu, E. M. Wolff, Y. C. Tsai, W. Ye, X. Zhou and P. A. Jones, “The Putative Tumor Suppressor microRNA-101 Modulates the Cancer Epigenome by Repressing the Polycomb Group Protein EZH2,” Cancer Research, Vol. 69, No. 6, 2009, pp. 2623-2629. Hdoi:10.1158/0008-5472.CAN-08-3114
- S. Varambally, Q. Cao, R. S. Mani, S. Shankar, X. Wang, B. Ateeq, B. Laxman, X. Cao, X. Jing, K. Ramnarayanan, et al., “Genomic Loss of microRNA-101 Leads to Overexpression of Histone Methyltransferase EZH2 in Cancer,” Science, Vol. 322, No. 5908, 2008, pp. 1695-1699. Hdoi:10.1126/science.1165395
- V. Maselli, D. Di Bernardo and S. Banfi, “CoGemiR: A Comparative Genomics microRNA Database,” BMC Genomics, Vol. 9, No. 1, 2008, p. 457. Hdoi:10.1186/1471-2164-9-457
- J. H. Yang, P. Shao, H. Zhou, Y. Q. Chen, L. H. Qu, “DeepBase: A Database for Deeply Annotating and Mining Deep Sequencing Data,” Nucleic Acids Research, Vol. 38, No. 1, 2010, pp. D123-D130. Hdoi:10.1093/nar/gkp943
- M. Maragkakis, M. Reczko, V. A. Simossis, P. Alexiou, G. L. Papadopoulos, T. Dalamagas, G. Giannopoulos, G. Goumas, E. Koukis, K. Kourtis, et al., “DIANA-microT Web Server: Elucidating microRNA Functions through Target Prediction,” Nucleic Acids Research, Vol. 37, No. 2, 2009, pp. W273-W276. Hdoi:10.1093/nar/gkp292
- F. Xiao, Z. Zuo, G. Cai, S. Kang, X. Gao and T. Li, “miRecords: An Integrated Resource for microRNATarget Interactions,” Nucleic Acids Research, Vol. 37, No. 1, 2009, pp. D105-D110.Hdoi:10.1093/nar/gkn851
- K. C. Miranda, T. Huynh, Y. Tay, Y. S. Ang, W. L. Tam, A. M. Thomson, B. Lim and I. Rigoutsos, “A PatternBased Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes,” Cell, Vol. 126, No. 6, 2006, pp. 1203-12017. Hdoi:10.1016/j.cell.2006.07.031
- M. Kertesz, N. Iovino, U. Unnerstall, U. Gaul and E. Segal, “The Role of Site Accessibility in microRNA Target Recognition,” Nature Genetics, Vol. 39, No. 10, 2007, pp. 1278-1284. Hdoi:10.1038/ng2135
- D. Betel, M. Wilson, A. Gabow, D. S. Marks and C. Sander, “The microRNA.org resource: Targets and expression,” Nucleic Acids Research, Vol. 36, No. 1, 2008, pp. D149-D153. Hdoi:10.1093/nar/gkm995
- J. H. Yang, J. H. Li, P. Shao, H. Zhou, Y. Q. Chen and L. H. Qu, “StarBase: A Database for Exploring microRNAmRNA Interaction Maps from Argonaute CLIP-Seq and Degradome-Seq Data,” Nucleic Acids Research, Vol. 39, No. 1, 2011, pp. D202-D209. Hdoi:10.1093/nar/gkq1056
- A. Stark, J. Brennecke, R. B. Russell and S. M. Cohen, “Identification of Drosophila MicroRNA targets, ” PLOS Biology, Vol. 1, No. 3, 2003, p. E60. Hdoi:10.1371/journal.pbio.0000060
- N. Rajewsky and N. D. Socci, “Computational Identification of microRNA Targets,” Developmental Biology, Vol. 267, No. 2, 2004, pp. 529-535. Hdoi:10.1016/j.ydbio.2003.12.003
- B. John, A. J. Enright, A. Aravin, T. Tuschl, C. Sander, D. S. Marks, “Human MicroRNA targets,” PLOS Biology, Vol. 2, No. 11, 2004, p. e363. Hdoi:10.1371/journal.pbio.0020363
- A. J. Enright, B. John, U. Gaul, T. Tuschl, C. Sander and D. S. Marks, “MicroRNA Targets in Drosophila,” Genome Biology, Vol. 5, No. 1, 2003, p. R1. Hdoi:10.1186/gb-2003-5-1-r1
- B. P. Lewis, C. B. Burge and D. P. Bartel, “Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are microRNA Targets,” Cell, Vol. 120, No. 1, 2005, pp. 15-20. Hdoi:10.1016/j.cell.2004.12.035
- M. Kiriakidou, P. T. Nelson, A. Kouranov, P. Fitziev, C. Bouyioukos, Z. Mourelatos and A. Hatzigeorgiou, “A Combined Computational-Experimental Approach Predicts Human microRNA Targets,” Genes & Development, Vol. 18, No. 10, 2004, pp. 1165-1178. Hdoi:10.1101/gad.1184704
- A. Krek, D. Grün, M. N. Poy, R. Wolf, L. Rosenberg, E. J. Epstein, P. MacMenamin, I. da Piedade, K. C. Gunsalus, M. Stoffel and N. Rajewsky, “Combinatorial microRNA Target Predictions,” Nature Genetics, Vol. 37, No. 5, 2005, pp. 495-500. Hdoi:10.1038/ng1536
- M. Rehmsmeier, P. Steffen, M. Hochsmann and R. Giegerich, “Fast and Effective Prediction of microRNA/ Target Duplexes,” RNA Society, Vol. 10, No. 10, 2004, pp. 1507-1517. Hdoi:10.1261/rna.5248604
- J. C. Huang, T. Babak, T. W. Corson, G. Chua, S. Khan, B. L. Gallie, T. R. Hughes, B. J. Blencowe, B. J. Frey, Q. D. Morris, “Using Expression Profiling Data to Identify Human microRNA Targets,” Nature Methods, Vol. 4, No. 12, 2007, pp. 1045-1049. Hdoi:10.1038/nmeth1130
- X. Wang and I. M. El Naqa, “Prediction of Both Conserved and Nonconserved microRNA Targets in Animals,” Bioinformatics, Vol. 24, No. 3, 2008, pp. 325-332. Hdoi:10.1093/bioinformatics/btm595
- P. Sethupathy, M. Megraw and A. G. Hatzigeorgiou, “A Guide through Present Computational Approaches for the Identification of Mammalian microRNA Targets,” Nature Methods, Vol. 3, No. 11, 2006, pp. 881-886. Hdoi:10.1038/nmeth954
- G. Tzur, A. Israel, A. Levy, H. Benjamin, E. Meiri, Y. Shufaro, K. Meir, E. Khvalevsky, Y. Spector, N. Rojansky, et al., “Comprehensive Gene and microRNA Expression Profiling Reveals a Role for microRNAs in Human Liver Development,” PLoS One, Vol. 4, No. 10, 2009, p. e7511. Hdoi:10.1371/journal.pone.0007511
- K. P. Porkka, M. J. Pfeiffer, K. K. Waltering, R. L. Vessella, T. L. Tammela and T. Visakorpi, “MicroRNA Expression Profiling in Prostate Cancer,” Cancer Research, Vol. 67, No. 13, 2007, pp. 6130-6135. Hdoi:10.1158/0008-5472.CAN-07-0533
- F. Petrocca, A. Vecchione and C. M. Croce, “Emerging Role of miR-106b-25/miR-17-92 Clusters in the Control of Transforming Growth Factor Beta Signaling,” Cancer Research, Vol. 68, No. 20, 2008, pp. 8191-8194. Hdoi:10.1158/0008-5472.CAN-08-1768
- C. M. Croce, “Causes and Consequences of microRNA Dysregulation in Cancer,” Nature Reviews Genetics, Vol. 10, No. 10, 2009, pp. 704-714. Hdoi:10.1038/nrg2634
- N. Li, C. Wei, A. F. Olena and J. G. Patton, “Regulation of Endoderm Formation and Left-Right Asymmetry by miR-92 during Early Zebrafish Development,” Development, Vol. 138, No. 9, 2011, pp. 1817-1826. Hdoi:10.1242/dev.056697
- Y. S. Lee and A. Dutta, “The Tumor Suppressor microRNA Let-7 Represses the HMGA2 Oncogene,” Genes & Development, Vol. 21, No. 9, 2007, pp. 1025-1030. Hdoi:10.1101/gad.1540407
- B. Boyerinas, S. M. Park, N. Shomron, M. M. Hedegaard, J. Vinther, J. S. Andersen, C. Feig, J. Xu, C. B. Burge and M. E. Peter, “Identification of Let-7-Regulated Oncofetal Genes,” Cancer Research, Vol. 68, No. 8, 2008, pp. 2587-2591. Hdoi:10.1158/0008-5472.CAN-08-0264
- H. Xu, J. H. He, Z. D. Xiao, Q. Q. Zhang, Y. Q. Chen, H. Zhou and L. H. Qu, “Liver-Enriched Transcription Factors Regulate microRNA-122 that Targets CUTL1 during Liver Development,” Hepatology, Vol. 52, No. 4, 2010, pp. 1431-1442. Hdoi:10.1002/hep.23818
- J. Hou, L. Lin, W. Zhou, Z. Wang, G. Ding, Q. Dong, L. Qin, X. Wu, Y. Zheng, Y. Yang, et al., “Identification of miRNomes in Human Liver and Hepatocellular Carcinoma Reveals miR-199a/b-3p as Therapeutic Target for Hepatocellular Carcinoma,” Cancer Cell, Vol. 19, No. 2, 2011, pp. 232-243. Hdoi:10.1016/j.ccr.2011.01.001
- N. Raschzok, W. Werner, H. Sallmon, N. Billecke, C. Dame, P. Neuhaus and I. M. Sauer, “Temporal Expression Profiles Indicate a Primary Function for microRNA during the Peak of DNA Replication after Rat Partial Hepatectomy,” American Journal of Physiology—Regulatory, Integrative and Comparative Physiology, Vol. 300, No. 6, 2011, pp. R363-R372. Hdoi:10.1152/ajpregu.00632.2010
- D. Liu, J. Fan, W. Zeng, Y. Zhou, S. Ingvarsson and H. Chen, “Quantitative Analysis of miRNA Expression in Several Developmental Stages of Human Livers,” Hepatology Research, Vol. 40, No. 8, 2010, pp. 813-822. Hdoi:10.1111/j.1872-034X.2010.00683.x
- S. Sõber, M. Laan and T. Annilo, “MicroRNAs miR-124 and miR-135a are Potential Regulators of the Mineralocorticoid Receptor Gene (NR3C2) Expression,” Biochemical and Biophysical Research Communications, Vol. 391, No. 1, 2010, pp. 727-732. Hdoi:10.1016/j.bbrc.2009.11.128
- P. P. Medina and F. J. Slack, “MicroRNAs and Cancer: An Overview,” Cell Cycle, Vol. 7, No. 16, 2008, pp. 2485-2492. Hdoi:10.4161/cc.7.16.6453
- L. Agnelli, L. Mosca, S. Fabris, M. Lionetti, A. Andronache, I. Kwee, K. Todoerti, D. Verdelli, C. Battaglia, F. Bertoni, et al., “A SNP Microarray and FISH-Based Procedure to Detect Allelic Imbalances in Multiple Myeloma: An Integrated Genomics Approach Reveals a Wide Gene Dosage Effect,” Genes Chromosomes Cancer, Vol. 48, No. 7, 2009, pp. 603-614. Hdoi:10.1002/gcc.20668
- K. Sakurai, C. Furukawa, T. Haraguchi, K. Inada, K. Shiogama, T. Tagawa, S. Fujita, Y. Ueno, A. Ogata, M. Ito, Y. Tsutsumi and H. Iba, “MicroRNAs miR-199a-5p and -3p Target the Brm Subunit of SWI/SNF to Generate a Double-Negative Feedback Loop in a Variety of Human Cancers,” Cancer Research, Vol. 71, 2011, pp. 1680-1689. Hdoi:10.1158/0008-5472.CAN-10-2345
- A. A. Dar, S. Majid, D. de Semir, M. Nosrati, V. Bezrookove and M. Kashani-Sabet, “miR-205 Suppresses Melanoma Cell Proliferation and Induces Senescence via Regulation of E2F1,” The Journal of Biological Chemistry, Vol. 286, No. 19, 2011, pp. 16606-16614. Hdoi:10.1074/jbc.M111.227611
- B. K. Dey, J. Gagan and A. Dutta, “miR-206 and -486 Induce Myoblast Differentiation by Downregulating Pax7,” Molecular and Cellular Biology, Vol. 31, 2011, pp. 203- 214. Hdoi:10.1128/MCB.01009-10
- W. P. Tsang and T. T. Kwok, “The miR-18a microRNA Functions as a Potential Tumor Suppressor by Targeting on K-Ras,” Carcinogenesis, Vol. 30, 2009, pp. 953-959. Hdoi:10.1093/carcin/bgp094
- T. Ohgawara, S. Kubota, H. Kawaki, S. Kondo, T. Eguchi, N. Kurio, E. Aoyama, A. Sasaki and M. Takigawa, “Regulation of Chondrocytic Phenotype by Micro RNA 18a: Involvement of Ccn2/Ctgf as a Major Target Gene,” FEBS Letters, Vol. 583, 2009, pp. 1006-1010. Hdoi:10.1016/j.febslet.2009.02.025
- J. B. Patel, H. N. Appaiah, R. M. Burnett, P. BhatNakshatri, G. Wang, R. Mehta, S. Badve, M. J. Thomson, S. Hammond, P. Steeg, Y. Liu and H. Nakshatri, “Control of EVI-1 Oncogene Expression in Metastatic Breast Cancer Cells through microRNA miR-22,” Oncogene, Vol. 30, 2011, pp. 1290-1301. Hdoi:10.1038/onc.2010.510
- W. Liu, O. Zabirnyk, H. Wang, Y. H. Shiao, M. L. Nickerson, S. Khalil, L. M. Anderson, A. O. Perantoni, J. M. Phang, “miR-23b Targets Proline Oxidase, a Novel Tumor Suppressor Protein in Renal Cancer,” Oncogene, Vol. 29, No. 35, 2010, pp. 4914-4924. Hdoi:10.1038/onc.2010.237
- A. Fort, C. Borel, E. Migliavacca, S. E. Antonarakis, R. J. Fish and M. Neerman-Arbez, “Regulation of Fibrinogen Production by microRNAs,” Blood, Vol. 116, No. 14, 2010, pp. 2608-2615. Hdoi:10.1182/blood-2010-02-268011
- Z. L. Chen, X. H. Zhao, J. W. Wang, B. Z. Li, Z. Wang, J. Sun, F. W. Tan, D. P. Ding, X. H. Xu, F. Zhou, et al., “MicroRNA-92a Promotes Lymph Node Metastasis of Human Esophageal Squamous Cell Carcinoma via ECadherin,” The Journal of Biological Chemistry, Vol. 286, No. 12, 2011, pp. 10725-10734. Hdoi:10.1074/jbc.M110.165654
- M. R. Benakanakere, Q. Li, M. A. Eskan, A. V. Singh, J. Zhao, J. C. Galicia, P. Stathopoulou, T. B. Knudsen, D. F. Kinane, “Modulation of TLR2 Protein Expression by miR-105 in Human Oral Keratinocytes,” The Journal of Biological Chemistry, Vol. 284, No. 34, 2009, pp. 23107- 23115. Hdoi:10.1074/jbc.M109.013862
- S. A. Georges, M. C. Biery, S. Y. Kim, J. M. Schelter, J. Guo, A. N. Chang, A. L. Jackson, M. O. Carleton, P. S. Linsley, M. A. Cleary, et al., “Coordinated Regulation of Cell Cycle Transcripts by p53-Inducible microRNAs, miR-192 and miR-215,” Cancer Research, Vol. 68, No. 24, 2008, pp. 10105-10112. Hdoi:10.1158/0008-5472.CAN-08-1846
- Y. Murakami, T. Yasuda, K. Saigo, T. Urashima, H. Toyoda, T. Okanoue and K. Shimotohno, “Comprehensive Analysis of microRNA Expression Patterns in Hepatocellular Carcinomaand Non-Tumorous Tissues,” Oncogene, Vol. 25, No. 17, 2006, pp. 2537-2545. Hdoi:10.1038/sj.onc.1209283
- Y. S. Huang, Y. Dai, X. F. Yu, S. Y. Bao, Y. B. Yin, M. Tang and C. X. Hu, “Microarray Analysis of microRNA Expression in hepatocellular Carcinoma and Non-Tumorous Tissues without Viral Hepatitis,” Journal of Gastroenterology and Hepatology, Vol. 23, No. 1, 2008, pp. 87-94. Hdoi:10.1111/j.1440-1746.2007.05223.x
- M. Furuta, K. I. Kozaki, S. Tanaka, S. Arii, I. Imoto and J. Inazawa, “MiR-124 and miR-203 are Epigenetically Silenced Tumor-Suppressive microRNAs in Hepatocellular Carcinoma,” Carcinogenesis, Vol. 31, No. 5, 2010, pp. 766-776. Hdoi:10.1093/carcin/bgp250
- G. E. Chung, J. H. Yoon, S. J. Myung, J. H. Lee, S. H. Lee, S. M. Lee, S. J. Kim, S. Y. Hwang, H. S. Lee and C. Y. Kim, “High Expression of microRNA-15b Predicts a Low Risk of Tumor Recurrence Following Curative Resection of Hepatocellular Carcinoma,” Oncology Reports, Vol. 23, No. 1, 2010, pp. 113-119.
- I. Barshack, E. Meiri, S. Rosenwald, D. Lebanony, M. Bronfeld, S. Aviel Ronen, K. Rosenblatt, S. PolakCharcon, I. Leizerman, M. Ezagouri, et al., “Differential Diagnosis of Hepatocellular Carcinoma from Metastatic Tumors in the Liver Using microRNA Expression,” The International Journal of Biochemistry & Cell Biology, Vol. 42, No. 8, 2010, pp. 1355-1362. Hdoi:10.1016/j.biocel.2009.02.021
- M. Shigoka, A. Tsuchida, T. Matsudo, Y. Nagakawa, H. Saito, Y. Suzuki, T. Aoki, Y. Murakami, H. Toyoda, T. Kumada, et al., “Deregulation of miR-92a Expression is Implicated in Hepatocellular Carcinoma Development,” Pathology International, Vol. 60, No. 5, 2010, pp. 351- 357. Hdoi:10.1111/j.1440-1827.2010.02526.x
- Q. W. Wong, A. K. Ching, A. W. Chan, K. W. Choy, K. F. To, P. B. Lai and N. Wong, “MiR-222 Overexpression Confers Cell Migratory Advantages in Hepatocellular Carcinoma through Enhancing AKT Signaling,” Clinical Cancer Research, Vol. 16, No. 3, 2010, pp. 867-875. Hdoi:10.1158/1078-0432.CCR-09-1840
- L. Gramantieri, F. Fornari, E. Callegari, S. Sabbioni, G. Lanza, C. M. Croce, L. Bolondi and M. Negrini, “MicroRNA Involvement in Hepatocellular Carcinoma,” Journal of Cellular and Molecular Medicine, Vol. 12, No. 6, 2008, pp. 2189-2204. Hdoi:10.1111/j.1582-4934.2008.00533.x
- Y. Wang, A. T. Lee, J. Z. Ma, J. Wang, J. Ren, Y. Yang, E. Tantoso, K. B. Li, L. L. Ooi, P. Tan, et al., “Profiling microRNA Expression in Hepatocellular Carcinoma Reveals microRNA-224 Up-Regulation and Apoptosis Inhibitor-5 as a microRNA-224-Specific Target,” The Journal of Biological Chemistry, Vol. 283, No. 19, 2008, pp. 13205-13215. Hdoi:10.1074/jbc.M707629200
- F. Fornari, M. Milazzo, P. Chieco, M. Negrini, G. A. Calin, G. L. Grazi, D. Pollutri, C. M. Croce, L. Bolondi and L. Gramantieri, “MiR-199a-3p Regulates mTOR and c-Met to Influence the Doxorubicin Sensitivity of Human Hepatocarcinoma Cells,” Cancer Research, Vol. 70, No. 12, 2010, pp. 5184-5193. Hdoi:10.1158/0008-5472.CAN-10-0145
- S. Toffanin, Y. Hoshida, A. Lachenmayer, A. Villanueva, L. Cabellos, B. Minguez, R. Savic, S. C. Ward, S. Thung, D. Y. Chiang, et al., “MicroRNA-Based Classification of Hepatocellular Carcinoma and Oncogenic Role of miR- 517a,” Gastroenterology, Vol. 140, No. 5, 2011, pp. 1618-1628. Hdoi:10.1053/j.gastro.2011.02.009
- J. Kota, R. R. Chivukula, K. A. O’Donnell, E. A. Wentzel, C. L. Montgomery, H. W. Hwang, T. C. Chang, P. Vivekanandan, M. Torbenson, K. R. Clark, et al., “Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model,” Cell, Vol. 137, 2009, No. 1005-1017. Hdoi:10.1016/j.cell.2009.04.021
- L. Gramantieri, M. Ferracin, F. Fornari, A. Veronese, S. Sabbioni, C. G. Liu, G. A. Calin, C. Giovannini, E. Ferrazzi, G. L. Grazi, et al., “Cyclin G1 is a Target of miR- 122a, a microRNA Frequently Down-Regulated in Human Hepatocellular Carcinoma,” Cancer Research, Vol. 67, No. 13, 2007, pp. 6092-6099. Hdoi:10.1158/0008-5472.CAN-06-4607
- T. Xu, Y. Zhu, Y. Xiong, Y. Y. Ge, J. P. Yun and S. M. Zhuang, “MicroRNA-195 Suppresses Tumorigenicity and Regulates G1/S Transition of Human Hepatocellular Carcinoma Cells,” Hepatology, Vol. 50, No. 1, 2009, pp. 113-121. Hdoi:10.1002/hep.22919
- F. F. Lan, H. Wang, Y. C. Chen, C. Y. Chan, S. S. Ng, K. Li, D. Xie, M. L. He, M. C. Lin and H. F. Kung, “Hsa-let-7g Inhibits Proliferation of Hepatocellular Carcinoma Cells by Down-Regulation of c-Myc and UpRegulation of p16(INK4A),” International Journal of Cancer, Vol. 128, No. 2, 2010, pp. 319-331. Hdoi:10.1002/ijc.25336
- C. le Sage, R. Nagel, D. A. Egan, M. Schrier, E. Mesman, A. Mangiola, C. Anile, G. Maira, N. Mercatelli, S. A. Ciafre, et al., “Regulation of the p27(Kip1) Tumor Suppressor by miR-221 and miR-222 Promotes Cancer Cell Proliferation,” The EMBO Journal, Vol. 26, No. 15, 2007, pp. 3699-3708. Hdoi:10.1038/sj.emboj.7601790
- F. Fornari, L. Gramantieri, M. Ferracin, A. Veronese, S. Sabbioni, G. A. Calin, G. L. Grazi, C. Giovannini, C. M. Croce, L. Bolondi, et al., “MiR-221 Controls CDKN1C/ p57 and CDKN1B/p27 Expression in Human Hepatocellular Carcinoma,” Oncogene, Vol. 27, 2008, pp. 5651- 5661. Hdoi:10.1038/onc.2008.178
- I. Ivanovska, A. S. Ball, R. L. Diaz, J. F. Magnus, M. Kibukawa, J. M. Schelter, S. V. Kobayashi, L. Lim, J. Burchard, A. L. Jackson, et al., “MicroRNAs in the miR-106b Family Regulate p21/CDKN1A and Promote Cell Cycle Progression,” Molecular and Cellular Biology, Vol. 28, No. 7, 2008, pp. 2167-2174. Hdoi:10.1128/MCB.01977-07
- N. Wu, X. Liu, X. Xu, X. Fan, M. Liu, X. Li, Q. Zhong and H. Tang, “MiR-373, a Novel Regulator of PPP6C, Functions as an Oncogene in Hepatocellular Carcinoma,” FEBS Journal, Vol. 278, 2011, pp. 2044-2054. Hdoi:10.1111/j.1742-4658.2011.08120.x
- C. J. Lin, H. Y. Gong, H. C. Tseng, W. L. Wang and J. L. Wu, “miR-122 Targets an Anti-Apoptotic Gene, Bcl-w, in Human Hepatocellular Carcinoma Cell Lines,” Biochemical and Biophysical Research Communications, Vol. 375, No. 3, 2008, pp. 315-320. Hdoi:10.1016/j.bbrc.2008.07.154
- P. Pineau, S. Volinia, K. McJunkin, A. Marchio, C. Battiston, B. Terris, V. Mazzaferro, S. W. Lowe, C. M. Croce and A. Dejean, “miR-221 Overexpression Contributes to Liver Tumorigenesis,” Proceedings of the National Academy of Sciences USA, Vol. 107, No. 1, 2010, pp. 264-269. Hdoi:10.1073/pnas.0907904107
- S. Bai, M. W. Nasser, B. Wang, S. H. Hsu, J. Datta, H. Kutay, A. Yadav, G. Nuovo, P. Kumar and K. Ghoshal, “MicroRNA-122 Inhibits Tumorigenic Properties of Hepatocellular Carcinoma Cells and Sensitizes These Cells to Sorafenib,” The Journal of Biological Chemistry, Vol. 284, No. 46, 2009, pp. 32015-32027. Hdoi:10.1074/jbc.M109.016774
- C. Coulouarn, V. M. Factor, J. B. Andersen, M. E. Durkin and S. S. Thorgeirsson, “Loss of miR-122 Expression in Liver Cancer Correlates with Suppression of the Hepatic Phenotype and Gain of Metastatic Properties,” Oncogene, Vol. 28, No. 40, 2009, pp. 3526-3536. Hdoi:10.1038/onc.2009.211
- F. Fornari, L. Gramantieri, C. Giovannini, A. Veronese, M. Ferracin, S. Sabbioni, G. A. Calin, G. L. Grazi, C. M. Croce, S. Tavolari, et al., “MiR-122/cyclin G1 Interaction Modulates p53 Activity and Affects Doxorubicin Sensitivity of Human Hepatocarcinoma Cells,” Cancer Research, Vol. 69, No. 14, 2009, pp. 5761-5767. Hdoi:10.1158/0008-5472.CAN-08-4797
- W. C. Tsai, P. W. Hsu, T. C. Lai, G. Y. Chau, C. W. Lin, C. M. Chen, C. D. Lin, Y. L. Liao, J. L. Wang, Y. P. Chau, et al., “MicroRNA-122, a Tumor Suppressor Microrna that Regulates Intrahepatic Metastasis of Hepatocellular Carcinoma,” Hepatology, Vol. 49, No. 5, 2009, pp. 1571-1582. Hdoi:10.1002/hep.22806
- L. Ma, J. Liu, J. Shen, L. Liu, J. Wu, W. Li, J. Luo, Q. Chen and C. Qian, “Expression of miR-122 Mediated by Adenoviral Vector Induces Apoptosis and Cell Cycle Arrest of Cancer Cells,” Cancer Biology and Therapy, Vol. 9, No. 7, 2010, pp. 554-561. Hdoi:10.4161/cbt.9.7.11267
- R. Zhang, L. Wang, G. R. Yu, X. Zhang, L. B. Yao, A. G. Yang, “MicroRNA-122 Might be a Double-Edged Sword in Hepatocellular Carcinoma,” Hepatology, Vol. 50, 2009, pp. 1322-1323. Hdoi:10.1002/hep.23108
- J. Ji, L. Zhao, A. Budhu, M. Forgues, H. L. Jia, L. X. Qin, Q. H. Ye, J. Yu, X. Shi, Z. Y. Tang, et al., “Let-7g Targets Collagen Type I Alpha2 and Inhibits Cell Migration in Hepatocellular Carcinoma,” Journal of Hepatology, Vol. 52, No. 5, 2010, pp. 690-697. Hdoi:10.1016/j.jhep.2009.12.025
- S. Shimizu, T. Takehara, H. Hikita, T. Kodama, T. Miyagi, A. Hosui, T. Tatsumi, H. Ishida, T. Noda, H. Nagano, et al., “The Let-7 Family of microRNAs Inhibits Bcl-xL Expression and Potentiates Sorafenib-Induced Apoptosis in Human Hepatocellular Carcinoma,” Journal of Hepatology, Vol. 52, No. 6, 2010, pp. 698-704. Hdoi:10.1016/j.jhep.2009.12.024
- M. Garofalo, G. Di Leva, G. Romano, G. Nuovo, S. S. Suh, A. Ngankeu, C. Taccioli, F. Pichiorri, H. Alder, P. Secchiero, et al., “miR-221 & 222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation,” Cancer Cell, Vol. 16, 2009, No. 498-509. Hdoi:10.1016/j.ccr.2009.10.014
- L. Gramantieri, F. Fornari, M. Ferracin, A. Veronese, S. Sabbioni, G. A. Calin, G. L. Grazi, C. M. Croce, L. Bolondi and M. Negrini, “MicroRNA-221 Targets Bmf in Hepatocellular Carcinoma and Correlates with Tumor Multifocality,” Clinical Cancer Research, Vol. 15, No. 18, 2009, pp. 5073-5081. Hdoi:10.1158/1078-0432.CCR-09-0092
- Y. Li, W. Tan, T. W. Neo, M. O. Aung, S. Wasser, S. G. Lim, T. M. Tan, “Role of the miR-106b-25 microRNA Cluster in Hepatocellular Carcinoma,” Cancer Science, Vol. 100, 2009, pp. 1234-1242. Hdoi:10.1111/j.1349-7006.2009.01164.x
- I. Ivanovska, A. S. Ball, R. L. Diaz, J. F. Magnus, M. Kibukawa, J. M. Schelter, S. V. Kobayashi, L. Lim, J. Burchard, A. L. Jackson, et al., “MicroRNAs in the miR-106b Family Regulate p21/CDKN1A and Promote Cell Cycle Progression,” Molecular and Cellular Biology, Vol. 28, No. 7, 2008, pp. 2167-2174. Hdoi:10.1128/MCB.01977-07
- F. Meng, R. Henson, H. Wehbe-Janek, K. Ghoshal, S. T. Jacob and T. Patel, “MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer,” Gastroenterology, Vol. 133, No. 2, 2007, pp. 647-658. Hdoi:10.1053/j.gastro.2007.05.022
- Q. W. Wong, R. W. Lung, P. T. Law, P. B. Lai, K. Y. Chan, K. F. To and N. Wong, “MicroRNA-223 is Commonly Repressed in Hepatocellular Carcinoma and Potentiates Expression of Stathmin1,” Gastroenterology, Vol. 135, No. 1, 2008, pp. 257-269. Hdoi:10.1053/j.gastro.2008.04.003
- L. Wu, C. Cai, X. Wang, M. Liu, X. Li and H. Tang, “MicroRNA-142-3p, a New Regulator of RAC1, Suppresses the Migration and Invasion of Hepatocellular Carcinoma Cells,” FEBS Letters, Vol. 585, No. 9, 2011, pp. 1322-1330. Hdoi:10.1016/j.febslet.2011.03.067
- J. Ding, S. Huang, S. Wu, Y. Zhao, L. Liang, M. Yan, C. Ge, J. Yao, T. Chen, D. Wan, et al., “Gain of miR-151 on Chromosome 8q24.3 Facilitates Tumour Cell Migration and Spreading through Downregulating RhoGDIA,” Nature Cell Biology, Vol. 12, No. 4, 2010, pp. 390-399. Hdoi:10.1038/ncb2039
- B. Wang, S. H. Hsu, S. Majumder, H. Kutay, W. Huang, S. T. Jacob and K. Ghoshal, “TGFbeta-Mediated Upregulation of Hepatic miR-181b Promotes Hepatocarcinogenesis by Targeting TIMP3,” Oncogene, Vol. 29, No. 12, 2010, pp. 1787-1797. Hdoi:10.1038/onc.2009.468
- J. Yao, L. Liang, S. Huang, J. Ding, N. Tan, Y. Zhao, M. Yan, C. Ge, Z. Zhang, T. Chen, et al., “MicroRNA-30d Promotes Tumor Invasion and Metastasis by Targeting Galphai2 in Hepatocellular Carcinoma,” Hepatology, Vol. 51, No. 3, 2010, pp. 846-856.
- D. R. Hurst, M. D. Edmonds and D. R. Welch, “Metastamir: The Field of Metastasis-Regulatory microRNA is Spreading,” Cancer Research, Vol. 69, No. 19, 2009, pp. 7495-7498. Hdoi:10.1158/0008-5472.CAN-09-2111
- M. Garofalo, G. Di Leva, G. Romano, G. Nuovo, S. S. Suh, A. Ngankeu, C. Taccioli, F. Pichiorri, H. Alder, P. Secchiero, et al., “miR-221 & 222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation,” Cancer Cell, Vol. 16, 2009, pp. 498-509. Hdoi:10.1016/j.ccr.2009.10.014
- D. M. Dykxhoorn, “MicroRNAs and Metastasis: Little RNAs Go a Long Way,” Cancer Research, Vol. 70, No. 16, 2010, pp. 6401-6406. Hdoi:10.1158/0008-5472.CAN-10-1346
- H. Yang, T. W. Li, J. Peng, X. Tang, K. S. Ko, M. Xia, M. A. Aller, “A Mouse Model of Cholestasis-Associated Cholangiocarcinoma and Transcription Factors Involved in Progression,” Gastroenterology, Vol. 141, No. 1, 2011, pp. 378-388. Hdoi:10.1053/j.gastro.2011.03.044
- L. Chen, H. X. Yan, W. Yang, L. Hu, L. X. Yu, Q. Liu, L. Li, D. D. Huang, J. Ding, F. Shen, et al., “The Role of microRNA Expression Pattern in Human Intrahepatic Cholangiocarcinoma,” Journal of Hepatology, Vol. 50, No. 2, 2009, pp. 358-369. Hdoi:10.1016/j.jhep.2008.09.015
- Y. Kawahigashi, T. Mishima, Y. Mizuguchi, Y. Arima, S. Yokomuro, T. Kanda, O. Ishibashi, H. Yoshida, T. Tajiri and T. Takizawa, “MicroRNA Profiling of Human Intrahepatic Cholangiocarcinoma Cell Lines Reveals Biliary Epithelial Cell-Specific microRNAs,” Journal of Nippon Medical School, Vol. 76, No. 4, 2009, pp. 188-197. Hdoi:10.1272/jnms.76.188
- C. Braconi, N. Huang and T. Patel, “MicroRNA-Dependent Regulation of DNA Methyltransferase-1 and Tumor Suppressor Gene Expression by Interleukin-6 in Human Malignant Cholangiocytes,” Hepatology, Vol. 51, 2010, pp. 881-890.
- F. Meng, R. Henson, M. Lang, H. Wehbe, S. Maheshwari, J. T. Mendell, J. Jiang, T. D. Schmittgen and T. Patel, “Involvement of Human Micro-RNA in Growth and Response to Chemotherapy in Human Cholangiocarcinoma Cell Lines,” Gastroenterology, Vol. 130, No. 7, 2006, pp. 2113-2129. Hdoi:10.1053/j.gastro.2006.02.057
- J. L. Mott, S. Kobayashi, S. F. Bronk and G. J. Gores, “Mir-29 Regulates Mcl-1 Protein Expression and Apoptosis,” Oncogene, Vol. 26, No. 42, 2007, pp. 6133-6140. Hdoi:10.1038/sj.onc.1210436
- F. Meng, H. Wehbe-Janek, R. Henson, H. Smith and T. Patel, “Epigenetic Regulation of MicroRNA-370 by Interleukin-6 in Malignant Human Cholangiocytes,” Oncogene, Vol. 27, 2007, pp. 378-386. Hdoi:10.1038/sj.onc.1210648
- H. Yang, T. W. Li, K. S. Ko, M. Xia and S. C. Lu, “Switch from Mnt-Max to Myc-Max Induces p53 and Cyclin D1 Expression and Apoptosis during Cholestasis in Mouse and Human Hepatocytes,” Hepatology, Vol. 49, No. 3, 2009, pp. 860-870. Hdoi:10.1002/hep.22720
- T. Masyuk, A. Masyuk and N. LaRusso, “MicroRNAs in Cholangiociliopathies,” Cell Cycle, Vol. 8, 2009, pp. 1324-1328. Hdoi:10.4161/cc.8.9.8253
- R. T. Moon, A. D. Kohn, G. V. De Ferrari and A. Kaykas, “WNT and Beta-Catenin Signalling: Diseases and Therapies,” Nature Reviews Genetics, Vol. 5, No. 9, 2004, pp. 691-701. Hdoi:10.1038/nrg1427
- K. S. Zaret, “Genetic Programming of Liver and Pancreas Progenitors: Lessons for Stem-Cell Differentiation,” Nature Reviews Genetics, Vol. 9, No. 5, 2008, pp. 329-340. Hdoi:10.1038/nrg2318
- N. Barker and H. Clevers, “Mining the Wnt Pathway for Cancer Therapeutics,” Nature Reviews Drug Discovery, Vol. 5, No. 12, 2006, pp. 997-1014. Hdoi:10.1038/nrd2154
- Y. Nakanuma, M. Sasaki, Y. Sato, X. Ren, H. Ikeda and K. Harada, “Multistep Carcinogenesis of Perihilar Cholangiocarcinoma Arising in the Intrahepatic Large Bile Ducts,” World journal of hepatology, Vol. 1, No. 1, 2009, pp. 35-42. Hdoi:10.4254/wjh.v1.i1.35
- S. Boyault, D. S. Rickman, A. de Reyniès, C. Balabaud, S. Rebouissou, E. Jeannot, A. Hérault, J. Saric, J. Belghiti, D. Franco, et al., “Transcriptome Classification of HCC is Related to Gene Alterations and to New Therapeutic Targets,” Hepatology, Vol. 45, No. 1, 2007, pp. 42-52. Hdoi:10.1002/hep.21467
- K. Itatsu, Y. Zen, J. Yamaguchi, S. Ohira, A. Ishikawa, H. Ikeda, Y. Sato, K. Harada, M. Sasaki, et al., “Expression of Matrix Metalloproteinase 7 is an Unfavorable Postoperative Prognostic Factor in Cholangiocarcinoma of the Perihilar, Hilar, and Extrahepatic Bile Ducts,” Human Pathology, Vol. 39, No. 5, 2008, pp. 710-719. Hdoi:10.1016/j.humpath.2007.09.016
- K. Sugimachi, K. Taguchi, S. Aishima, S. Tanaka, M. Shimada, K. Kajiyama, K. Sugimachi and M. Tsuneyoshi, “Altered Expression of Beta-Catenin without Genetic Mutation in Intrahepatic Cholangiocarcinoma,” Modern Pathology, Vol. 14, No. 9, 2001, pp. 900-905. Hdoi:10.1038/modpathol.3880409
- S. T. Hashimi, J. A. Fulcher, M. H. Chang, L. Gov, S. Wang and B. Lee, “MicroRNA Profiling Identifies miR-34a and miR-21 and Their Target Genes JAG1 and WNT1 in the Coordinate Regulation of Dendritic Cell Differentiation,” Blood, Vol. 114, 2009, pp. 404-414. Hdoi:10.1182/blood-2008-09-179150
- Y. Chen, W. Liu, T. Chao, Y. Zhang, X. Yan, Y. Gong, B. Qiang, J. Yuan, M. Sun and X. Peng, “MicroRNA-21 Down-Regulates the Expression of Tumor Suppressor PDCD4 in Human Glioblastoma Cell T98G,” Cancer Letters, Vol. 272, No. 2, 2008, pp. 197-205. Hdoi:10.1016/j.canlet.2008.06.034
- S. T. Hashimi, J. A. Fulcher, M. H. Chang, L. Gov, S. Wang and B. Lee, “MicroRNA Profiling Identifies miR-34a and miR-21 and Their Target Genes JAG1 and WNT1 in the Coordinate Regulation of Dendritic Cell Differentiation,” Blood, Vol. 114, 2009, No. 404-414. Hdoi:10.1182/blood-2008-09-179150
- Y. Chen, W. Liu, T. Chao, Y. Zhang, X. Yan, Y. Gong, B. Qiang, J. Yuan, M. Sun, X. Peng, “MicroRNA-21 DownRegulates the Expression of Tumor Suppressor PDCD4 in Human Glioblastoma Cell T98G,” Cancer Letters, Vol. 272, No. 2, 2008, No. 197-205. Hdoi:10.1016/j.canlet.2008.06.034
- Q. Yao, H. Xu, Q. Q. Zhang, H. Zhou, L. H. Qu, “MicroRNA-21 Promotes Cell Proliferation and Down-Regulates the Expression of Programmed Cell Death 4 (PDCD4) in HeLa Cervical Carcinoma Cells,” Biochemical and Biophysical Research Communications, Vol. 388, No. 3, 2009, pp. 539-542. Hdoi:10.1016/j.bbrc.2009.08.044
- Z. Zhang, Z. Li, C. Gao, P. Chen, J. Chen, W. Liu, S. Xiao and H. Lu, “miR-21 Plays a Pivotal Role in Gastric Cancer Pathogenesis and Progression,” Laboratory Investigation, Vol. 88, No. 12, 2008, pp. 1358-1366. Hdoi:10.1038/labinvest.2008.94
- Y. J. Kim, S. J. Hwang, Y. C. Bae and J. S. Jung, “MiR-21 Regulates Adipogenic Differentiation through the Modulation of TGF-beta Signaling in Mesenchymal Stem Cells Derived from Human Adipose Tissue,” Stem Cells, Vol. 27, No. 12, 2009, pp. 3093-3102.
- T. Papagiannakopoulos, A. Shapiro and K. S. Kosik, “MicroRNA-21 Targets a Network of Key Tumor-Suppressive Pathways in Glioblastoma Cells,” Cancer Research, Vol. 68, No. 19, 2008, pp. 8164-8172. Hdoi:10.1158/0008-5472.CAN-08-1305
- J. Zheng, H. Xue, T. Wang, Y. Jiang, B. Liu, J. Li, Y. Liu, W. Wang, B. Zhang and M. Sun, “MiR-21 Downregulates the Tumor Suppressor P12 CDK2AP1 and Stimulates Cell Proliferation and Invasion,” Journal of Cellular Biochemistry, Vol. 112, No. 3, 2011, pp. 872-880. Hdoi:10.1002/jcb.22995
- M. Korpal, E. S. Lee, G. Hu and Y. Kang, “The MiR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-Cadherin Transcriptional Repressors ZEB1 and ZEB2,” The Journal of Biological Chemistry, Vol. 283, No. 22, 2008, pp. 14910-14914. Hdoi:10.1074/jbc.C800074200
- U. Burk, J. Schubert, U. Wellner, O. Schmalhofer, E. Vincan, S. Spaderna and T. Brabletz, “A Reciprocal Repression between ZEB1 and Members of the miR-200 Family Promotes EMT and Invasion in Cancer Cells,” EMBO Reports, Vol. 9, No. 6, 2008, 582-589. Hdoi:10.1038/embor.2008.74
- P. A. Gregory, A. G. Bert, E. L. Paterson, S. C. Barry, A. Tsykin, G. Farshid, M. A. Vadas, Y. Khew-Goodall and G. J. Goodall, “The miR-200 Family and miR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1,” Nature Cell Biology, Vol. 10, No. 5, 2008, 593-601. Hdoi:10.1038/ncb1722
- D. Kong, Y. Li, Z. Wang, S. Banerjee, A. Ahmad, H. R. Kim and F. H. Sarkar, “MiR-200 Regulates PDGF-DMediated Epithelial-Mesenchymal Transition, Adhesion, and Invasion of Prostate Cancer Cells,” Stem Cells, Vol. 27, No. 8, 2009, pp. 1712-1721. Hdoi:10.1002/stem.101
- S. M. Park, A. B. Gaur, E. Lengyel and M. E. Peter, “The miR-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-cadherin Repressors ZEB1 and ZEB2,” Genes & Development, Vol. 22, No. 7, 2008, pp. 894-907. Hdoi:10.1101/gad.1640608
- V. P. Tryndyak, F. A. Beland and I. P. Pogribny, “E-Cadherin Transcriptional Down-Regulation by Epigenetic and MicroRNA-200 Family Alterations is Related to Mesenchymal and Drug-Resistant Phenotypes in Human Breast Cancer Cells,” International Journal of Cancer, Vol. 126, 2010, pp. 2575-2583.
- O. Saydam, Y. Shen, T. Würdinger, O. Senol, E. Boke, M. F. James, B. A. Tannous, A. O. Stemmer-Rachamimov, M. Yi, R. M. Stephens, et al., “Downregulated Microrna-200a in Meningiomas Promotes Tumor Growth by Reducing E-Cadherin and Activating the Wnt/beta-Catenin Signaling Pathway,” Molecular and Cellular Biology, Vol. 29, No. 21, 2009, pp. 5923-5940. Hdoi:10.1128/MCB.00332-09
- M. Korpal, E. S. Lee, G. Hu and Y. Kang, “The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting Of E-Cadherin Transcriptional Repressors ZEB1 and ZEB2,” The Journal of Biological Chemistry, Vol. 283, No. 22, 2008, pp. 14910-14914. Hdoi:10.1074/jbc.C800074200
- U. Burk, J. Schubert, U. Wellner, O. Schmalhofer, E. Vincan, S. Spaderna and T. Brabletz, “A Reciprocal Repression between ZEB1 and Members of the miR-200 Family Promotes EMT and Invasion in Cancer Cells,” EMBO Reports, Vol. 9, 2008, pp. 582-589. Hdoi:10.1038/embor.2008.74
- P. A. Gregory, A. G. Bert, E. L. Paterson, S. C. Barry, A. Tsykin, G. Farshid, M. A. Vadas, Y. Khew-Goodall and G. J. Goodall, “The miR-200 Family and miR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1,” Nature Cell Biology, Vol. 10, No. 5, 2008, pp. 593-601. Hdoi:10.1038/ncb1722
- D. Kong, Y. Li, Z. Wang, S. Banerjee, A. Ahmad, H. R. Kim and F. H. Sarkar, “MiR-200 Regulates PDGF-DMediated Epithelial-Mesenchymal Transition, Adhesion, and Invasion of Prostate Cancer Cells,” Stem Cells, Vol. 27, No. 8, 2009, pp. 1712-1721. Hdoi:10.1002/stem.101
- S. M. Park, A. B. Gaur, E. Lengyel and M. E. Peter, “The miR-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-Cadherin Repressors ZEB1 and ZEB2,” Genes & Development, Vol. 22, No. 7, 2008, pp. 894-907. Hdoi:10.1101/gad.1640608
- V. P. Tryndyak, F. A. Beland and I. P. Pogribny, “Ecadherin Transcriptional Down-Regulation by Epigenetic and microRNA-200 Family Alterations is Related to Mesenchymal and Drug-Resistant Phenotypes in Human Breast Cancer Cells,” International Journal of Cancer, Vol. 126, No. 11, 2010, pp. 2575-2583.
- O. Saydam, Y. Shen, T. Würdinger, O. Senol, E. Boke, M. F. James, B. A. Tannous, A. O. Stemmer-Rachamimov, M. Yi, R. M. Stephens, et al., “Downregulated microRNA-200a in Meningiomas Promotes Tumor Growth by Reducing E-Cadherin and Activating the Wnt/betaCatenin Signaling Pathway,” Molecular and Cellular Biology, Vol. 29, No. 21, 2009, pp. 5923-5940. Hdoi:10.1128/MCB.00332-09
- C. J. Sherr, “Cancer cell cycles,” Science, Vol. 274, No. 5293, 1996, pp. 1672-1677. Hdoi:10.1126/science.274.5293.1672
- A. B. Pardee, “G1 Events and Regulation of Cell Proliferation,” Science, Vol. 246, No. 4930, 1989, pp. 603-608. Hdoi:10.1126/science.2683075
- H. Matsushime, M. E. Ewen, D. K. Strom, J. Y. Kato, S. K. Hanks, M. F. Roussel and C. J. Sherr, “Identification and Properties of an Atypical Catalytic Subunit (p34PSKJ3/cdk4) for Mammalian D Type G1 Cyclins,” Cell, Vol. 71, 1992, pp. 323-334. Hdoi:10.1016/0092-8674(92)90360-O
- M. Serrano, G. J. Hannon and D. Beach, “A New Regulatory Motif in Cell-Cycle Control Causing Specific Inhibition of Cyclin D/CDK4,” Nature, Vol. 366, No. 6456, 1993, pp. 704-707. Hdoi:10.1038/366704a0
- S. Waga, G. J. Hannon, D. Beach and B. Stillman, “The p21 Inhibitor of Cyclin-Dependent Kinases Controls DNA Replication by Interaction with PCNA,” Nature, Vol. 369, 1994, pp. 574-578. Hdoi:10.1038/369574a0
- W. S. el-Deiry, J. W. Harper, P. M. O’Connor, V. E. Velculescu, C. E. Canman, J. Jackman, J. A. Pietenpol, M. Burrell, D. E. Hill and Y. Wang, “WAF1/CIP1 is Induced in p53-Mediated G1 Arrest and Apoptosis,” Cancer Research, Vol. 54, No. 5, 1994, No. 1169-1174.
- A. Ishikawa, M. Sasaki, Y. Sato, S. Ohira, M. F. Chen, S. F. Huang, K. Oda, Y. Nimura and Y. Nakanuma, “Frequent p16ink4a Inactivation is an Early and Frequent Event of Intraductal Papillary Neoplasm of the Liver Arising in Hepatolithiasis,” Human Pathology, Vol. 35, No. 12, 2004, pp. 1505-1514. Hdoi:10.1016/j.humpath.2004.08.014
- Y. Nakanishi, Y. Zen, S. Kondo, T. Itoh, K. Itatsu and Y. Nakanuma, “Expression of Cell Cycle-Related Molecules in Biliary Premalignant Lesions: Biliary Intraepithelial Neoplasia and Biliary Intraductal Papillary Neoplasm,” Human Pathology, Vol. 39, No. 8, 2008, pp. 1153-1161. Hdoi:10.1016/j.humpath.2007.11.018
- K. Itatsu, M. Sasaki, K. Harada, J. Yamaguchi, H. Ikeda, Y. Sato, T. Ohta, H. Sato, M. Nagino, Y. Nimura, et al., “Phosphorylation of Extracellular Signal-Regulated Kinase 1/2, p38 Mitogen-Activated Protein Kinase and Nuclear Translocation of Nuclear Factor-kappaB are Involved in Upregulation of Matrix Metalloproteinase-9 by Tumour Necrosis Factor-alpha,” Liver International, Vol. 29, No. 2, 2009, pp. 291-298. Hdoi:10.1111/j.1478-3231.2008.01858.x
- C. Welch, Y. Chen and R. L. Stallings, “MicroRNA-34a Functions as a Potential Tumor Suppressor by Inducing Apoptosis in Neuroblastoma Cells,” Oncogene, Vol. 26, 2007, pp. 5017-5022. Hdoi:10.1038/sj.onc.1210293
- H. Tazawa, N. Tsuchiya, M. Izumiya and H. Nakagama, “Tumor-Suppressive miR-34a Induces Senescence-Like Growth Arrest through Modulation of the E2F Pathway in Human Colon Cancer Cells,” Proceedings of the National Academy of Sciences USA, Vol. 104, No. 39, 2007, pp. 15472-15477. Hdoi:10.1073/pnas.0707351104
- O. Merkel, D. Asslaber, J. D. Piñón, A. Eglea and R. Greil, “Interdependent Regulation of p53 and miR-34a in Chronic Lymphocytic Leukemia,” Cell Cycle, Vol. 9, 2010, pp. 2764-2768. Hdoi:10.4161/cc.9.14.12267
- T. C. Chang, E. A. Wentzel, O. A. Kent, K. Ramachandran, M. Mullendore, K. H. Lee, G. Feldmann, M. Yamakuchi, M. Ferlito, C. J. Lowenstein, et al., “Transactivation of miR-34a by p53 Broadly Influences Gene Expression and Promotes Apoptosis,” Molecular Cell, Vol. 26, No. 5, 2007, pp. 745-752. Hdoi:10.1016/j.molcel.2007.05.010
- N. Raver-Shapira, E. Marciano, E. Meiri, Y. Spector, N. Rosenfeld, N. Moskovits, Z. Bentwich and M. Oren, “Transcriptional Activation of miR-34a Contributes to p53-Mediated Apoptosis,” Molecular Cell, Vol. 26, 2007, pp. 731-743. Hdoi:10.1016/j.molcel.2007.05.017
- V. Tarasov, P. Jung, B. Verdoodt, D. Lodygin, A. Epanchintsev, A. Menssen, G. Meister and H. Hermeking, “Differential Regulation of microRNAs by p53 Revealed by Massively Parallel Sequencing: miR-34a is a p53 Target that Induces Apoptosis and G1-arrest,” Cell Cycle, Vol. 6, 2007, pp. 1586-1593. Hdoi:10.4161/cc.6.13.4436
- M. Fabbri, A. Bottoni, M. Shimizu, R. Spizzo, M. S. Nicoloso, S. Rossi, E. Barbarotto, A. Cimmino, B. Adair, S. E. Wojcik, et al., “Association of a microRNA/TP53 Feedback Circuitry with Pathogenesis and Outcome of B-cell Chronic Lymphocytic Leukemia,” The Journal of the American Medical Association, Vol. 305, No. 1, 2011, pp. 59-67. Hdoi:10.1001/jama.2010.1919
- Y. Wang, C. G. Lee, “MicroRNA and Cancer—Focus on Apoptosis,” Journal of Cellular and Molecular Medicine, Vol. 13, No. 1, 2009, pp. 12-23. Hdoi:10.1111/j.1582-4934.2008.00510.x
- S. W. Lowe and A. W. Lin, “Apoptosis in cancer,” Carcinogenesis, Vol. 21, No. 3, 2000, pp. 485-495. Hdoi:10.1093/carcin/21.3.485
- M. Sasaki, J. Yamaguchi, K. Itatsu, H. Ikeda and Y. Nakanuma, “Over-Expression of Polycomb Group Protein EZH2 Relates to Decreased Expression of p16 INK4a in Cholangiocarcinogenesis in Hepatolithiasis,” The Journal of Pathology, Vol. 215, No. 2, 2008, pp. 175-183. Hdoi:10.1002/path.2345
- Y. Zen, K. Harada, M. Sasaki, K. Tsuneyama, K. Katayanagi, Y. Yamamoto and Y. Nakanuma, “Lipopolysaccharide Induces Overexpression of MUC2 and MUC5AC in Cultured Biliary Epithelial Cells: Possible Key Phenomenon of Hepatolithiasis,” American Journal of Pathology, Vol. 161, No. 4, 2002, pp. 1475-1484. Hdoi:10.1016/S0002-9440(10)64423-9
- S. Dangi-Garimella, M. J. Strouch, P. J. Grippo, D. J. Bentrem and H. G. Munshi, “Collagen Regulation of Let- 7 in Pancreatic Cancer Involves TGF-β1-Mediated Membrane Type 1-Matrix Metalloproteinase Expression,” Oncogene, Vol. 30, No. 8, 2011, pp. 1002-1008. Hdoi:10.1038/onc.2010.485
- Y. Kodama, M. Hijikata, R. Kageyama, K. Shimotohno, T. Chiba, “The Role of Notch Signaling in the Development of Intrahepatic Bile Ducts,” Gastroenterology, Vol. 127, No. 6, 2004, pp. 1775-1786. Hdoi:10.1053/j.gastro.2004.09.004
- V. Bolos, J. Grego-Bessa and J. L. de la Pompa, “Notch Signaling in Development and Cancer,” Endocrine Reviews, Vol. 28, No. 3, 2007, pp. 339-363. Hdoi:10.1210/er.2006-0046
- N. Ishimura, S. F. Bronk and G. J. Gores, “Inducible Nitric Oxide Synthase Up-Regulates Notch-1 in Mouse Cholangiocytes: Implications for Carcinogenesis,” Gastroenterology, Vol. 128, No. 5, 2005, pp. 1354-1368. Hdoi:10.1053/j.gastro.2005.01.055
- H. Francis, G. Alpini and S. DeMorrow, “Recent Advances in the Regulation of Cholangiocarcinoma Growth,” American Journal of Physiology-Gastrointestinal and Liver Physiology, Vol. 299, No. 1, 2010, pp. G1-G9. Hdoi:10.1152/ajpgi.00114.2010
- F. Guessous, Y. Zhang, A. Kofman, A. Catania, Y. Li, D. Schiff, B. Purow and R. Abounader, “MicroRNA-34a is Tumor Suppressive in Brain Tumors and Glioma Stem Cells,” Cell Cycle, Vol. 9, No. 6, 2010, pp. 1031-1036. Hdoi:10.4161/cc.9.6.10987
- P. W. Ingham and A. P. McMahon, “Hedgehog Signaling in Animal Development: Paradigms and Principles,” Genes & Development, Vol. 15, No. 23, 2001, pp. 3059- 3087. Hdoi:10.1101/gad.938601
- K. Koebernick and T. Pieler, “Gli-Type Zinc Finger Proteins as Bipotential Transducers of Hedgehog Signaling,” Differentiation, Vol. 70, No. 2-3, 2002, pp. 69-76. Hdoi:10.1046/j.1432-0436.2002.700201.x
- N. Oishi and X. W. Wang, “Novel Therapeutic Strategies for Targeting Liver Cancer Stem Cells,” International Journal of Biological Sciences, Vol. 7, No. 5, 2011, pp. 517-535. Hdoi:10.7150/ijbs.7.517
- C. Osipo and L. Miele, “Hedgehog Signaling in Hepatocellular Carcinoma: Novel Therapeutic Strategy Targeting Hedgehog Signaling in HCC,” Cancer Biology and Therapy, Vol. 5, No. 2, 2006, pp. 238-239. Hdoi:10.4161/cbt.5.2.2566
- L. Yang, G. Xie, Q. Fan and J. Xie, “Activation of the Hedgehog-Signaling Pathway in Human Cancer and the Clinical Implications” Oncogene, Vol. 29, No. 4, 2010, pp. 469-481. Hdoi:10.1038/onc.2009.392
- T. Uziel, F. V. Karginov, S. Xie, J. S. Parker, Y. D. Wang, A. Gajjar, L. He, D. Ellison, R. J. Gilbertson, G. Hannon and M. F. Roussel, “The miR-17~92 Cluster Collaborates with the Sonic Hedgehog Pathway in Medulloblastoma,” Proceedings of the National Academy of Sciences USA, Vol. 106, No. 8, 2009, pp. 2812-2817. Hdoi:10.1073/pnas.0809579106
- A. Omenetti, A. Porrello, Y. Jung, L. Yang, Y. Popov, S.S. Choi, et al., “Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition during Biliary Fibrosis in Rodents and Humans,” The Journal of Clinical Investigation, Vol. 118, No. 10, 2008, pp. 3331-3342.
- A. Omenetti, Y. Popov, Y. Jung, S. S. Choi, R. P. Witek, L. Yang, et al., “The Hedgehog Pathway Regulates Remodeling Responses to Biliary Obstruction in Rats,” International Journal of Gastroenterology & Hepatology, Vol. 57, No. 9, 2008, pp. 1275-1282. Hdoi:10.1136/gut.2008.148619
- A. Omenetti, L. Yang, Y. X. Li, S. J. McCall, Y. Jung, J. K. Sicklick, et al., “Hedgehog-Mediated MesenchymalEpithelial Interactions Modulate Hepatic Response to Bile Duct Ligation,” Laboratory Investigation, Vol. 87, No. 5, 2007, pp. 499-514.
- Y. Jung, S. J. McCall, Y. X. Li and A. M. Diehl, “Bile Ductules and Stromal Cells Express Hedgehog Ligands and/or Hedgehog Target Genes in Primary Biliary Cirrhosis,” Hepatology, Vol. 45, No. 5, 2007, pp. 1091-1096. Hdoi:10.1002/hep.21660
- A. Omenetti and A. M. Diehl, “Hedgehog Signaling in Cholangiocytes,” Current Opinion in Gastroenterology, Vol. 27, 2011, pp. 268-275. Hdoi:10.1097/MOG.0b013e32834550b4
- A. S. Flynt, N. Li, E. J. Thatcher, L. Solnica-Krezel and J. G. Patton, “Zebrafish miR-214 Modulates Hedgehog Signaling to Specify Muscle Cell Fate,” Nature Genetics, Vol. 39, No. 2, 2007, pp. 259-263. Hdoi:10.1038/ng1953
- H. Yang, W. Kong, L. He, et al., “MicroRNA Expression Profiling in Human Ovarian Cancer: miR-214 Induces Cell Survival and Cisplatin Resistance by Targeting PTEN,” Cancer Research, Vol. 68, No. 2, 2008, pp. 425- 433. Hdoi:10.1158/0008-5472.CAN-07-6426
- R. Amin and L. Mishra, “Liver Stem Cells and tgf-Beta in Hepatic Carcinogenesis,” Gastrointest Cancer Research, Vol. 2, 2008, pp. S27-S30.
- Y. Tang, K. Kitisin, W. Jogunoori, C. Li, C. X. Deng and S. C. Mueller, et al., “Progenitor/Stem Cells Give Rise to Liver Cancer due to Aberrant TGF-beta and IL-6 Signaling,” Proceedings of the National Academy of Sciences USA, Vol. 105, No. 7, 2008, pp. 2445-2450. Hdoi:10.1073/pnas.0705395105
- L. Mishra, R. Derynck and B. Mishra, “Transforming Growth Factor-beta Signaling in Stem Cells and Cancer,” Science, Vol. 310, No. 5745, 2005, pp. 68-71. Hdoi:10.1126/science.1118389
- K. Breuhahn, T. Longerich and P. Schirmacher, “Dysregulation of Growth Factor Signaling in Human Hepatocellular Carcinoma,” Oncogene, Vol. 25, 2006, pp. 3787-3800. Hdoi:10.1038/sj.onc.1209556
- H. G. Kim, Y. H. Chung, B. C. Song, J. Kim, S. H. Yang, Y. S. Lee and D. J. Suh, “Expression of Transforming Growth Factor beta-1 in Chronic Hepatitis and Hepatocellular Carcinoma Associated with Hepatitis C Virus Infection,” The Korean Journal of Internal Medicine, Vol. 15, No. 3, 2000, pp. 165-170.
- J. F. Tsai, J. E. Jeng, L. Y. Chuang, et al., “Elevated Urinary Transforming Growth Factor-beta1 Level as a Tumour Marker and Predictor of Poor Survival in Cirrhotic Hepatocellular Carcinoma,” British Journal of Cancer, Vol. 76, No. 2, 1997, pp. 244-250. Hdoi:10.1038/bjc.1997.369
- Y. Lu, L. Q. Wu, C. S. Li, et al., “Expression of Transforming Growth Factors in Hepatocellular Carcinoma and Its Relations with Clinicopathological Parameters and Prognosis,” Hepatobiliary & Pancreatic Diseases International, Vol. 7, 2008, pp. 174-178.
- M. Ikeguchi, A. Iwamoto, K. Taniguchi, et al., “The Gene Expression Level of Transforming Growth Factor-beta (TGF-beta) as a Biological Prognostic Marker of Hepatocellular Carcinoma,” Journal of Experimental & Clinical Cancer Research, Vol. 24, No. 3, 2005, pp. 415- 421.
- Y. Shirai, S. Kawata, S. Tamura, N. Ito, H. Tsushima, K. Takaishi, S. Kiso and Y. Matsuzawa, “Plasma Transforming Growth Factor-beta 1 in Patients with Hepatocellular Carcinoma. Comparison with Chronic Liver Diseases,” Cancer, Vol. 73, No. 9, 1994, pp. 2275-2279. Hdoi:10.1002/1097-0142(19940501)73:9<2275::AID-CNCR2820730907>3.0.CO;2-T
- J. P. Lu, J. Q. Mao, M. S. Li, S. L. Lu, X. Q. Hu, S. N. Zhu and S. Nomura, “In situ Detection of TGF Betas, TGF Beta Receptor II mRNA and Telomerase Activity in Rat Cholangiocarcinogenesis,” World Journal of Gastroenterology, Vol. 9, No. 3, 2003, pp. 590-594.
- C. Benckert, S. Jonas, T. Cramer, Z. Von Marschall, G. Schafer, M. Peters, K. Wagner, C. Radke, B. Wiedenmann, P. Neuhaus, et al., “Transforming Growth Factor Beta 1 Stimulates Vascular Endothelial Growth Factor Gene Transcription in Human Cholangiocellular Carcinoma Cells,” Cancer Research, Vol. 63, No. 5, 2003, pp. 1083-1092.
- Y. Zen, K. Harada, M. Sasaki, T. Chen, M. Chen, T. Yeh, Y. Jan, S. Huang, Y. Nimura and Y. Nakamura, “Intrahepatic Cholangiocarcinoma Escapes from Growth Inhibitory Effect of Transforming Growth Factor-beta1 by Overexpression of Cyclin D1,” Laboratory Investigation, Vol. 85, No. 4, 2005, pp. 572-581. Hdoi:10.1038/labinvest.3700236H
- M. E. Burlone and A. Budkowska, “Hepatitis C Virus Cell Entry Role of Lipoproteins and Cellular Receptors,” Journal of General Virology, Vol. 90, No. 5, 2009, pp. 1055-1070. Hdoi:10.1099/vir.0.008300-0
- M. Nassal and B. Hepatitis, “Viruses: Reverse Transcription a Different Way,” Virus Research, Vol. 134, No. 1-2, 2008, pp. 235-249. Hdoi:10.1016/j.virusres.2007.12.024
- Z. H. Liu and J. L. Hou, “Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Dual Infection,” International Journal of Medical Sciences, Vol. 3, No. 2, 2006, pp. 57- 62. Hdoi:10.7150/ijms.3.57
- D. P. Bartel, “MicroRNAs: Genomics, Biogenesis, Mechanism, and Function,” Cell, Vol. 116, 2004, pp. 281- 297. Hdoi:10.1016/S0092-8674(04)00045-5
- N. Bushati and S. M. Cohen, “MicroRNA functions,” Annual Review of Cell and Developmental Biology, Vol. 23, 2007, pp. 175-205. Hdoi:10.1146/annurev.cellbio.23.090506.123406
- J. M. Thomson, M. Newman, J. S. Parker, E. M. Morin-Kensicki, T. Wright and S. M. Hammond, “Extensive Post-Transcriptional Regulation of microRNAs and Its Implications for Cancer,” Genes & Development, Vol. 20, No. 16, 2006, pp. 2202-2207. Hdoi:10.1101/gad.1444406H
- M. L. Yeung, Y. Bennasser, T. G. Myers, G. J. Jiang, M. Benkirane and K. T. Jeang, “Changes in microRNA Expression Profiles in HIV 1 Transfected,” Retrovirology, Vol. 2, No. 2, 2005, p. 81. Hdoi:10.1186/1742-4690-2-S1-S81
- A. Gupta, J. J. Gartner, P. Sethupathy, A. G. Hatzigeorgiou, N. W. Fraser, “Anti-Apoptotic Function of a microRNA Encoded by the HSV1 Latency-Associated Transcript,” Nature, Vol. 442, No. 7098, 2006, pp. 82-85.
- Z. Ghosh, B. Mallick and J. Chakrabarti, “Cellular versus Viral microRNAs in Host-Virus Interaction,” Nucleic Acids Research, Vol. 37, No. 4, 2009, No. 1035-1048. Hdoi:10.1093/nar/gkn1004
- O. Voinnet, “Induction and Suppression of RNA Silencing: Insights from Viral Infections,” Nature Reviews Genetics, Vol. 6, No. 3, 2005, pp. 206-220. Hdoi:10.1038/nrg1555
- R. L. Skalsky and B. R. Cullen, “Viruses, microRNAs, and Host Interactions,” Annual Review of Microbiology, Vol. 64, 2010, pp. 123-141. Hdoi:10.1146/annurev.micro.112408.134243
- Y. Bennasser, M. L. Yeung and K. T. Jeang, “HIV-1 TAR RNA Subverts RNA Interference in Transfected Cells through Sequestration of TAR RNA-binding Protein TRBP,” The Journal of Biological Chemistry, Vol. 281, No. 38, 2006, pp. 27674-27678. Hdoi:10.1074/jbc.C600072200
- S. Pfeffera and T. F. Baumert, “Impact of microRNAs for Pathogenesis and Treatment of Hepatitis C Virus Infection,” Gastroentérologie Clinique et Biologique, Vol. 34, No. 8-9, 2010, pp. 431-435. Hdoi:10.1016/j.gcb.2010.04.010
- P. Sarnow, C. L. Jopling, K. L. Norman, S. Schütz and K. A.Wehner, “MicroRNAs Expression, Avoidance and Subversion by Vertebrate Viruses,” Nature Reviews, Vol. 4, No. 9, 2006, pp. 651-659. Hdoi:10.1038/nrmicro1473
- E. Gottwein and B. R. Cullen, “Viral and Cellular microRNAs as Determinants of Viral Pathogenesis and Immunity,” Cell Host & Microbe, Vol. 3, No. 6, 2008, pp. 375-387. Hdoi:10.1016/j.chom.2008.05.002
- W. B. Jin and F. L. Wu, “HBV-Encoded microRNA Candidate and Its Target,” Computational Biology and Chemistry, Vol. 31, No. 2, 2007, pp. 124-126. Hdoi:10.1016/j.compbiolchem.2007.01.005
- M. Honda, T. Yamashita, T. Ueda, H. Takatori, R. Nishino and S. Kaneko, “Different Signaling Pathways in the Livers of Patients with Chronic Hepatitis B or Chronic Hepatitis C,” Hepatology, Vol. 44, No. 5, 2006, pp. 1122-1138. Hdoi:10.1002/hep.21383
- C. Braconi, N. Valeri, P. Gasparini, N. Y. Huang, C. Taccioli, G. Nuovo, T. Suzuki, C. M. Croce and T. Patel, “Hepatitis C Virus Proteins Modulate MicroRNA Expression and Chemosensitivity in Malignant Hepatocytes,” Clinical Cancer Research, Vol. 16, No. 3, 2010, pp. 957-966. Hdoi:10.1158/1078-0432.CCR-09-2123H
- X. X. Peng, Y. Li, K. A. Walters, E. R. Rosenzweig, S. L. Lederer, L. D. Aicher, S. Proll and M. G. Katze, “Computational Identification of Hepatitis C Virus Associated microRNA-mRNA Regulatory Modules in Human Livers,” BMC Genomics, Vol. 10, 2009, p. 373. Hdoi:10.1186/1471-2164-10-373
- X. Y. Liu, T. Y. Wang, T. Wakita and W. Yang, “Systematic Identification of microRNA and Messenger RNA Profiles in Hepatitis C Virus-Infected Human Hepatoma Cells,” Virology, Vol. 398, No. 1, 2010, pp. 57-67. Hdoi:10.1016/j.virol.2009.11.036
- N. M. Steuerwald, J. C. Parsons, K. Bennett, T. C. Bates and H. L. Bonkovsky, “Parallel microRNA and mRNA Expression Profiling of (genotype 1b) Human Hepatoma Cells Expressing Hepatitis C Virus,” Liver International, Vol. 30, No. 10, 2010, pp. 1490-1504. Hdoi:10.1111/j.1478-3231.2010.02321.x
- Y. Liu, J. J. Zhao, C. M. Wang, M. Y. Li, P. Hang, L. Wang, Y. Q. Chen, F. Zoulim, X. Ma and D. P. Xu, “Altered Expression Profiles of microRNAs in a Stable Hepatitis B Virus-Expressing Cell Line,” Chinese Medical Journal, Vol. 122, No. 1, 2009, pp. 10-14. Hdoi:10.3901/JME.2009.11.010
- H. T. Zhu, Q. Z. Dong, G. Wang, H. J. Zhou, N. Ren, H. L. Jia, Q. H. Ye and L. X. Qin, “Identification of Suitable Reference Genes for qRT-PCR Analysis of Circulating microRNAs in Hepatitis B Virus-Infected Patients,” Molecular Biotechnology, Vol. 50, No. 1, 2012, pp. 49-56. Hdoi:10.1007/s12033-011-9414-6
- S. Ura, M. Honda, T. Yamashita, T. Ueda, H. Takatori, R. Nishino, H. Sunakozaka, Y. Sakai, K. Horimoto and S. Kaneko, “Differential microRNA Expression between Hepatitis B and Hepatitis C Leading Disease Progression to Hepatocellular Carcinoma,” Hepatology, Vol. 49, No. 4, 2009, pp. 1098-1112. Hdoi:10.1002/hep.22749
- M. Lagos-Quintana, R. Rauhut, A. Yalcin, J. Meyer, W. Lendeckel and T. Tuschl, “Identification of Tissue-Specific microRNAs from Mouse,” Current Biology, Vol. 12, No. 9, 2002, pp. 735-739. Hdoi:10.1016/S0960-9822(02)00809-6
- C. L. Jopling, M. K. Yi, A. M. Lancaster, S. M. Lemon and P. Sarnow, “Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific micro-RNA,” Science, Vol. 309, No. 5740, 2005, pp. 1577-1581. Hdoi:10.1126/science.1113329
- C. L. Jopling, S. Schütz and P. Sarnow, “Position-Dependent Function for a Tandem microRNA miR-122- binding Site Located in the Hepatitis C Virus RNA Genome,” Cell Host & Microbe, Vol. 4, 2008, pp. 77-85. Hdoi:10.1016/j.chom.2008.05.013
- K. Shruti, K. Shrey and R. Vibha, “Micro RNAs: Tiny Sequences with Enormous Potential,” Biochemical and Biophysical Research Communications, Vol. 407, No. 4, 2011, pp. 445-449. Hdoi:10.1016/j.bbrc.2011.03.058
- E. S. Machlin, P. Sarnow and S. M. Sagan, “Masking the 5′ Terminal Nucleotides of the Hepatitis C Virus Genome by an Unconventional microRNA-target RNA Complex,” Proceedings of the National Academy of Sciences USA, Vol. 108, No. 8, 2011, pp. 3193-3198. Hdoi:10.1073/pnas.1012464108
- J. A. Wilson, C. Zhang, A. Huys and C. D. Richardson, “Human Ago2 Is Required for Efficient MicroRNA 122 Regulation of Hepatitis C Viru s RNA Accumulation and Translation,” Journal of Virology, Vol. 85, No. 5, 2011, pp. 2342-2350. Hdoi:10.1128/JVI.02046-10
- J. I. Henke, D. Goergen, J. Zheng, Y. Song, C. G. Schüttler, C. Fehr, C. Jünemann and M. Niepmann, “MicroRNA-122 Stimulates Translation of Hepatitis C Virus RNA,” EMBO Journal, Vol. 27, No. 24, 2008, pp. 3300- 3310. Hdoi:10.1038/emboj.2008.244
- M. Niepmann, “Activation of Hepatitis C Virus Translation by a Liver-Specific micro-RNA,” Cell Cycle, Vol. 8, No. 10, 2009, pp. 1473-1477. Hdoi:10.4161/cc.8.10.8349
- R. K. Jangra, M. K. Yi and S. M. Lemon, “Regulation of Hepatitis C Virus Translation and Infectious Virus Production,” Journal of Virology, Vol. 84, No. 13, 2010, pp. 6615-6625. Hdoi:10.1128/JVI.00417-10
- Y. Shan, J. Y. Zheng, R. W. Lambrecht and H. L. Bonkovsky, “Reciprocaleffects of micro-RNA-122 on Expression of Heme Oxygenase-1 and Hepatitis C Virus Genes in Human Hepatocytes,” Gastroenterology, Vol. 133, No. 4, 2007, pp. 1166-1174. Hdoi:10.1053/j.gastro.2007.08.002H
- L. P. Qiu, H. X. Fan, W. S. Jin, B. Zhao, Y. Z. Wang, Y. Ju, L. Z. Chen, Y. Chen, Z. P. Duan and S. D. Meng, “MiR-122-Induced Down-Regulation of HO-1 Negatively Affects miR-122-Mediated Suppression of HBV,” Biochemical and Biophysical Research Communications, Vol. 398, No. 4, 2010, pp. 771-777. Hdoi:10.1016/j.bbrc.2010.07.021
- Y. Murakami, H. H. Aly, A. Tajima, I. Inoue and K. Shimotohno, “Regulation of the Hepatitis C Virus Genome Replication by miR-199a,” Journal of Hepatology, Vol. 50, No. 3, 2009, pp. 453-460. Hdoi:10.1016/j.jhep.2008.06.010
- G. L. Zhang, Y. X. Li, S. Q. Zheng, M. Liu, X. Li, H. Tang, “Suppression of Hepatitis B Virus Replication by microRNA-199a-3p and microRNA-210,” Antiviral Research, Vol. 88, No. 2, 2010, pp. 169-175. Hdoi:10.1016/j.antiviral.2010.08.008
- W. Hou, Q. Tian, J. Zheng and H. L. Bonkovsky, “Micro-RNA-196 Represses Bach1 Protein and Hepatitis C Virus Gene Expression in Human Hepatoma Cells Expressing Hepatitis C Viral Proteins,” Hepatology, Vol. 51, No. 5, 2010, pp. 1494-1504. Hdoi:10.1002/hep.23401
- N. M. Steuerwald, J. C. Parsons, K. Bennett, T. C. Bates and H. L. Bonkovsky, “Parallel microRNA and mRNA Expression Profiling of (genotype 1b) Human Hepatoma cells Expressing Hepatitis C Virus,” Liver International, Vol. 30, No. 10, 2010, pp. 1490-1504. Hdoi:10.1111/j.1478-3231.2010.02321.x
- K. Banaudha, M. Kaliszewski, T. Korolnek, L. Florea, M. L. Yeung, K. T. Jeang and A. Kumar, “MicroRNA Silencing of Tumor Suppressor DLC-1 Promotes Efficient Hepatitis C Virus Replication in Primary Human Hepatocytes,” Hepatology, Vol. 53, No. 1, 2011, pp. 53-61. Hdoi:10.1002/hep.24016
- X. Zhang, E. Zhang, Z. Ma, R. Pei, M. Jiang, J. F. Schlaak, M. Roggendorf and M. Lu, “Modulation of HBV Replication and Hepatocyte Differentiation by microRNA-1,” Hepatology, Vol. 53, No. 5, 2011, pp. 1476- 1485. Hdoi:10.1002/hep.24195
- N. Potenza, U. Papa, N. Mosca, F. Zerbini, V. Nobile and A. Russo, “Human microRNA hsa-miR-125a-5p Interferes with Expression of Hepatitis B Virus Surface Antigen,” Nucleic Acids Research, Vol. 39, No. 12, 2011, pp. 5157-5163. Hdoi:10.1093/nar/gkr067
- F. L. Wu, W. B. Jin, J. H. Li and A. G. Guo, “Targets for Human Encoded microRNAs in HBV Genes,” Virus Genes, Vol. 42, No. 2, 2011, pp. 157-161. Hdoi:10.1007/s11262-010-0555-7
- A. Tan, S. H. Yeh, C. J. Liu, C. Cheung and P. J. Chen, “Viral Hepatocarcinogenesis: From Infection to Cancer,” Liver International, Vol. 28, No. 2, 2008, pp. 175-188. Hdoi:10.1111/j.1478-3231.2007.01652.x
- Y. Ladeiro, G. Couchy, C. Balabaud, P. Bioulac-Sage, L. Pelletier, S. Rebouissou and J. Zucman-Rossi, “MicroRNA Profiling in Hepatocellular Tumors is Associated with Clinical Features and Oncogene/Tumor Suppressor Gene Mutations,” Hepatology, Vol. 47, No. 6, 2008, pp. 1955-1963. Hdoi:10.1002/hep.22256
- R. E. Lanford, E. S. Hildebrandt-Eriksen, A. Petri, R. Persson, M. Lindow, M. E. Munk, S. Kauppinen and H. Orum, “Therapeutic Silencing of microRNA-122 in Primates with Chronic Hepatitis C Virus Infection,” Science, Vol. 327, No. 5962, 2010, pp. 198-201. Hdoi:10.1126/science.1178178
- A. Tan, S. H. Yeh, C. J. Liu, C. Cheung and P. J. Chen, “Viral Hepatocarcinogenesis: From Infection to Cancer,” Liver Internatioanl, Vol. 28, No. 2, 2008, pp. 175-188. Hdoi:10.1111/j.1478-3231.2007.01652.x
- I. M. Pedersen, G. Cheng, S. Wieland, S. Volinia, C. M. Croce, F. V. Chisari and M. David, “Interferon Modulation of Cellular microRNAs as an Antiviral Mechanism,” Nature, Vol. 449, No. 7164, 2007, pp. 919-922. Hdoi:10.1038/nature06205
- C. Scagnolari, P. Zingariello, J. Vecchiet, C. Selvaggi, D. Racciatti, G. Taliani, E. Riva, E. Pizzigallo and G. Antonelli, “Differential Expression of Interferon-Induced microRNAs in Patients with Chronic Hepatitis C Virus Infection Treated with Pegylated Interferon Alpha,” Virology Journal, Vol. 7, No. 1, 2010, p. 311. Hdoi:10.1186/1743-422X-7-311
- Y. Murakami, M. Tanaka, H. Toyoda, K. Hayashi, M. Kuroda, A. Tajima and K. Shimotohno, “Hepatic microRNA Expression is Associated with the Response to Interferon Treatment of Chronic Hepatitis C,” BMC Medical Genomics, Vol. 3, No. 1, 2010, p. 48. Hdoi:10.1186/1755-8794-3-48
- V. S. Mahajan, A. Drake and J. Chen, “Virus-Specific Host miRNAs Antiviral Defenses or Promoters of Persistent Infection,” Trends in Immunology, Vol. 30, No. 1, 2009, pp. 1-7. Hdoi:10.1016/j.it.2008.08.009
- W. Li, L. Xie, X. He, J. Li, K. Tu, L. Wei, et al., “Diagnostic and Prognostic Implications of microRNAs in Human Hepatocellular Carcinoma,” International Journal of Cancer, Vol. 123, No. 7, 2008, pp. 1616-1622. Hdoi:10.1002/ijc.23693
- J. Jiang, Y. Gusev, I. Aderca, T. A. Mettler, D. M. Nagorney, D. J. Brackett, et al., “Association of MicroRNA Expression in Hepatocellular Carcinomas with Hepatitis Infection, Cirrhosis and Patient Survival,” Clinical Cancer Research, Vol. 14, No. 2, 2008, pp. 419-427. Hdoi:10.1158/1078-0432.CCR-07-0523H
- H. Su, J. R. Yang, T. Xu, J. Huang, L. Xu, Y. Yuan and S. M. Zhuang, “MicroRNA-101, Downregulated in Hepatocellular Carcinoma, Promotes Apoptosis and Suppresses Tumorigenicity,” Cancer Research, Vol. 69, No. 3, 2009, pp. 1135-1142. Hdoi:10.1158/0008-5472.CAN-08-2886
- J. Ji, T. Yamashita, A. Budhu, M. Forgues, H. L. Jia, C. Li, et al., “Identification of microRNA-181 by GenomeWide Screening as a Critical Player in EpCAM-Positive Hepatic Cancer Stem Cells,” Hepatology, Vol. 50, No. 2, 2009, pp. 472-480. Hdoi:10.1002/hep.22989
- H. Varnholt, U. Drebber, F. Schulze, I. Wedemeyer, P. Schirmacher, H. P. Dienes and M. Odenthal, “MicroRNA Gene Expression Profile of Hepatitis C Virus-Associated Hepatocellular Carcinoma,” Hepatology, Vol. 47, No. 4, 2008, pp. 1223-1232. Hdoi:10.1002/hep.22158
- A. Budhu, H. L. Jia, M. Forgues, C. G. Liu, D. Goldstein, A. Lam, K. A. Zanetti, Q. H. Ye, L. X. Qin, et al., “Identification of Metas-Tasis-Related microRNAs in Hepatocellular Carcinoma,” Hepatology, Vol. 47, No. 3, 2008, pp. 897-907. Hdoi:10.1002/hep.22160
- K. Z. Qu, K. Zhang, H. Li, N. H. Afdhala and M. Albitar, “Circulating microRNAs as Biomarkers for Hepatocellular Carcinoma,” Journal of Clinical Gastroenterology, Vol. 45, No. 4, 2011, pp. 355-360. Hdoi:10.1097/MCG.0b013e3181f18ac2
- Y. Yamamoto, N. Kosaka, M. Tanaka, et al., “MicroRNA-500 as a Potential Diagnostic Marker for Hepatocellular Carcinoma,” Biomarkers, Vol. 14, No. 7, 2009, pp. 529-538 Hdoi:10.3109/13547500903150771
- J. Li, Y. Wang, W. Yu, J. Chen and J. Luo, “Expression of Serum miR-221 in Human Hepatocellular Carcinoma and Its Prognostic Significance,” Biochemical and Biophysical Research Communications, Vol. 406, No. 1, 2011, pp. 70-73. Hdoi:10.1016/j.bbrc.2011.01.111
- B. S. Sun, Q. Z. Dong, Q. H. Ye, H. J. Sun, H. L. Jia, X. Q. Zhu, et al., “Lentiviral-Mediated miRNA against Osteopontin Suppresses Tumor Growth and Metastasis of Human Hepatocellular Carcinoma,” Hepatology, Vol. 48, No. 6, 2008, pp. 1834-1842. Hdoi:10.1002/hep.22531
NOTES
*Corresponding author.