Open Journal of Physical Chemistry
Vol.4 No.3(2014), Article ID:48242,17 pages
DOI:10.4236/ojpc.2014.43012
A Stereochemically-Bent β-Hairpin: Scrutiny of Folding by Comparing a Heteropolypeptide and Cognate Oligoalanine
Kinshuk Raj Srivastava1,2*, Susheel Durani2
1Department of Physics and Astronomy, Michigan State University, East Lansing, USA
2Department of Chemistry, Indian Institute of Technology Bombay, Mumbai, India
Email: *kinshukraj2@gmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/
![]()
![]()

Received 23 May 2014; revised 20 June 2014; accepted 17 July 2014
ABSTRACT
A poly-L β-hairpin bent stereochemically as a boat-shaped protein of mixed-L,D structure is scrutinized in basis of ordering as minimum of energy specific for its sequenceand solvent. The model suitable for the scrutiny is accomplished by design. A terminally-blocked oligoalanine is nucleated over DPro6-Gly7 and DPro6-LAsp7 dipeptide structures as a twelve-residue β-hairpin and bent stereochemically as a boat-shaped fold. The structure is inverse designed with side chains suitable to bind substrate p-nitophenyl phosphate, a surrogate substrate of acetyl choline and CO2. The designed sequences were proven by spectroscopy and molecular dynamics to order with solvent effects of water and display high binding affinity for the substrate. One of the proteins and a cognate oligoalanine are evolved with molecular dynamics to equilibrium in a solvent bath of water. Molecular dynamics studies establish that heteropolypeptide well ordered as β-hairpin fold and cognate oligoalanine as an ensemble of hairpin-like folds in water. The ordering of cognate oligoalanine as ensembles of hairpin-like folds manifests combined role of water as strong dielectric and weak dipolar solvent of peptides. The roles of stereochemistry and chemical details of sequence in defining polypeptides as energy minima under specific effect of solvent are illuminated and have been discussed.
Keywords:Protein Folding, Protein Stereochemistry, β-Hairpin, Polyalanine Model, Solvent Effects

1. Introduction
Protein folding presents in determination of the basis of specificity for sequences a formidable challenge [1] -[3] . The challenge is in the size of protein molecule and of the system that orders the structure as minima of energy. The challenge is in characterizing the minima in its defining interactions over main chain, side chains, and solvent. Quantum mechanics is relevant but, being computationally expensive, will only apply to greatly simplified models and typically under vacuum [4] -[14] . Force fields of structures and their interactions developed empirically [15] -[17] will allow equilibria to be computed, provided the models are small enough structures to allow the computation [18] -[20] . Critical in this study are the models that will allow both observing and computing equilibria [21] -[23] . Critical can be the questions posed and the models examined.
Critical in protein folding is the basis for sequence and solvent roles. The basis has been addressed with oligoalanines as main chain models [24] -[31] . The models are scrutinized for effects of structure modifications like extending main chains and mutating side chains [32] [33] . A promising scrutiny, implied in the studies Flory reported in 1967 [34] -[36] , was pursued in our lab [37] [38] . The models were modified stereochemically and the effect was scrutinized with specific reporter solvents [37] [39] . It has thus been established that poly-L structure will fold with energetic frustration, hydrogen bonds local in poly-L chain enforce intervening peptides to unfavorable electrostatics of a conformation. Specificity of folding thus is a critical function of screening effects in electrostatics of a conformation. Consequently, folding of protein main chain was proven to manifest two complementary solvent effects, dielectric, to determine if local chain segments fold to a conformation or unfold to β conformation, and dipolar solvation, to determine if peptides hydrogen bond or solvate and thus fold or unfold the chain [39] .
Extending the approach, oligoalanines are now targeted for addressing sequence role. The models are scrutinized for effect of mutation as specific heteropolypeptides in water. Equilibria are computed and the folds are scrutinized, over the microstates populated, in the basis of ordering as sequence specific folds. Crucial for the study will be the ability to compute equilibria, which will be easy for “folded” heteropolypeptide but tough for “unfolded” oligoalanine. Thus we implement the studies with the models that are in conformational diversity as oligoalanines restricted with effects of chain length and D residues. β-hairpins are smallest protein models amenable to further minimization and articulation with stereochemistry [40] . With this approach, a twelve-residue model is examined as a stereochemically bent β-hairpin protein in this report. The heteropolypeptides proven to order and display good affinity for the substrate and thus act as receptor though exceptionally simple and small in size. Evolving heteropolypeptides and cognate oligoalanines to equilibrium, the ensembles are resolved to contributing microstates, which are investigated in interactions of main chain and side chains. Contribution of main chain stereochemistry, side chain interactions and peptide-solvent interactions responsible for ordering specific conformation of peptide have been discussed.
2. Results
2.1. Design
The design of the protein examined in this study is illustrated in Figure 1. Terminally blocked oligoalanine is nucleated over DPro6-Gly7 and DPro6-LAsp7 dipeptide structures as Type II’ β-hairpin. The sequence positions 3 and 10 were mutated stereochemically and conformationally as D residues; this furnishes the desired boatshaped fold. Ala residues are mutated to possible other protein residues, except Pro and Gly. Thus minimum energy sequences specific for the desired fold are generated computationally. Sequences were optimized with β-sheet favoring residues compatible with targeted fold. Polar residues were planned over solvent-accessible base of the fold and neutral-aromatic residues were planned in solvent-sequestered molecular cleft. The two sequences H1 and H2 are differ in having Gly7 or LAsp7 as the second corner residue in their β-turns. Hydrogen bonds, ion-pairs, hydrophobic contacts, p-p, and cation-p interactions, recruited for possibility of locking the heteropolypeptide as a protein, are listed in Table1 Neutral-aromatic and cation-aromatic residues provided in molecular pocket of the fold are for possible interaction with pNPP (p-nitophenyl phosphate) as the binding ligand based on p-p, cation-p, and ion-pair interactions. H1 and H2 were synthesized and evaluated for folding, and ligand binding function. A1 is oligoalanine analogous to H1. In addressing DPro6-LAsp7 dipeptide for possible role in folding of H1 and A1, the structure is mutated to LPro6-LAla7 in the oligoalanine analog A2.
A1:Ac-Ala1-Ala2-DAla3-Ala4-Ala5-DPro6-Asp7-Ala8-Ala9-DAla10-Ala11-Ala12-NH2
H1:Ac-Arg1-Glu2-DAla3-Ser4-Tyr5-DPro6-Asp7-His8-Asn9-DThr10-Lys11-Trp12-NH2
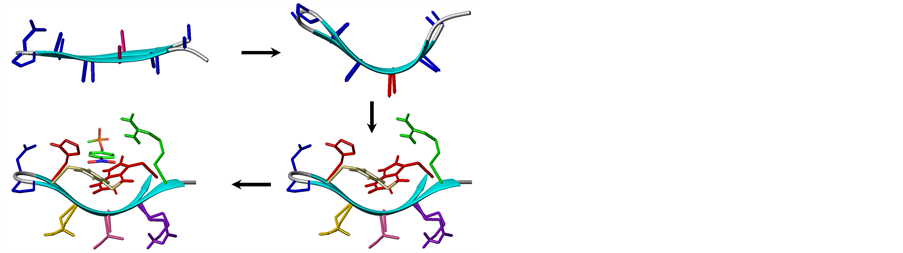
Figure 1. Stepwise design involving stereochemical mutation of β-hairpin as boat-shaped fold and inverse optimization of H1 as receptor for p-nitrophenyl phosphate.
H2:Ac-Arg1-Glu2-DAla3-Ser4-Tyr5-DPro6-Gly7-His8-Asn9-DThr10-Lys11-Trp12-NH2
A2:Ac-Ala1-Ala2-DAla3-Ala4-Ala5-LPro6-Ala7-Ala8-Ala9-DAla10-Ala11-Ala12-NH2

Table 1. Specific interactions folding H1 as a protein.
2.2. Synthesis and Experimental Studies
H1 and H2 were synthesized by solid-phase peptide synthesis. Requisite ion peaks appearing in MALDI mass spectra, as noted in Figure S1 (Supplementary Material), characterize the structures. The peptides display 1H NMR resonances broadly in accordance with their structures as is noted in Figure S2 (Supplementary Material). In concentration regime of the NMR experiment (0.25 - 2.5 mM), peptides were soluble and manifested no noteworthy changes in chemical shifts or line widths on tenfold dilution (results not shown). The structures thus appear to be freely soluble contrasted with poly-L hairpins, which tend to aggregate. The stereochemical modification of hairpin by bending the structure appears to have suppressed tendency of natural hairpin to aggregate to a diminished aqueous solubility.
CD spectra recorded for peptide H1 Figure 2. A maximum of ellipticity at ~198 nm and a coupled minimum at ~208 nm suggests ordering of the peptides as β-hairpins. Another minimum of ellipticity at ~215 coupled with a maximum at ~228 nm appears and could be coupled exciton due to involvement of aromatic residues in p-p interaction. As is noted in Figure 1, the boat-shaped fold has aromatic side chains clustered in its molecular cleft, which may promote aromatic-aromatic interactions within or between strands of the bent hairpin. The bend in artificial hairpin has provided for the interactions that are atypical for canonical poly-L β-hairpin. Ordering of the structures was tested in a thermal unfolding experiment, which was monitored with CD. Ellipticity at 228 nm diminishes with increase of temperature and recovers fully on cooling, as is noted in the inset of Figure 2. This suggest that Peptide H1 may fold and unfold like proteins.
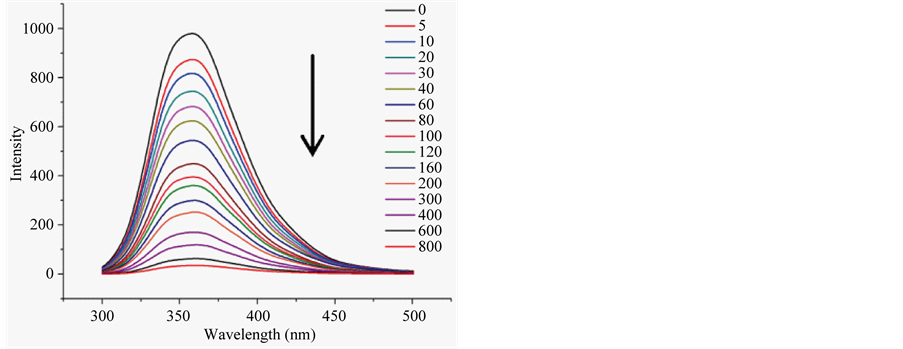
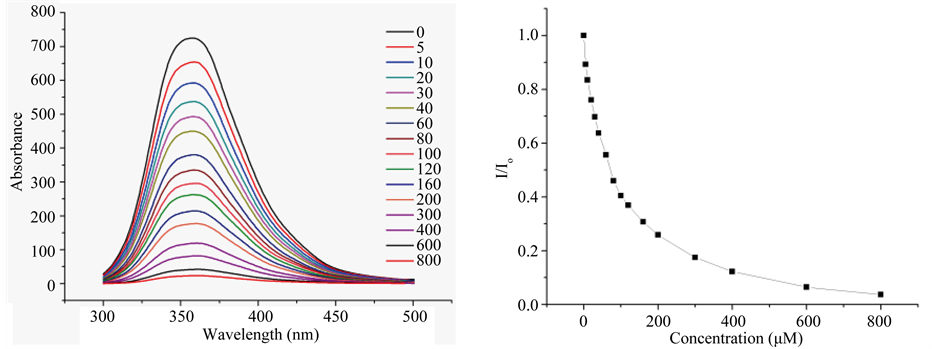
H1 and H2 were tested for binding with p-nitrophenyl phosphate (pNPP). On titration with pNPP, both peptides manifest strong quenching of fluorescence as is noted in Figure 3 and Figure S3 of Supplementary material. Analysis of the data with Stern-Volmer equation gave the binding constants given in Table2 Peptides were evaluated for ligand binding with AutoDock. The evaluation was undertaken with central member of the largest cluster in H1 populating equilibrium modeled with MD, as we shall discuss. The result of the calculation is in Table2 The calculated binding energy is in reasonable agreement with the experimentally determined value. Aromatic side chains in molecular cleft provide a specific site for ligand binding conforming to quenching of fluorescence on ligand binding. On combined evidence of reversible folding, and ligand binding, H1 and
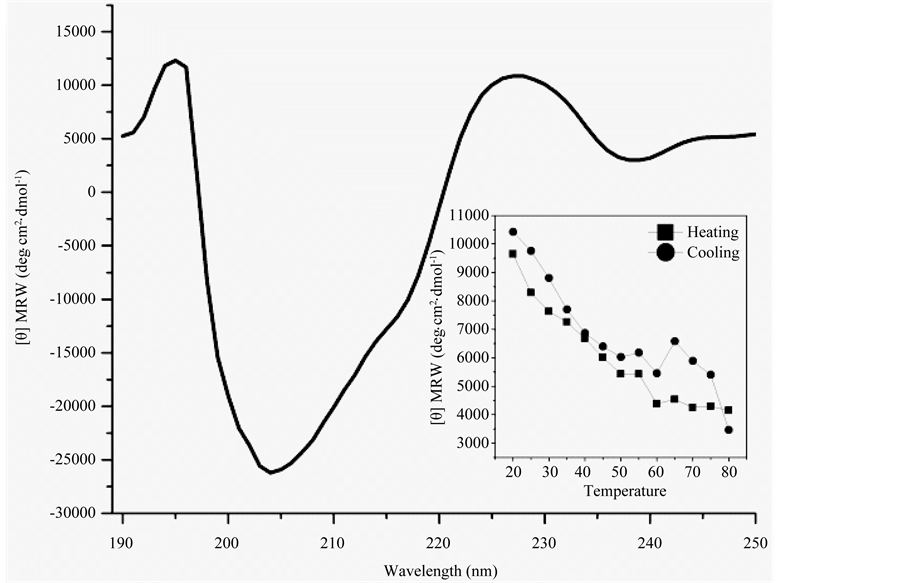
Figure 2. CD spectra of heteropolypeptides H1 in water at 25˚C. CD thermal melting curves of heteropolypeptides H1 recorded at 228 nm presented as inset.
 (a)
(a)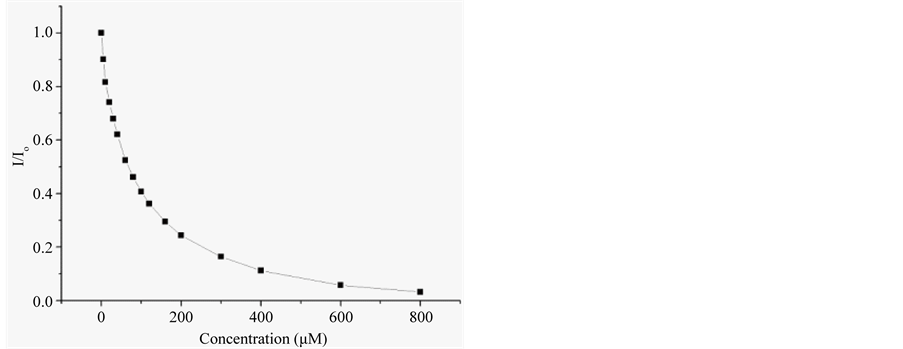 (b)
(b)
Figure 3. Quenching of tryptophan fluorescence of peptide H1 (20 μM) in 20 mM Tris-HCl buffer at pH 7.5, on progressive titration with increasing titration with pNPP (panel (a)), and plot of relative fluorescence intensity as a function of pNPP concentration (panel (b)).
H2 are justified as protein models.
2.3. Modeling of Folding
H1, A1, and A2 were submitted to molecular dynamics in a solvent bath of water to sample their conformational ensemble. Gromos 96 force field modified for accommodation of D residues is implemented at 298 K using SPC-water model as the solvent. Trajectories were monitored in conformational phase space of polypeptide structure, which was mapped to discrete microstates using 0.15 nm RMSD cut-off over backbone atoms to dis
Table 2. Observed binding energy of peptide varients for pNPP.
tinguish conformationally specific clusters. The clustering was implemented with Duara et al. algorithm [41] ; the central member of each cluster was taken to model a specific microstate, viz., as a discrete fold populating equilibrium. Time evolution of population in microstates during MD is compared in Figure 4. H1 equilibrates early and saturates as an ensemble of 6 microstates. A1 and A2 evolve slowly and do not manifest a robust asymptote in evolution of microstates even after 250 ns of MD. However, assuming equilibria to be reasonably approximated, the simulations were concluded at this point in time. Compared to 6 microstates in H1, A1 and A2 are noted to populate in, respectively, 523 and 983 microstates (see Table 3). Clearly, sequence length is critical for the ability to compute equilibria. Side chains and β-turn fold H1. β-turn in DPro6-LAsp7 structure also restricts conformational diversity in A1; mutated to LPro6-LAla7 structure, A2 is nearly two-fold greater in density of microstates than A1. According to mole fraction in minima of energy, 0.96 in H1, free energy change in ordering of the structure in its ensemble is −7.8 kJ∙M−1. Likewise, 0.13 and 0.21 in mole fraction, A1 and A2 have their energy minima ordered with free energy change of, respectively, 4.7 kJ∙M−1 and 3.3 kJ∙M−1. Relative to A1, the energy minima of H1 manifest net free energy change of −12.5 kJ∙M−1; this reflects sequence contribution in ordering of H1.
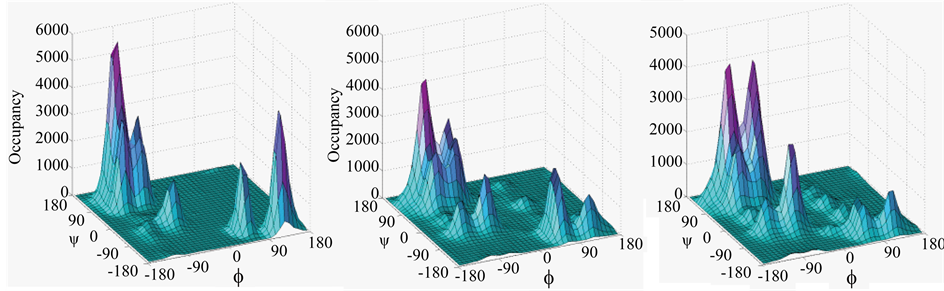
H1, A1, and A2 were assessed in distribution of Rg over the conformers populating macrostates, in percentage occupancy of specific f, y basins, and in percentage occurrence of specific main chain hydrogen bonds, short (SR), medium (MR), and long ranged (LR). The data are summarized in Table 4 and Figure S4 of Supplementary Material. Appearnece of Rg maxima of entire macrostate at 0.54 nm for H1 and at 0.52 for A1; and maxima of first microstate at 0.55 nm for H1 and at 0.52 nm for A1, suggest more compaction of A1 than H1. Macrostate in H1 is ~92 % in β + PPII basin and ~6% in a basin, while in A1 it changes to ~80% occupancy in β + PPII basin and ~10% in a basin, as is illustrated in Figures S5 of Supplementary Material. According to residue-level basin occupancies (Figure S6 of Supplementary Material), Pro6 and Asp7 are locked in Type II’ β-turn and only marginally dispersed in A1. LPro6 and LAla7 in A2 are considerably dispersed and accordingly, stereochemical effect of D-proline and interactions of Asp side chain contribute in ordering both A1 and H1.
According to the data in Table 4, H1 is locked with 8.7 hydrogen bonds per molecule of which 5.0 are with in backbone, 2.8 are with backbone-sidechain (mc-sc), and 0.9 within different side chains. A1 lacks hydrogen bonding groups in Ala side chains and has mc-mc hydrogen bonds diminished to 3.4 from 5.0 in H1. In H1, ~61% of mc-mc hydrogen bonds are LR type (n-n ≥ 6), implying an ordered β-sheet structure, while ~32% are MR type (n-n ± 3), conforming to its β-turn structure. In its conformational dispersal, A1 diminishes in LR hydrogen bonds to 55% from ~61% in H1, and increases in MR hydrogen bonds to ~35% from ~32% in H1.
2.4. Interactions Ordering H1
Interactions ordering H1 were assessed by monitoring distribution of Rg over specific side chain pairs. Specific pairs with Rg distribution peaking at ≤0.3 nm, noted in Figure 5, are strongly associated. Specifically, Trp12- Tyr5, His8-Tyr5, and Trp12-His8 are closely interacting, and could be involved in p-p interaction, and Lys11-Glu2 and Ser4-Asn9 are closely interacting, and could be involved in mutual hydrogen bonds. The appearance of peak at 0.25 nm in the Rg distribution of DAla3-DThr10 indicate the presence of hydrogen bond between DThr10 side chain hydroxyl and DAla3 main chain carbonyl. Arg1-Tyr5, a case of potential cation-p interaction, and Asp9-His8, a case of potential hydrogen bond, are bimodal in Rg distribution (Figure 5), which implies that the side chains only interact transiently. Arg1-Trp12 pair, a possible case for p-p and cation-p interaction, has Rg distribution peaking at 0.65 nm; clearly the side chains do not interact.
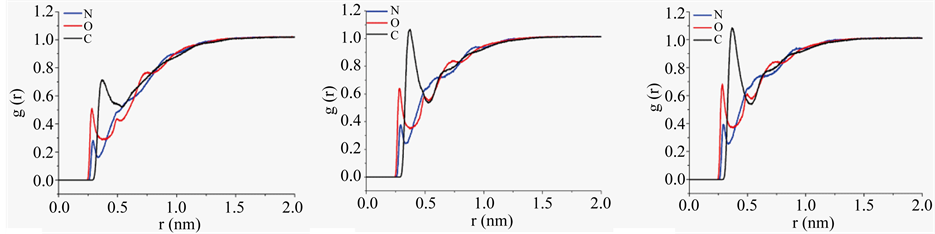
H1 was assessed in its interactions with solvent. The radial distribution of water oxygen against specific backbone and Cβ atoms are presented in Figure 6 and against other specific side chain atoms are presented in Figure S7 of Supplementary material. The overall spatial distribution plots of water oxygen are presented in Figure S8 of Supplementary material. As evidenced in reduction of radial distribution function (RDF) magni-
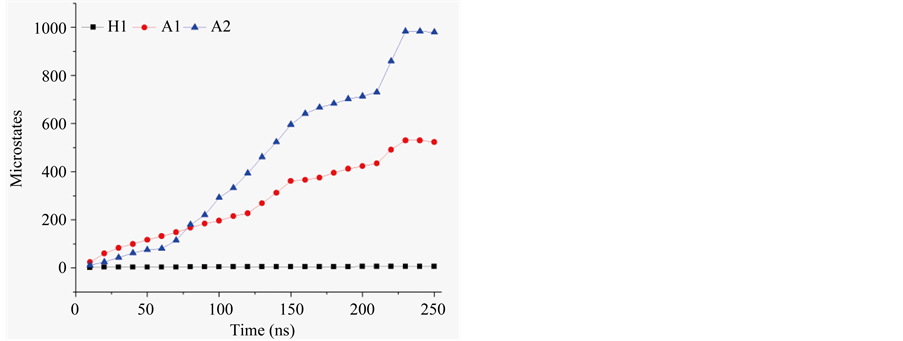
Figure 4. Evolution of microstates during molecular dynamics.
Table 3. Microstate populating equilibria, showing population statistics and radius of gyration distribution.
Table 4. Conformational parameters of microstates.
aHydrogen bonds are short (SR; i → i ± 2), medium (MR; i → i ± 3 + i → i ± 4) and long ranged (LR; i → i ± 5 + i → i ± ³6 according to sequence separation between donor and acceptor residue. bBasin definitions are, α: L/Df = −/+ 20 to −/+ 100, L/Dy = −/+ 20 to −/+ 80; β: L/Df = −/+ 90 to −/+ 170, L/Dy = +/− 80 to +/− 180; PPII: L/Df = −/+ 30 to −/+ 90, L/Dy = +/− 80 to +/− 170. mc-mc indicate main chain-main chain; sc-sc indicate side chain-side chain; mc-sc indicate main chain-side chain hydrogen bonds whereas p-p indicate the total hydrogen bonds observed in the entire peptide.
tudes relative to equilibrium solvent density of 1, H1 and A1 have specific backbone atoms sequestered from solvent possibly in intramolecular hydrogen bonds. The greater density of main chain-main chain hydrogen bonds in H1 than in A1 and A2 support the observation of radial distribution. RDF peaks of water oxygen against oxygen and nitrogen atoms of specific side chains display maxima typically at ~0.3 nm, indicating the strong solvation of polar atoms of side chains. RDF maxima of water oxygen are diminished against β carbons of aromatic and aliphatic side chains, conformed to solvent sequestered nature of non polar side chain groups and their participation in p-p interaction and hydrophobic clustering.
2.5. Specific Microstates Populating the Equilibria
Figure 7 shows ribbon representation of top five microstates in each ensemble. The individual populations, shown in parenthesis, add up to ~99% in H1 and ~40% in A1 and A2. The microstates in H1 and A1 have the general appearance of bent hairpins, which are well locked in H1 and floppy in A1. From f, y plots, shown alongside, H1 and A1 are noted to have DPro and LAsp locked in Type II’ β-turn. Remarkably, many microstates
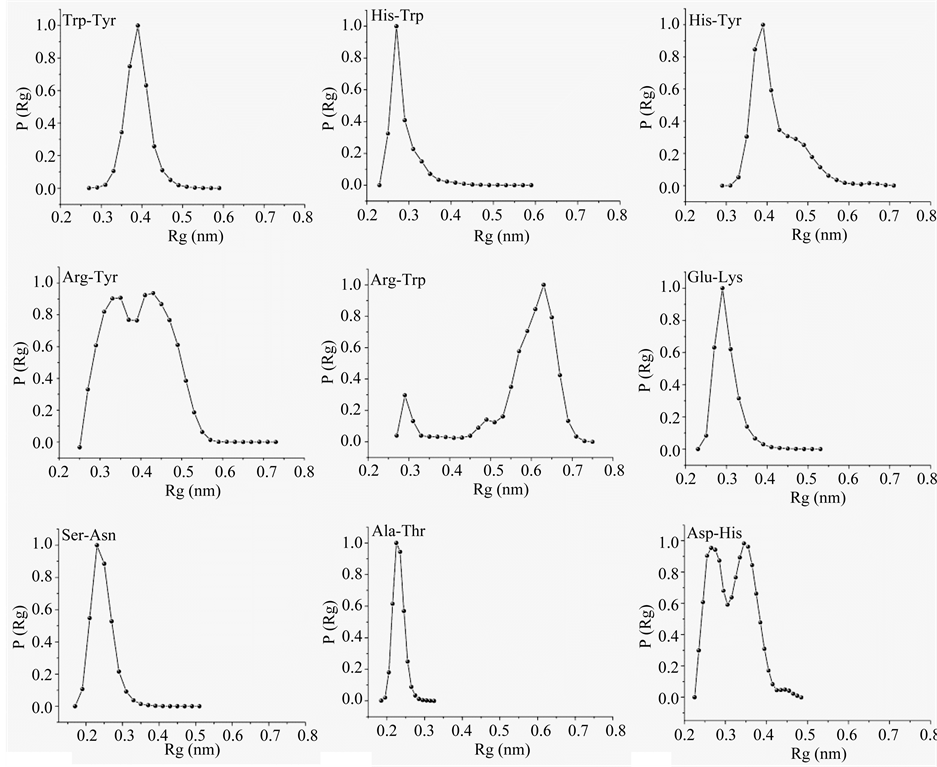
Figure 5. Radius of gyration (Rg) distribution of specific side chain pairs of H1 over its macrostate.
 (a) (b) (c)
(a) (b) (c)
Figure 6. Radial distribution of water oxygen atoms against main-chain nitrogen (blue trace), oxygen (red trace), and side chain Cβ of H1 (panel (a)), A1 (panel (b)) and A2 (panel (c)) over macrostates.
in A2 also appear to be in hairpin-like folds. Presumably LPro induces chain reversal to promote the folds. An effect contributing specific folds may involve L, D, L segments ordering to a curved morphology due to Lβ, Dβ, Lβ conformation, in practically all the microstates, according to f, y plots. Identical in curvature, the strand sections are well poised for mutual antiparallel β-sheet hydrogen bonds in curved hairpins. Indeed, A2 is comparable to A1 in the number of backbone hydrogen bonds at ~3.5 (see Table 4), and is significantly higher in LR hydrogen bonds, at ~70%, implying more extensive β-sheet structure, compared to 60% in H1 and only
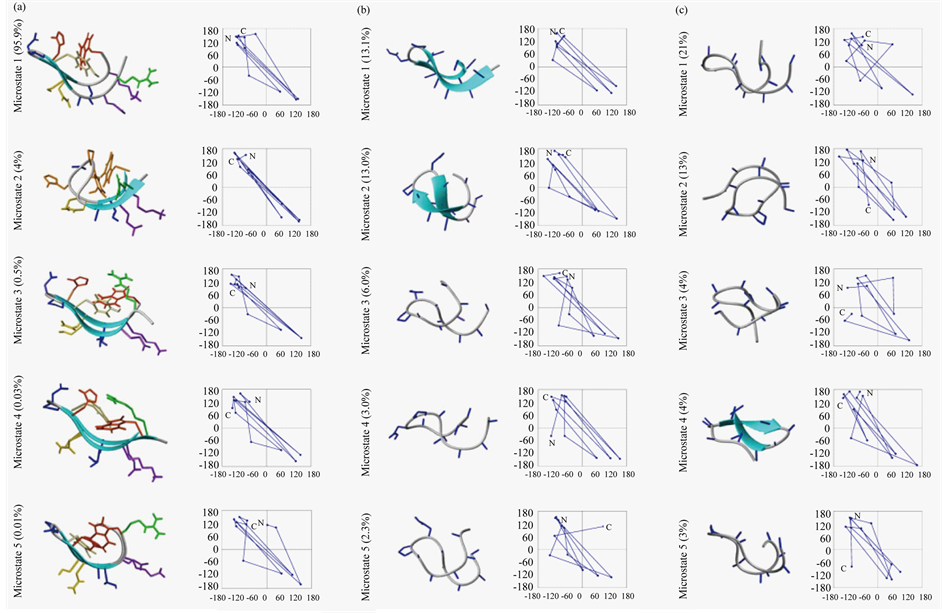
Figure 7. Ribbon representation of central members of top microstates of H1 (panel (a)), A1 (panel (b)) and A2 (panel (c)) showing populations in parenthesis and f, ψ plots underneath.
55% in A1. Thus the identical curved morphology of alternating L, D, L structures may constrain conformational diversity in H1, A1, and A2 due to intramolecular main chain hydrogen bonds between strands facilitated by chain reversal in the models. H1 is well locked in its first microstate with aromatic interactions, interactions of several cross-strand side chains, and in intramolecular main chain hydrogen bonds. Aromatics are clustered in all the microstates of H1; polydispersity of microstates involves considerable fraying of Arg as N-terminal residue. Due to a local twist in main chain, Asp7 is pushed out of a basin uniquely in microstate 2 of the ensemble.
3. Discussion
Proteins fold over a complex interplay of interactions of main chain, side chains, and solvent [1] -[3] . In addressing the interplay, oligoalanines have been fruitful models [4] [5] [23] [26] [27] [30] [32] [39] . A fruitful enquiry of the models has involved mutating stereochemical structure and probing the effect with specific reporter solvents [37] -[39] . Consequently, stereochemistry has been implicated in critical role: poly-L folds order to local hydrogen bonds of main chain under conflict with unfavorable electrostatics of a conformation. Accordingly, solvents fold main chain with two complementary effects, screening of electrostatics, to allow or disallow a conformation, and dipolar solvation of peptide, to allow or disallow hydrogen bonds [39] . Crucial to unmasking of the folding model has been mutation of stereochemistry and examination of solvent effect of water.
The approach has now been extended to scrutiny of sequence role. Model proteins have been mutated as cognate oligoalanines and the effect examined with reporter solvents. Applying water as solvent, side chain and main chain structures were probed in the critical interactions involved. The fold probed with water in this study was designed and validated as receptor protein against targeted ligand. Applied as reporters against homochiral and heterochiral models, water and DMSO illuminated critical effects of main chain [37] [39] . Comparison of heteropolypeptides and cognate oligoalanines in water have illuminated critical effects of sequences.
The critical effect of stereochemistry was exemplified in this study on the effect of D-proline-locked β-turn structure. DPro-LAsp structure restricted conformation in “unfolded” oligoalanine considerably relative to LPro-LAla structure. DPro and Asp side chain at turn position locked the structure as an ensemble of hairpin-like folds. The ordering of ologoalanines as an ensemble of hairpin-like folds manifest the role of set ereochemistry in nucleating the turn and ordering specific β-hairpin. The stereochemical effect of a turn in nucleating structure, and main chain hydrogen bonds were noted to be the important factor for locking the structure as hairpin-like folds. Indeed, ordering of oligoalanines to ~3.5 main chain hydrogen bonds, only marginally less than 5 in H1, suggests that water could be a significant fold promoting solvent directly at level of main chain. This observation manifest a role for solvent water as a weak solvent of peptides and thus as a promoter of folds passively by allowing formation of main chain hydrogen bonds. Thus, the present study establishes the folding of protein in water involves combined effect one, dielectric effect in screening of electrostatics of poly-L peptides, and, two, weak dipolar solvation of peptides [39] .
With effects of side chains, heteropolypeptide structure were found to fold with solvents effects of water. The folding involved ordering of main chain with increased sampling of peptide hydrogen bonds and increased sampling of β conformation. We found the sequence complement of side chains order the first microstate in A1 to the first microstate in H1 with a free energy gain of modest ~12 kJ∙M−1. The magnitude includes entropic cost of ordering ~500 microstates in A1 to only 6 in H1 and the gain in the interactions of specific side chains. Interactions of side chains compensate not only for unfavorable entropy of conformational ordering, but also for unfavorable enthalpy of desolvation of peptides and unfavorable electrostatics of a conformation. Thus heteropolypeptides order with synergy of main chain and side chain effects. Idiosyncrasies of specific folds may be involved but generic effects of main chain and side chain structures are likely to be important. Broadly, similar interactions involving side chains were observed in folding of heteropolypeptides, but the mix of physical effects is expected to be specific for the solvent role as screen of main chain electrostatics and solvation of peptides. Water as a solvent manifested in this study close interactions mainly of His-Trp, Trp-Tyr, Glu-Lys, and Ser-Asn side chains and weaker interaction of several other side chains. In general, hydrogen bonds of polar groups and hydrophobic aggregation of nonpolar groups, could be more critical effects in interaction of the structure with water as solvent.
4. Conclusion
Ordering of proteins as energy minima specific for their sequences was investigated with combination of experiment and computation. A stereochemically bent β-hairpin is designed as receptor protein for scrutiny of the forces ordering proteins. The heteopolypeptide found to order as sequence-specific folds under the influence of position specific interactions over several side chains. The ordering of cognate polyalanine as an ensemble of hairpin-like folds manifests the combined role of water. The water dielectric effect screens the electrostatics of poly-L peptides, and water being weak dipolar solvent passively promoter of the main chain hydrogen bond. The success of achieving high affinity for targeted ligand in exceptionally small peptide illustrates the power of the proposed design principles and affirms stereochemistry as a valuable aid in customizing molecular morphological plans. The proteins, being small in size, facilitated the analysis of its folding and ligand binding with molecular dynamics, which allow scrutinizing the individual roles of backbone and side chain and solvent roles in protein folding.
5. Material and Methods
5.1. Peptide Modeling
Peptides were modeled either with the in-house software package CAPM (Computer Aided Peptide Modeling), capable of handling D-amino acid effectively. In-house program PDB make was used for generation of PDB coordinates of CAPM modeled structure. Sequences were designed with help of in-house sequence optimization program IDeAS, capable of handling D-amino acid effectively.
5.2. Preparation of Equilibrium Ensembles
Molecular dynamics were performed with gromos-96 43A1 force field in GROMACS 3.3.3 [42] [43] in a periodic box of with water as explicit solvent. The simulation was performed under NVT condition [38] . We used 1.4 nm cut off for Non-bonded list with 0.8 nm shift and used 2 fs as integration step. Initial velocities were drawn from Maxwellian distribution. Temperature was coupled to an external bath with relaxation time constant of 0.1 ps. Bond lengths were constrained with SHAKE [44] to geometric accuracy 10−4. Electrostatics were treated with Particle Mesh Ewald [45] [46] for charged system whereas SHIFT was used for neutral peptides. We implemented a 1.4 nm coulomb cutoff, 0.12 nm fourier spacing, and 4 as an interpolation order. Peptides constrained to the center of the periodic cubic box were surrounded by solvent water to 1 atm density at 298 K. We first minimize the energy of solute followed by minimization of solvent energies while restraining solute, and finally both were energy minimized after removing restraint. We started the molecular dynamics simulation and sampled the trajectory at 10 ps intervals. We have discarded initial 3 ns trajectory as pre-equilibration period, before analyzing the data.
5.3. Characterization of Macrostate and Polypeptide Microstates
We used Daura et al. Algorithm for clustering peptide conformers in cartesian space with ≤0.15 nm RMSD cutoff over backbone atoms. This out put different microstates in their diminishing population, viz., diminishing thermodynamic stability. We calculated the free energy of first microstate (most populous) using equation DG = −RT ln K, where K = p1/ptotal − p1, where is R gas constant, and T is temperature, and p1 is the population of the first microstate. We considered the most populated first microstate as ordered state and evaluated its stability with respect to the remaining microstate considered as unordered state.
5.4. Solvation Shell Analysis
We calculated the radial distribution and spatial distribution of specific solvent atoms around peptide for first microstate with g_rdf and g_spatial functions in GROMACS.
5.5. Molecular Docking
For docking the ligand to peptide, we used in build flexible docking algorithm of AutoDock 4.0 [47] . The representative structure of first microstate was obtained by clustering the three aromatic residues over entire trajectory. This structure was used as a receptor structure for docing with ligand. Using genetic algorithm with RMSD tolerance of 2 Å, structurally distinct conformational clusters of the ligand were ranked in terms of increasing energy. The observed the lowest energy of peptide-ligand complex were reported as the binding energy.
5.6. Peptide Synthesis
Pepetides were synthesized by solid phase peptide synthesis using Fmoc chemistry on Rink Amide AM resin with HOBt/DIC as coupling reagents [48] . Each coupling, monitored with Kaiser and chloranil tests were periodically performed to check the coupling of each amino acid. 30% (v/v) piperidine-DMF was used for deprotection in each step of synthesis. Acetylation of N-terminus were achieved with Ac2O:DIPEA:DMF in 1:2:20 ratio. Reagents K (82.5% TFA/5% dry-phenol/5% thioanisole/2.5% ethandithiol/5% water) was used for simultaneous deprotection of side chain and final cleavage of peptide chain from resin. The finally cleaved peptides were precipitated in anhydrous diethyl ether. The precipitated peptides were then lyophilized in 1:4 H2O:tBuOH solution, resultant peptides were stored in freeze. Peptide purity was assessed with HPLC over RP-C18 (10 mM, 10 mm × 250 mm; Merck) eluting with CH3CN\H2O (0.1%TFA) 0% - 100% gradients.
5.7. Mass Spectrometry
Mass spectra were recorded either by MALDI-TOF (Matrix Assisted Laser Desorption Ionization-Time of Flight) mode on AXIMA-CFR Kratos instrument.
5.8. Nuclear Magnetic Resonance Spectroscopy
1H NMR spectra were recorded on 800 MHz Bruker instrument at 298 K in 90% H2O/10% D2O in citrate buffer at pH ~3 with 2.5 mM and 0.25 mM concentrations of peptides. Solvent was suppressed with pre-saturation or WATERGATE sequence, as provided in Bruker softwares.
5.9. Circular Dichroism
Far-UV Circular Dichroism (CD) spectrum were recorded on JASCO J-810 CD instrument at 298 K in 0.2 cm path length quartz cell. Using 2 nm bandwidth and scanning speed 100 nm/min with 1.0 s time constant in 1 nm steps, we record five scans and averaged them. Each spectra was corrdcted for solvent absorbance. We finally report the values in molar residue ellipticity [qMRW] by converting the observed values in millidegrees using well reported equation.
5.10. Spectrofluorometry
Fluorescence spectra were recorded on a Perkin Elmer LS-55 spectrofluorimeter. We collectec the data at 298 K in 1 mL cell by exiting the sample at 280 nm and recording the emission in the wavelength range of 300 - 500 nm range, with 5 nm excitation and emission slits width. A scan rate of 100 mm/min with 1 nm steps were used. We kept the fixed concentrations of peptide 20 mM and varied the substrate (pNPP and pNPA) concentration in the range of 0 - 400 mM. All experiments were performed in 20 mM Tris-HCl buffer at ~7.5 pH. We calculated Stern-Volmer constant (KSV) for the external quencher i.e. pNPP using the following biomolecular quenching equation.
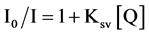 (1)
(1)
where I0 = fluorescence intensity in the absence of external quencher, I = fluorescence intensity in the presence of quencher, Q = concentration of the quencher, and KSV = Stern-Volmer constant calculated from the slope of line. The emission maximum intensities of tryptophan were fit as a function of pNPP concentration to the described 1:1 binding isotherm and Kd and hence binding energy were estimated.
Acknowledgements
We acknowledge DST (09DST028), Government of India, for financial support and IIT Bombay for the computing facility “Corona”. KRS is recipients of fellowships from Council of Scientific and Industrial Research (CSIR).
References
- Dill, K.A., Ozkan, S.B., Shell, M.S. and Weikl, T.R. (2008) The Protein Folding Problem. Annual Review of Biophysics, 37, 289-316. http://dx.doi.org/10.1146/annurev.biophys.37.092707.153558
- Onuchic, J.N. and Wolynes, P.G. (2004) Theory of Protein Folding. Current Opinion in Structural Biology, 14, 70-75. http://dx.doi.org/10.1016/j.sbi.2004.01.009
- Daggett, V. and Fersht, A. (2003) The Present View of the Mechanism of Protein Folding. Nature Reviews Molecular Cell Biology, 4, 497-502. http://dx.doi.org/10.1038/nrm1126
- Dannenberg, J.J. (2005) The Importance of Cooperative Interactions and a Solid-State Paradigm to Proteins: What Peptide Chemists Can Learn from Molecular Crystals. Advances in Protein Chemistry, 72, 227-273. http://dx.doi.org/10.1016/S0065-3233(05)72009-X
- Hua, S., Xu, L., Li, W. and Li, S. (2011) Cooperativity in Long Alphaand 3(10)-Helical Polyalanines: Both Electrostatic and van der Waals Interactions Are Essential. Journal of Physical Chemistry B, 115, 11462-11469. http://dx.doi.org/10.1021/jp203423w
- Elstner, M., Jalkanen, K.J., Knapp-Mohammady, M., Frauenheim, T. and Suhai, S. (2000) DFT Studies on Helix Formation in N-Acetyl-(L-Alanyl)n-N’-Methylamide for n = 1 - 20. Chemical Physics, 256, 15-27. http://dx.doi.org/10.1016/S0301-0104(00)00100-2
- Rossi, M., Blum, V., Kupser, P., von Helden, G., Bierau, F., Pagel, K., Meijer, G. and Scheffler, M. (2010) Secondary Structure of Ac-Alan-LysH+ Polyalanine Peptides (n = 5, 10, 15) in Vacuo: Helical or Not? Journal of Physical Chemistry Letters, 1, 3465-3470. http://dx.doi.org/10.1021/jz101394u
- Salvador, P., Asensio, A. and Dannenberg, J.J. (2007) The Effect of Aqueous Solvation upon Alpha-Helix Formation for Polyalanines. Journal of Physical Chemistry B, 111, 7462-7466. http://dx.doi.org/10.1021/jp071899a
- Topol, I.A., Burt, S.K., Deretey, E., Tang, T.H., Perczel, A., Rashin, A. and Csizmadia, I.G. (2001) Alphaand 3(10)-Helix Interconversion: A Quantum-Chemical Study on Polyalanine Systems in the Gas Phase and in Aqueous Solvent. Journal of the American Chemical Society, 123, 6054-6060. http://dx.doi.org/10.1021/ja0038934
- Tsai, M.I., Xu, Y. and Dannenberg, J.J. (2005) Completely Geometrically Optimized DFT/ONIOM Triple-Helical Collagen-Like Structures Containing the ProProGly, ProProAla, ProProDAla, and ProProDSer Triads. Journal of the American Chemical Society, 127, 14130-14131. http://dx.doi.org/10.1021/ja053768y
- Tsai, M.I., Xu, Y. and Dannenberg, J.J. (2009) Ramachandran Revisited. DFT Energy Surfaces of Diastereomeric Trialanine Peptides in the Gas Phase and Aqueous Solution. Journal of Physical Chemistry B, 113, 309-318. http://dx.doi.org/10.1021/jp8063646
- Tsemekhman, K., Goldschmidt, L., Eisenberg, D. and Baker, D. (2007) Cooperative Hydrogen Bonding in Amyloid Formation. Protein Science, 16, 761-764. http://dx.doi.org/10.1110/ps.062609607
- Han, W.G., Jalkanen, K.J., Elstner, M. and Suhai, S. (1998) Theoratical Study of Aqueous N-Acetyl-L-Alanine N’-Methyl amide: Structure and Raman, VCD, and ROA Spectra. Journal of Physical Chemistry B, 102, 2587-2602. http://dx.doi.org/10.1021/jp972299m
- Wieczorek, R. and Dannenberg, J.J. (2005) Enthalpies of Hydrogen-Bonds in Alpha-Helical Peptides. An ONIOM DFT/AM1 Study. Journal of the American Chemical Society, 127, 14534-14535. http://dx.doi.org/10.1021/ja053839t
- Brooks, B.R., Brooks III, C.L., Mackerell Jr., A.D., Nilsson, L., Petrella, R.J., Roux, B., Won, Y., Archontis, G., Bartels, C., Boresch, S., Caflisch, A., Caves, L., Cui, Q., Dinner, A.R., Feig, M., Fischer, S., Gao, J., Hodoscek, M., Im, W., Kuczera, K., Lazaridis, T., Ma, J., Ovchinnikov, V., Paci, E., Pastor, R.W., Post, C.B., Pu, J.Z., Schaefer, M., Tidor, B., Venable, R.M., Woodcock, H.L., Wu, X., Yang, W., York, D.M. and Karplus, M. (2009) CHARMM: The Bio-molecular Simulation Program. Journal of Computational Chemistry, 30, 1545-1614. http://dx.doi.org/10.1002/jcc.21287
- Guvench, O. and MacKerell Jr., A.D. (2008) Comparison of Protein Force Fields for Molecular Dynamics Simulations. Molecular Modeling of Proteins, 443, 63-88. http://dx.doi.org/10.1007/978-1-59745-177-2_4
- van Gunsteren, W.F., Dolenc, J. and Mark, A.E. (2008) Molecular Simulation as an Aid to Experimentalists. Current Opinion in Structural Biology, 18, 149-153. http://dx.doi.org/10.1016/j.sbi.2007.12.007
- Wang, D., Friedmann, M., Gattin, Z., Jaun, B. and van Gunsteren, W.F. (2010) The Propensity of α-Aminoisobutyric Acid (=2-Methylalanine; Aib) to Induce Helical Secondary Structure in an α-Heptapeptide: A Computational Study. Helvetica Chimica Acta, 93, 1513-1531. http://dx.doi.org/10.1002/hlca.200900420
- Yu, H., Ramseier, M., Burgi, R. and van Gunsteren, W.F. (2004) Comparison of Properties of Aib-Rich Peptides in Crystal and Solution: A Molecular Dynamics Study. ChemPhysChem, 5, 633-641. http://dx.doi.org/10.1002/cphc.200301026
- Zagrovic, B., Lipfert, J., Sorin, E.J., Millett, I.S., van Gunsteren, W.F., Doniach, S. and Pande, V.S. (2005) Unusual Compactness of a Polyproline Type II Structure. Proceedings of the National Academy of Sciences of the United States of America, 102, 11698-11703. http://dx.doi.org/10.1073/pnas.0409693102
- Brooks III, C.L. (2002) Protein and Peptide Folding Explored with Molecular Simulations. Accounts of Chemical Research, 35, 447-454. http://dx.doi.org/10.1021/ar0100172
- Shea, J.E. and Brooks III, C.L. (2001) From Folding Theories to Folding Proteins: A Review and Assessment of Simulation Studies of Protein Folding and Unfolding. Annual Review of Physical Chemistry, 52, 499-535. http://dx.doi.org/10.1146/annurev.physchem.52.1.499
- Karanicolas, J. and Brooks III, C.L. (2004) An Evolution of Minimalist Models for Protein Folding: From the Behavior of Protein-Like Polymers to Protein Function. Biosilico, 2, 127-133.
- Chakrabartty, A. and Baldwin, R.L. (1995) Stability of α-Helices. Advances in Protein Chemistry, 46, 141-176. http://dx.doi.org/10.1016/S0065-3233(08)60334-4
- Head-Gordon, T., Stillinger, F.H., Wright, M.H. and Gay, D.M. (1992) Poly(L-Alanine) as a Universal Reference Material for Understanding Protein Energies and Structures. Proceedings of the National Academy of Sciences of the United States of America, 89, 11513-11517. http://dx.doi.org/10.1073/pnas.89.23.11513
- Makowska, J., Liwo, A., ?mudzińska, W., Lewandowska, A., Chmurzynski, L. and Scheraga, H.A. (2012) Like-Charged Residues at the Ends of Oligoalanine Sequences Might Induce a Chain Reversal. Biopolymers, 97, 240-249. http://dx.doi.org/10.1002/bip.22013�
- Makowska, J., Rodziewicz-Motowidlo, S., Baginska, K., Makowski, M., Vila, J.A., Liwo, A., Chmurzynski, L. and Scheraga, H.A. (2007) Further Evidence for the Absence of Polyproline II Stretch in the XAO Peptide. Biophysical Journal, 92, 2904-2917. http://dx.doi.org/10.1529/biophysj.106.097550
- Pappu, R.V., Srinivasan, R. and Rose, G.D. (2000) The Flory Isolated-Pair Hypothesis Is Not Valid for Polypeptide Chains: Implications for Protein Folding. Proceedings of the National Academy of Sciences of the United States of America, 97, 12565-12570. http://dx.doi.org/10.1073/pnas.97.23.12565
- Ramakrishnan, V., Ranbhor, R. and Durani, S. (2004) Existence of Specific “Folds” in Polyproline II Ensembles of an “Unfolded” Alanine Peptide Detected by Molecular Dynamics. Journal of the American Chemical Society, 126, 16332-16333. http://dx.doi.org/10.1021/ja045787y
- Shi, Z., Olson, C.A., Rose, G.D., Baldwin, R.L. and Kallenbach, N.R. (2002) Polyproline II Structure in a Sequence of Seven Alanine Residues. Proceedings of the National Academy of Sciences of the United States of America, 99, 9190-9195. http://dx.doi.org/10.1073/pnas.112193999
- Shi, Z., Woody, R.W. and Kallenbach, N.R. (2002) Is Polyproline II a Major Backbone Conformation in Unfolded Proteins? Advances in Protein Chemistry, 62, 163-240. http://dx.doi.org/10.1016/S0065-3233(02)62008-X
- Cheng, R.P., Girinath, P., Suzuki, Y., Kuo, H.T., Hsu, H.C., Wang, W.R., Yang, P.A., Gullickson, D., Wu, C.H., Koyack, M.J., Chiu, H.P., Weng, Y.J., Hart, P., Kokona, B., Fairman, R., Lin, T.E. and Barrett, O. (2010) Positional Effects on Helical Ala-Based Peptides. Biochemistry, 49, 9372-9384. http://dx.doi.org/10.1021/bi101156j
- Wieczorek, R. and Dannenberg, J.J. (2003) Hydrogen-Bond Cooperativity, Vibrational Coupling, and Dependence of Helix Stability on Changes in Amino Acid Sequence in Small 310-Helical Peptides. A Density Functional Theory Study. Journal of the American Chemical Society, 125, 14065-14071. http://dx.doi.org/10.1021/ja034034t
- Brant, D.A., Miller, W.G. and Flory, P.J. (1967) Conformational Energy Estimates for Statistically Coiling Polypeptide Chains. Journal of Molecular Biology, 23, 47-65. http://dx.doi.org/10.1016/S0022-2836(67)80066-4
- Flory, P.J. and Schimmel, P.R. (1967) Dipole Moments in Relation to Configuration of Polypeptide Chains. Journal of the American Chemical Society, 89, 6807-6813. http://dx.doi.org/10.1021/ja01002a001
- Flory, P.J. (1969) Statistical Mechanics of Chain Molecules. InterScience Publishers, New York.
- Kumar, A., Ramakrishnan, V., Ranbhor, R., Patel, K. and Durani, S. (2009) Homochiral Stereochemistry: The Missing Link of Structure to Energetics in Protein Folding. The Journal of Physical Chemistry B, 113, 16435-16442. http://dx.doi.org/10.1021/jp906811k
- Ramakrishnan, V., Ranbhor, R., Kumar, A. and Durani, S. (2006) The Link between Sequence and Conformation in Protein Structures Appears to Be Stereochemically Established. The Journal of Physical Chemistry B, 110, 9314-9323. http://dx.doi.org/10.1021/jp056417e
- Srivastava, K.R., Kumar, A., Goyal, B. and Durani, S. (2011) Stereochemistry and Solvent Role in Protein Folding: Nuclear Magnetic Resonance and Molecular Dynamics Studies of Poly-L and Alternating-L,D Homopolypeptides in Dimethyl Sulfoxide. The Journal of Physical Chemistry B, 115, 6700-6708. http://dx.doi.org/10.1021/jp200743w
- Durani, S. (2008) Protein Design with Land D-Alpha-Amino Acid Structures as the Alphabet. Accounts of Chemical Research, 41, 1301-1308. http://dx.doi.org/10.1021/ar700265t
- Daura, X., van Gunsteren, W.F. and Mark, A.E. (1999) Folding-Unfolding Thermodynamics of a Beta-Heptapeptide from Equilibrium Simulations. Proteins: Structure, Function, and Bioinformatics, 34, 269-280. http://dx.doi.org/10.1002/(SICI)1097-0134(19990215)34:3<269::AID-PROT1>3.0.CO;2-3
- Lindahl, E., Hess, B. and van der Spoel, D. (2001) GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. Journal of Molecular Modeling, 7, 306-317.
- Van Gunsteren, W.F., Billeter, S.R., Eising, A.A., Hünenberger, P.H., Krüger, P., Mark, A.E., Scott, W.R.P. and Tironi, I.G. (1996) Biomolecular Simulation: The GROMOS96 Manual and User Guide. Hochschulverlag AG an der ETH Zürich, Zürich.
- Ryckaert, J., Ciccotti, G. and Berendsen, H. (1977) Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. Journal of Computational Physics, 23, 327-341. http://dx.doi.org/10.1016/0021-9991(77)90098-5
- Darden, T., York, D. and Pedersen, L. (1993) Particle Mesh Ewald: An N-Log(N) Method for Ewald Sums in Large Systems. The Journal of Chemical Physics, 98, 10089-10092. http://dx.doi.org/10.1063/1.464397
- Essmann, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H. and Pedersen, L.G. (1995) A Smooth Particle Mesh Ewald Method. The Journal of Chemical Physics, 103, 8577-8593. http://dx.doi.org/10.1063/1.470117
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K. and Olson, A.J. (1998) Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. Journal of Computational Chemistry, 19, 1639-1662. http://dx.doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
- Chan, W.C. and White, P.D. (1989) Fmoc Solid Phase Peptide Synthesis: A Practical Approach. IRL Press, Oxford.
Supplementary Material
 (a)
(a)
 (b)
(b)
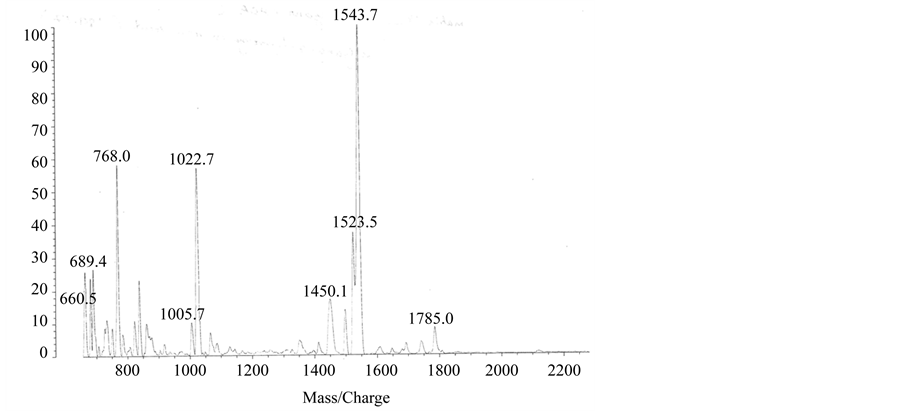
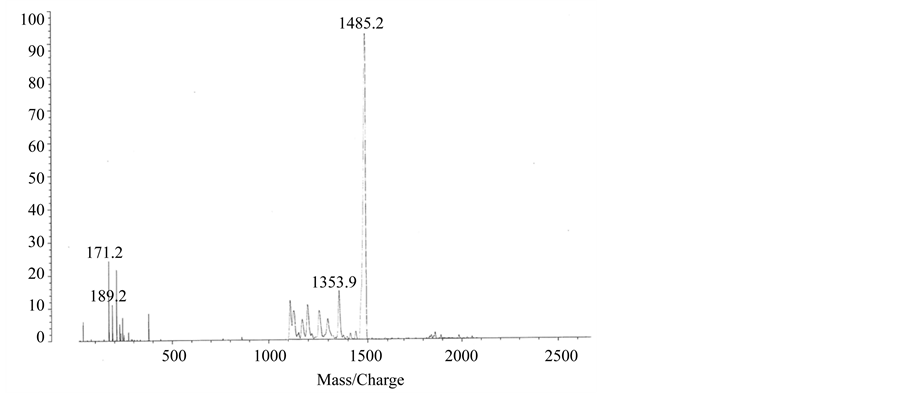
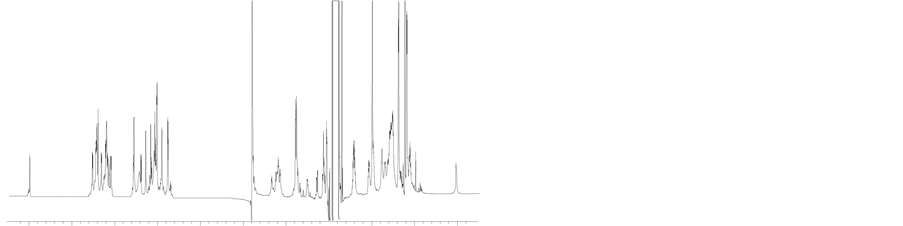
Figure S1. MALDI-Mass spectra of H1 (panel (a)) and H2 (panel (b)).
 (a) (b)
(a) (b)
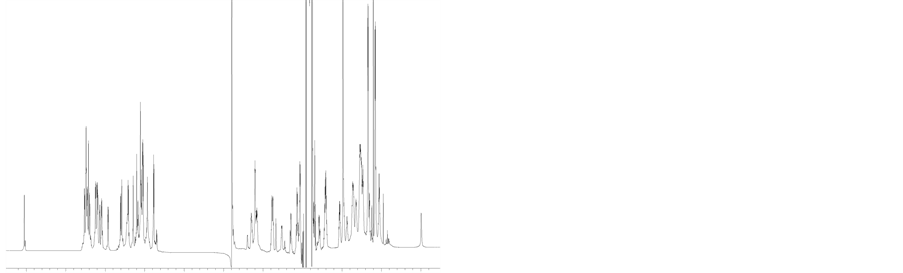
Figure S2. (a) 1H NMR spectra of H1 in citrate buffer (pH ~3) and (b) 1H NMR spectra of H2 in citrate buffer (pH ~3).
10 9 8 7 6 5 4 3 2 1 0 10 9 8 7 6 5 4 3 2 1 0
 (a) (b)
(a) (b)
Figure S3. Quenching of tryptophan fluorescence of peptide H2 (20 mM) in 20 mMTris-HCl buffer at pH 7.5, on progressive titration with increasing titration with pNPP (panel (a)), and plot of relative fluorescence intensity as a function of pNPP concentration (panel (b)).
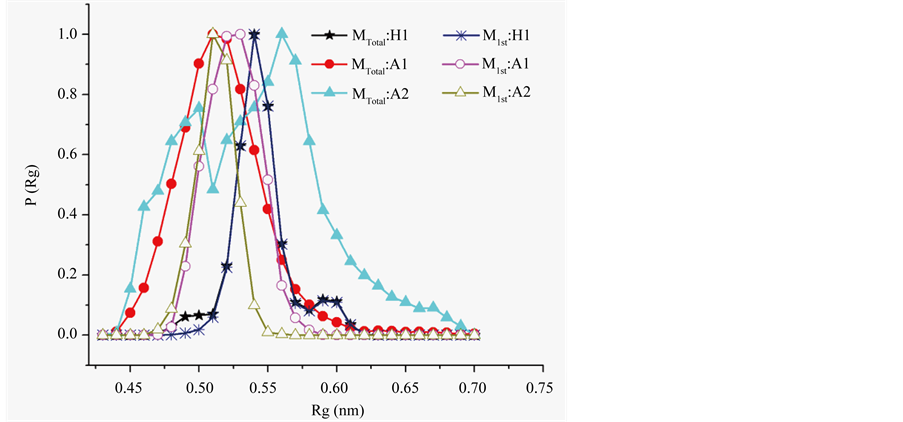
Figure S4. Radius of gyration (Rg) distribution of main chain atoms over conformers populating equilibrium.
 (a) (b) (c)
(a) (b) (c)
Figure S5. Occupancy of ensembles in H1 (panel (a)), A1 (panel (b)) and A2 (panel (c)) in specific basins of f, ψ.
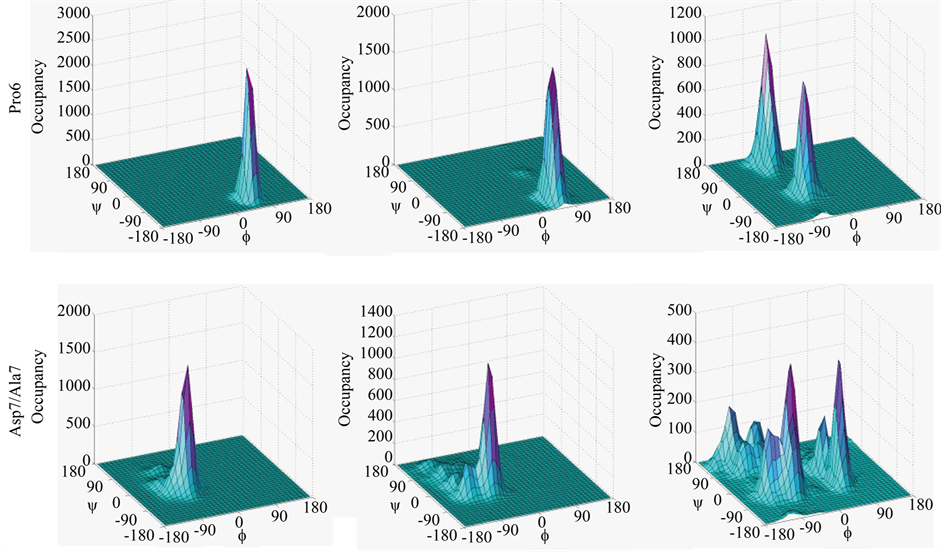
Figure S6. Specific β-turn residues in H1 (panel (a)), A1 (panel (b)), and A2 (panel (c)) are varied in f, ψ space.
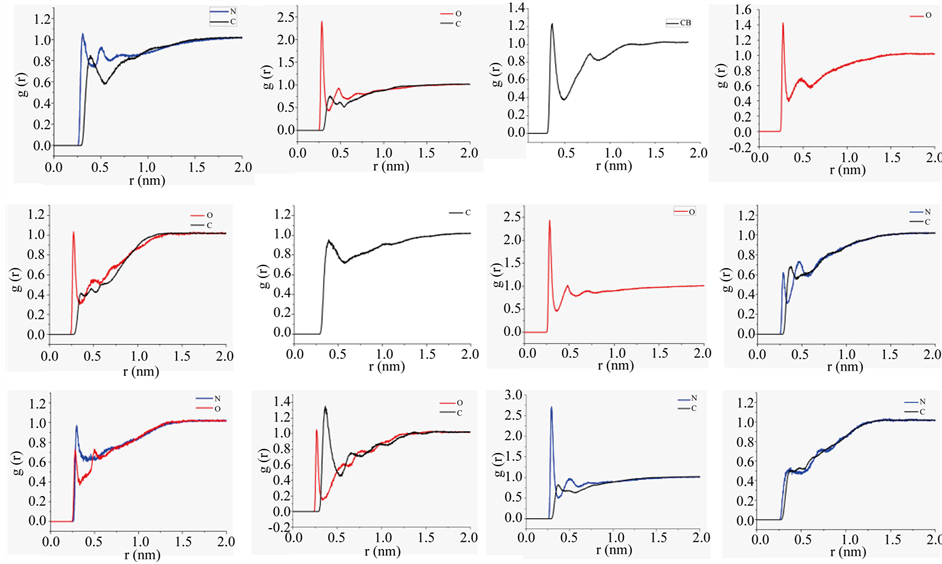
Figure S7. Radial distribution of water oxygen atoms against specific side chain nitrogen (blue trace), oxygen (red trace), and carbon (black trace) atoms of H1 over macrostate.


Figure S8. Spatial distribution of water oxygen atom around atoms of H1 (left panel) and A1 (right panel) over macrostate.
NOTES

*Corresponding author.