Open Journal of Physical Chemistry
Vol.05 No.01(2015), Article ID:53387,7 pages
10.4236/ojpc.2015.51001
Interconversion between Planar-Triangle, Trigonal-Pyramid and Tetrahedral Configurations of Boron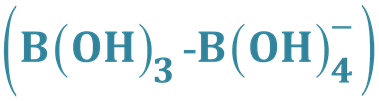 , Carbon
, Carbon
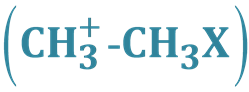 and for the Group 15 Elements as Nitrogen
and for the Group 15 Elements as Nitrogen . A Modelling Description with ab Initio Results and Pressure-Induced Experimental Evidence
. A Modelling Description with ab Initio Results and Pressure-Induced Experimental Evidence
Henk M. Buck
Kasteel Twikkelerf 94, Tilburg, The Netherlands
Email: h.m.buck@ziggo.nl
Copyright © 2015 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 30 December 2014; accepted 16 January 2015; published 20 January 2015

ABSTRACT
Recently a mechanistic understanding of the pressure- and/or temperature-induced coordination change of boron in a borosilicate glass has been demonstrated by Edwards et al. In situ high- pressure 11B solid-state NMR spectroscopy has been used in combination with ab initio calculations in order to obtain insight in the molecular geometry for the pressure-induced conversion. The results indicate a deformation of the B(OH)3 planar triangle, under isotropic stress, into a trigonal pyramid that serves as a precursor for the formation of a tetrahedral boron configuration. From our point of view, the deformation controlling the out-of-plane transition of boron accompanied with a D3h into C3v geometric change is an interesting transformation because it matches with our molecular description based on Van’t Hoff modelling for the tetrahedral change of carbon in CH3X by substitution of X with nucleophiles via a trigonal bipyramid state in which the transferred carbon is present as a methyl planar triangle “cation”. Van’t Hoff modelling and ab initio calculations have been also applied on the dynamics of the out-of-plane geometry of a transient positively charged carbon in a trigonal pyramidal configuration into a planar trivalent carbon cation. Finally the same model is also used for the C3v trigonal pyramidal configurations as NH3 of the group 15 elements in their nucleophilic abilities.
Keywords:
Pressure-Induced Coordination, Van’t Hoff Modelling, Ab Initio Calculations, Reaction Dynamics

1. Introduction
We introduced several groups as localized sites in organic molecular systems for accommodation of fundamental modes of bonding via intra- and inter-molecular reactions. Most of these interactions were focused on well- known reaction types, theoretically described with experimental evidence for complex models. In these studies, three-center four-, three-, and two-electron systems based on carbon-, boron-, hydrogen-, and halogen exchange reactions were studied along the lines of ab initio and Van’t Hoff (vtH) modelling studies [1] -[7] . In the case of a linear three-center model, it was possible with the dynamics of a regular tetrahedron to change its geometry into a trigonal bipyramidal transition- or intermediate complex, to predict the various bond lengths at specific locations on the reaction profile. Based on the numbers of electrons (4, 3 and 2, respectively) in the transient complex, all transfer or exchange reactions share the same ratio of the apical bond distances compared with the corresponding bonding distances in the initial state. These ratio numbers are then 1.333, 1.250, and 1.167, respectively. As illustration for this general concept, we describe the three-center four-electron transition state of the identity SN2 reaction X− + CH3-X ® [X-CH3-X]− ® X-CH3 + X− with X is a halogen. In the dynamics of this process, the methyl “cation” migrates between the partially negatively charged halogens in the trigonal bipyramid. This is illustrated in Figure 1.
In the vtH model, the tetrahedral angle of 109.47˚ results in a ratio number of 1 − cos109.47˚ = 1.333 (ratio between the apical bond length and the corresponding tetrahedral length). For the reaction shown in Figure 1, a good correspondence has been obtained between the ratio numbers defined as R(d) and R(θ):
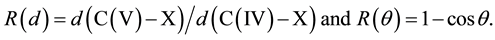
The results are given in Table 1 in combination with ab initio results [1] [3] [4] . The computations using relativistic DFT at the ZORA-OL0.YP/TZ2P level are obtained from Bento et al. [8] [9] , the other data from Glukhovtsev et al. [10] .
Within the scope of the vtH model, we used the experimental values of the bond distances and angles of the corresponding tetrahedral configuration for calculating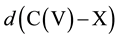 .
.
As mentioned before, this model has been used to describe the pressure-induced conversion of a BO3 triangle in planar B(OH)3 into a trigonal pyramid [11] . The latter structure may be considered as transition state in the formation of a tetrahedral configuration effectuated by local coordination environments via atomic oxygen sites

Figure 1. The identity substitution reaction of Cl− + CH3Cl via a pentavalent carbon.

Table 1. A comparison between R(d) and R(θ) values in combination with the pentavalent carbon state [XCH3X]−. The (ab initio) distances (d) are given in Å and the corresponding R(θ) values from the experimental angle data of their tetrahedral configurations.
in the crystalline lattice.
In our opinion, this type of reaction deserves a more fundamental appreciation because it demonstrates an electrophilic agent, generating an intrinsic conformational change without the formation of an intermediate bonding necessary for the ultimately tetrahedral configuration.
2. Results and Discussion
2.1. Combined Analysis of the Molecular Transformation for the Pressure-Induced B(OH)3 into (HO)3B-O-Lattice Conversion in the Crystal Material by ab Initio and Van’t Hoff Modelling
The intramolecular transformation of planar B(OH)3 (D3h symmetry) into a trigonal pyramid (C3v symmetry) following the vtH modelling under formation of the tetrahedral configuration as illustrated in Figure 2.
The corresponding geometric values for the various bond distances (in Å) and bond angles (in degrees) are given in Table 2. The site selected for the tetrahedral configuration of boron may be considered as an oxygen site. An interesting aspect of this specific isomerization by an intrinsic conformational change is that in the trigonal pyramid, boron chirality can be introduced through differentiation in the three B-O groups. Such an intermediate structure will be strongly dependent on the local orientation of surrounding sites. In this way a more precise information is obtained of the molecular bonding state on the principal reaction coordinate. The question remains whether this approach also gives a more definite answer for the mechanism of a “normal” encounter complex as e.g. between the electrophile B(OH)3 and the nucleophile OH−, vide infra.
Combination of the ab initio results and the vtH model results in a linear relationship between the O-B-axis angle (y) and the out-of-plane boron distance (x):
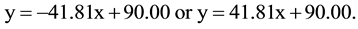
The latter expression corresponds with the O-B-axis angle values in parentheses.
For the (virtual) tetrahedral configuration a B-O distance of 1.397 Å is calculated with the critical value of 0.466 Å for the out-of-plane displacement. Both values correspond with 1.407 Å of the B-OH distance in the cage-like complex anion formed by sodium borate and 1,1,1-tris(hydroxymethyl) ethane: [Na(H2O)3]+[CH3C(CH2O)3B(OH)]− [12] . The other B-OCH2- bond lengths are 1.492 Å. The Na+ ion is octahedrally surrounded by six oxygens with one of the hydroxyl group on boron.
In the report of Edwards et al. [11] it was established that the out-of-plane displacement of the boron atom is consistent with the in situ high-pressure NMR spectra. This mode of deformation for the 11Bδiso value is given in

Figure 2. Out-of-plane displacement of boron in B(OH)3 following the characteristics of the Van’t Hoff (vtH) modelling under subsequent formation of a tetrahedral configuration.
Table 2. Geometry calculations of the vertical displacement of the boron out-of- plane orientation along the C3v symmetry axis starting from B(OH)3 with D3h symmetry. The bond lengths are given in Å, the angles in degrees.
aThese values correspond with the ultimate “out-of-plane” tetrahedral configuration as derived from the vtH model. The other values are derived from Ref. [11] .
Figure 3. In this figure a linear plot is given in combination with a curved line. The latter line has relevance because it demonstrates clearly the asymptotic character based on the maximum value of the out-of-plane orientation. A linear plot has been shown for the relation between11Bδiso and the variation in bond length of one of the B-O bonds in the planar B(OH)3. This representation conflicts with the absence of significant changes in the quadrupole parameters. The out-of-plane mode is in correspondence with the latter notification, demonstrating the effect of hybridization change in the dynamics with the corresponding symmetry change. The unique character of this intramolecular change will be demonstrated with relatively old descriptions as an illustration for the transition state in bimolecular reactions.
From the foregoing it is evident that simple addition reactions as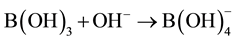 , the planar B(OH)3 changes its planar geometry into a tetrahedral configuration for collapse with the hydroxyl anion. Here it is experimentally demonstrated that the hypothetic model of Bell-Evans-Polanyi (BEP) is a fundamental principle in the construction of the progress of addition-substitution reactions [13] . The BEP model is shown in Figure 4 in a primitive way. Generally known, the crossing points correspond with the energy of the transient complexes on the reaction coordinate showing a shift from an early to a late transition state for that specific conversion.
, the planar B(OH)3 changes its planar geometry into a tetrahedral configuration for collapse with the hydroxyl anion. Here it is experimentally demonstrated that the hypothetic model of Bell-Evans-Polanyi (BEP) is a fundamental principle in the construction of the progress of addition-substitution reactions [13] . The BEP model is shown in Figure 4 in a primitive way. Generally known, the crossing points correspond with the energy of the transient complexes on the reaction coordinate showing a shift from an early to a late transition state for that specific conversion.
2.2. Ab Initio and Van’t Hoff Modelling for a Planar Trivalent Carbon Cation into a Trigonal Pyramidal Configuration. Experimental Evidence with a Diamond-Like Model as the in Situ 1-Adamantyl Cation
Recently, Fitzgibbons et al. published a paper on a high-pressure solid-state reaction of benzene [14] . There is strong experimental evidence based on various spectroscopic measurements that close-packed bundles of subnanometre-diameter sp3-bonded carbon threads (1.52 Å) are formed. The change in hybridization from a planar carbon into a tetrahedral configuration looks similar to the foregoing displacement. However, this conversion is radical-like that promotes an intermediate trigonal pyramidal conformational change.

Figure 3. Relation between 11B isotropic chemical shift in ppm versus the out-of-plane displacement of boron in B(OH)3 in Å (red: linear relation, blue: curved line following the data points). The 0.466 Å value is the maximum for the out-of- plane, indicated by the vertical line.
 Reaction coordinate
Reaction coordinate
Figure 4. The Bell-Evans-Polanyi energy profile for the reaction dynamics of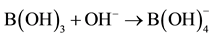 .
.
With the vtH model the relationship between the C-C-axis angle (y) and the out-of-plane carbon distance (x) is given by:
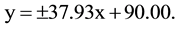
The ± sign corresponds with (0.513 Å, 109.47˚) and (0.513 Å, 70.53˚), respectively, for a C-C distance of 1.540 Å. Both (x, y) functions can be given a more general character via y = mx + 90:
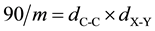
with dC-C = 1.540 Å and dX-Y is the bond distance in the tetrahedral configuration as can be seen by substitution of the corresponding m values.
The 1-adamantyl cation related to the diamond-like structure adamantane may be considered as an excellent cation for demonstrating a trigonal pyramidal configuration via in situ preparation. This aspect has been obtained attention by Jung et al. in their synthesis of highly substituted adamantanones from bicyclo [3.3.1] nonanes [15] . The in situ prepared 1-adamantyl cation without substituents necessary for the corresponding substituted one is given in Figure 5.
In the relaxed form this cation has a near planar geometry for the C1 as has been established by Harding et al. on the basis of 13C NMR data combined with sophisticated calculations [16] . Geometry calculations have been performed by Rasul et al. for this cation [17] . The bond distance of the planar C1 cation with the surrounding carbons has a value of 1.458 Å. This stringent reduction of the normal C-C distance (1.54 Å) of the in situ geometry promotes the increase of the three vertical bond lengths of 1.629 Å. The impact of the introduced stress shows some correspondence with the work of Schreiner et al. [18] . That study was directed on steric effects. They mentioned the importance of Van der Waals forces, including London dispersion forces, for the understanding of the stabilizing sterical interactions. X-ray evidence was obtained for the coupling products as e.g. diamantine-triamantane with a C-C distance of 1.704 Å. It seems realistic to assume that both aspects can be explained by (small) changes in the hybridization resulting in (small) angle variation concerning the crucial carbon locations [19] .
The in situ preparation of a corresponding adamantyl cation was performed by the reaction of trifluoromethanesulphonic acid and 1,5-dimethyl-3,7-dimethylenebicyclo [3.3.1] nonan-9-one. The cation formed is directly trapped by nucleophiles. The main point of the reaction may be considered as a cyclic three-center cyclopropenyl cation with a geometry corresponding with the intermediate transition structure of the unsymmetrically bridged 2-norpinyl cation. For details of the reaction see Ref. [6] .
2.3. Van’t Hoff Modelling of the C3v Trigonal Pyramidal Configurations as NH3 for the Group 15 Elements in Their Nucleophilic Abilities
The conceptualization as given in the aforementioned sections afford various possibilities to describe the nucleophilic abilities of the [N, P, As, ∙∙∙, Bi]H3 molecules of the group 15 elements. Going from nitrogen to the lower placed elements, the H-X-H angle (X = N, P, As, ∙∙∙, Bi) decreases from 107.8˚ to 90.48˚ for BiH3. This means that the out-of-plane location of X with regard to the triangle formed by the three hydrogens increases drastically from nitrogen towards phosphorus from 0.36d to 0.54d (0.56d, 0.56d and 0.57d) in which d is the XH bond length. Focused on nitrogen and phosphorus the difference between the C3v and the Td symmetry after protonation, the change in the out-of-plane location differs substantially for
 from 0.767 Å to 0.473 Å compared with the transition for NH3 to
from 0.767 Å to 0.473 Å compared with the transition for NH3 to
 from 0.366 Å to 0.368 Å1. This approach based on the vtH modelling gives an unique visualization of the drop in gas-phase proton affinity from NH3 to PH3. The influence of methyl substitution is significant. The exclusive difference in proton affinity between the methylated nitrogen and phosphorus compounds decreases under simultaneous increase of the proton affinity of the individual compounds. These changes in proton affinity correspond with the bond angles. Usually these effects are explained by hyperconjugation of the methyl group. From the foregoing model study it seems acceptable to assume that sterical effects play an important role. The role of these simple compounds is of importance specifically as an essential part of biological systems [20] [21] .
from 0.366 Å to 0.368 Å1. This approach based on the vtH modelling gives an unique visualization of the drop in gas-phase proton affinity from NH3 to PH3. The influence of methyl substitution is significant. The exclusive difference in proton affinity between the methylated nitrogen and phosphorus compounds decreases under simultaneous increase of the proton affinity of the individual compounds. These changes in proton affinity correspond with the bond angles. Usually these effects are explained by hyperconjugation of the methyl group. From the foregoing model study it seems acceptable to assume that sterical effects play an important role. The role of these simple compounds is of importance specifically as an essential part of biological systems [20] [21] .

Figure 5. In situ geometric configuration of 1-adamantyl cation (left) and the corresponding relaxed cation (right).
3. Conclusions
It has been established by Edwards et al., that in situ high-pressure 11B solid-state NMR spectroscopy for a borosilicate glass in combination with ab initio calculations provides insight in the molecular geometry for the pressure-induced conversion. The results indicate a deformation of the B(OH)3 planar triangle, under isotropic stress, into a trigonal pyramid that serves as a precursor for the formation of a tetrahedral boron configuration. The out-of-plane transition of boron with a D3h into C3v geometric change is an interesting transformation because it matches with the Van’t Hoff modelling originally based on the molecular description of the tetrahedral change of carbon by nucleophilic substitution reactions as CH3X (X is a leaving group) with Cl− via a trigonal bipyramid in which the transferred carbon is present as a methyl planar triangle “cation” linked to the two partially negatively charged apical positions Cl and X. Although the ultimately tetrahedral configuration for boron is a hypothetical case under the high-pressure conditions, this virtual state can be specified with ab initio calculations in combination with molecular modelling based on Van’t Hoff’s tetrahedron. It is our working model to give a more general support for this type of simple addition reactions which are of fundamental significance. Van’t Hoff modelling and ab initio calculations are also applied on the dynamics between a planar trivalent carbon cation and its trigonal pyramidal configuration. Experimental evidence is obtained with a diamond-like model as the in situ 1-adamantyl cation. Finally the Van’t Hoff modelling is also used for the C3v trigonal pyramidal configurations as NH3 for the group 15 elements in their nucleophilic abilities.
The combinations of sophisticated calculations and on the other hand modelling studies based on the Van’t Hoff tetrahedron model clearly demonstrate the fundamental significance for the understanding of the organization of bimolecular interactions via a specific intermediate state by extrapolation of the experimental results of Edwards et al. Reactions of this type are focused on symmetry changes as D3h ® C3v ® Td.
Acknowledgements
I thank my grandchildren Robin van Dorrestein and Martin Buck BSc for their valuable technical assistance.
References
- Buck, H.M. (2008) A Combined Experimental, Theoretical, and Van’t Hoff Model Study for Identity Methyl, Proton, Hydrogen Atom, and Hydride Exchange Reactions. Correlation with Three-Center Four-, Three-, and Two-Electron Systems. International Journal of Quantum Chemistry, 108, 1601-1614. http://dx.doi.org/10.1002/qua.21683
- Buck, H.M. (2010) A Linear Three-Center Four Electron Bonding Identity Nucleophilic Substitution at Carbon, Boron, and Phosphorus. A Theoretical Study in Combination with Van’t Hoff Modeling. International Journal of Quantum Chemistry, 110, 1412-1424. http://dx.doi.org/10.1002/qua.22252
- Buck, H.M. (2011) A Model Investigation of ab Initio Geometries for Identity and Nonidentity Substitution with Three-Center Four- and Three-Electron Transition States. International Journal of Quantum Chemistry, 111, 2242- 2250. http://dx.doi.org/10.1002/qua.22529
- Buck, H.M. (2012) Mechanistic Models for the Intramolecular Hydroxycarbene-Formaldehyde Conversion and Their Intermolecular Interactions: Theory and Chemistry of Radicals, Mono-, and Dications of Hydroxycarbene and Related Configurations. International Journal of Quantum Chemistry, 112, 3711-3719. http://dx.doi.org/10.1002/qua.24127
- Buck, H.M. (2013) An Adjusted Model for Simple 1,2-Dyotropic Reactions. Ab Initio MO and VB Considerations. Open Journal of Physical Chemistry, 3, 119-125. http://dx.doi.org/10.4236/ojpc.2013.33015
- Buck, H.M. (2014) Three-Center Configuration with Four, Three, and Two Electrons for Carbon, Hydrogen, and Halogen Exchange. A Model and Theoretical Study with Experimental Evidence. Open Journal of Physical Chemistry, 4, 33-43. http://dx.doi.org/10.4236/ojpc.2014.42006
- Yamashita, M., Yamamoto, Y., Akiba, K., Hashizume, D., Iwasaki, F., Takagi, N. and Nagase, S. (2005) Syntheses and Structures of Hypervalent Pentacoordinate Carbon and Boron Compounds Bearing an Anthracene Skeleton. Elucidation of Hypervalent Interaction Based on X-Ray Analysis and DFT Calculation. Journal of the American Chemical Society, 127, 4354-4371. http://dx.doi.org/10.1021/ja0438011
- Bento, A.P. and Bickelhaupt, F.M. (2008) Nucleophilicity and Leaving Group Ability in Frontside and Backside SN2 Reactions. Journal of Organic Chemistry, 73, 7290-7299. http://dx.doi.org/10.1021/jo801215z
- Bento, A.P. and Bickelhaupt, F.M. (2008) Frontside versus Backside SN2 Substitution of Group 14 Atoms. Origin of Reaction Barriers and Reasons for Their Absence. Chemistry―An Asian Journal, 3, 1783-1792. http://dx.doi.org/10.1002/asia.200800065
- Glukhovtsev, M.N., Pross, A. and Radom, L. (1995) Gas-Phase Identity SN2 Reactions of Halide Anions with Methyl Halides: A High-Level Computational Study. Journal of the American Chemical Society, 117, 2024-2032. http://dx.doi.org/10.1021/ja00112a016
- Edwards, T., Endo, T., Walton, J.H. and Sen, S. (2014) Observation of the Transition State for Pressure-Induced BO3 → BO4 Conversion in Glass. Science, 345, 1027-1029. http://dx.doi.org/10.1126/science.1256224
- Taylor, M.J., Grigg, J.A. and Rickard, C.E.F. (1992) The Structure of the Cage-Like Complex Anion Formed by Sodium Borate and 1,1,1-Tris(Hydroxymethyl)Ethane. Polyhedron, 11, 889-892. http://dx.doi.org/10.1016/S0277-5387(00)83337-9
- Dewar, M.J.S. (1969) The Molecular Orbital Theory of Organic Chemistry. Chapter 8, McGraw Hill Book Company, New York.
- Fitzgibbons, T.C., Guthrie, M., Xu, E.S., Crespi, V.H., Davidowski, S.K., Cody, G.D., Alem, N. and Badding, J.V. (2014) Benzene-Derived Carbon Nanothreads. Nature Materials, 14, 43-47. http://dx.doi.org/10.1038/nmat4088
- Jung, M.E. and Lee, G.S. (2014) Synthesis of Highly Substituted Adamantanones from Bicyclo[3.3.1]Nonanes. Journal of Organic Chemistry, 79, 10547-10552. http://dx.doi.org/10.1021/jo501368d
- Harding, M.E., Gauss, J. and von Ragué Schleyer, P. (2011) Why Benchmark-Quality Computations Are Needed to Reproduce 1-Adamantyl Cation NMR Chemical Shifts Accurately. Journal of Physical Chemistry A, 115, 2340-2344. http://dx.doi.org/10.1021/jp1103356
- Rasul, G., Olah, G.A. and Prakash, G.K.S. (2010) Density Functional Theory Study of Adamantanediyl Dications
![]() and Protio-Adamantyl Dications
and Protio-Adamantyl Dications![]() and Protio-Adamantyl Dications
and Protio-Adamantyl Dications![]() . Proceedings of the National Academy of Sciences of the United States of America, 101, 10868-10871.http://dx.doi.org/10.1073/pnas.0404137101
. Proceedings of the National Academy of Sciences of the United States of America, 101, 10868-10871.http://dx.doi.org/10.1073/pnas.0404137101 - Schreiner, P.R., Chernish, L.V., Gunchenko, P.A., Tikhonchuk, E.Y., Hausmann, H., Serafin, M., Schlecht, S., Dahl, J.E.P., Carlson, R.M.K. and Fokin, A.A. (2011) Overcoming Lability of Extremely Long Alkane Carbon-Carbon Bonds through Dispersion Forces. Nature, 477, 308-311. http://dx.doi.org/10.1038/nature10367
- Buck, H.M. (2000) Symmetry Restrictions as Starting Point for the Determination of Geometric Representations and the Dynamics of Cyclic π Systems. International Journal of Quantum Chemistry, 77, 641-650. http://dx.doi.org/10.1002/(SICI)1097-461X(2000)77:3<641::AID-QUA5>3.0.CO;2-R
- Buck, H.M. (2011) DNA Systems for B-Z Transition and Their Significance as Epigenetic Model: The Fundamental Role of the Methyl Group. Nucleosides, Nucleotides and Nucleic Acids, 30, 918-944. http://dx.doi.org/10.1080/15257770.2011.620580
- Buck, H.M. (2013) A Conformational B-Z DNA Study Monitored with Phosphatemethylated DNA as a Model for Epi- Genetic Dynamics Focused on 5-(Hydroxy)Methylcytosine. Journal of Biophysical Chemistry, 4, 37-46. http://dx.doi.org/10.4236/jbpc.2013.42005
 and Protio-Adamantyl Dications
and Protio-Adamantyl Dications . Proceedings of the National Academy of Sciences of the United States of America, 101, 10868-10871.
. Proceedings of the National Academy of Sciences of the United States of America, 101, 10868-10871.NOTES

1Bond angels and bond distances are taken from textbooks and literature.