Open Journal of Physical Chemistry
Vol.4 No.2(2014), Article ID:45079,11 pages DOI:10.4236/ojpc.2014.42006
Three-Center Configuration with Four, Three, and Two Electrons for Carbon, Boron, Hydrogen, and Halogen Exchange. A Model and Theoretical Study with Experimental Evidence
Henk M. Buck
Kasteel Twikkelerf 94, Tilburg, The Netherlands
Email: h.m.buck@ziggo.nl
Copyright © 2014 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 18 February 2014; revised 15 March 2014; accepted 22 March 2014
ABSTRACT
The introduction of specific sites in organic frames for accommodation of various modes of bonding has been focused on reaction types which are described by using different theoretical models with or without a definite experimental proof. In this study three-center four-, three-, and twoelectron systems based on carbon-, boron-, hydrogen-, and halogen exchange are under consideration. Based on the number of electrons in the transition state or transition complex it is shown that all transfer or exchange reactions share the same ratio numbers expressed as the quotient of the transitional bond distance under investigation and its normal bond length. With X-ray data of model systems it was even possible to give the ratio numbers for a three-center four-electron configuration experimental support with additional ab initio data. Furthermore a novel model type of substitution in organic chemistry is introduced through electrophilic insertion, informative for enzyme-substrate interactions based on the lock-and-key model. Reactions based on a three-center two-electron configuration mostly follow a nonlinear transition. In this alignment there will be a pursuit of cyclization for stabilization via homoaromaticity as homocyclopropenyl cation. The molecular dynamics of such a process is discussed based on recent X-ray crystallographic data of the symmetrically bridged, nonclassical geometry of the 2-norbornyl cation. In the present paper the focus is aimed at the transition intermediate of the (classical) 2-norbornyl cation involved in the isomerization into the nonclassical geometry. This model description is compared with a simple molecular rearrangement of the 1-propyl cation into the corner-protonated cyclopropane using the ab initio data. The exclusivity of the former isomerization compared with the latter one could be unambiguously demonstrated by the invention that the intramolecular electron shift can be expressed in a linear relationship between the concerned electron-donating and accepting bond lengths. Finally, the fluor transitions as divalent atoms in a three-center two-electron configuration are described. The role of fluor in comparison with the other halogens is striking. The attention was focused on an excellent correspondence between the recent chemical and theoretical evidence for a symmetrical fluoronium ion in solution. Simple dialkylfluoroniumions in contrast to the other halonium ions are not present in solution. Although the geometry of the fluoronium ion theoretically can be described as a real minimum, the C-F-C angle of 120˚ is apparently the borderline transition for dissociation in C+ and F-C.
Keywords:Three-Center Configuration, Frozen Transition State, Molecular Drone, Enigma, Classical and Nonclassical 2-Norbornyl Cation

1. Introduction
Recently, several groups introduced localized sites in organic frames for accommodation of fundamental modes of bonding via intraand intermolecular reactions. Most of these investigations are focused on well-known reaction types which are theoretically described with or without a definite proof for their experimental existence. In this study three-center four-, three-, and two-electron systems based on carbon-, boron-, hydrogen-, and halogen exchange are scrutinized. In the case of a linear three-center configuration, it is possible with the dynamics of a regular tetrahedron to change its geometry to a trigonal bipyramidal transition complex, to predict the bond lengths in the different states of the corresponding reaction profile. Based on the number of electrons in the transition state or transition complex it is then clear that all transfer or exchange reactions share the same ratio of the bond distances compared with its initial state. For the number of electrons (2, 3 and 4) in these transitions, the corresponding ratio numbers are 1.167, 1.250, and 1.333. This involves that these ratio numbers have no restrictions in their application concerning the migrating particle. As an example for the three-center four-electron transition state we select the well-known SN2 mechanism simplified by the substitution reaction X− + CH3-X → [X-CH3-X]− → X-CH3 + X− with X = halogen. In this case the methyl cation migrates between the negatively charged halogens. The transition state can be characterized as a trigonal bipyramidal configuration with X in the apical positions. In the most ideal configuration, the ratio number for the transition state i.e., the ratio between the apical distance and the corresponding bond length in the initial state is 1.333, visualized as 1-cos 109.47°. A similar result is obtained for the linear proton transfer between carbanions. In that case one is dealing with a divalent hydrogen, in the former case with a pentavalent carbon. The different aspects are supported by ab initio calculations. Both proton and methyl cation transfer reactions play an important role in biochemistry through their hydrogen bridging in DNA and proteins that results in efficient structural networks, and through methyl bridging controlling the interplay between the backbone of DNA and its bases. Specific attention has been given to the nonlinear three-center carbon two-electron system as recently experimentally described for the symmetrically bridged, nonclassical 2-norbornyl cation. The geometry of the latter cation characterized by calculations, is in excellent correspondence with its X-ray determined structure. The interest is focused on the dynamics of the formation of this cation starting from its ionized form. At the end halogen transfer reactions in a three-center bonding are considered because they are closely related to the variety of elementary particles in basic organic chemistry. A short introduction with relevant bond lengths is given in Figure 1, summarizing the aforementioned results for linear three-center four-, three-, and two-electron systems at the MP2/6-31 + G(d,p) level of theory [1] in combination with the model findings between parentheses [2] -[6] . The substituents are tert-butyl groups, and X represents H−, H∙, H+, 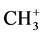 , and F−. For the C-H, C-F and C-C bond in the initial state, the selected values are 1.080Å, 1.420Å, and C-C 1.540Å, respectively.
, and F−. For the C-H, C-F and C-C bond in the initial state, the selected values are 1.080Å, 1.420Å, and C-C 1.540Å, respectively.
The results deal with a stationary point that is not an energy minimum on the reaction coordinate with the proton transfer in the cage model as exception. The distances for the symmetrically transitional intermediates can be obtained with d12 = d23= (1 + n/12) × dC-X, in which dC-X is the initial state and n is the number of electrons involved in the three-center bonding. In combination with a regular or distorted tetrahedral angle (δ) in the principal axis of the reaction coordinate, the ratio number R(p, δ) = 1-p × cos δ, with p = 1, 2, ... The distance in the
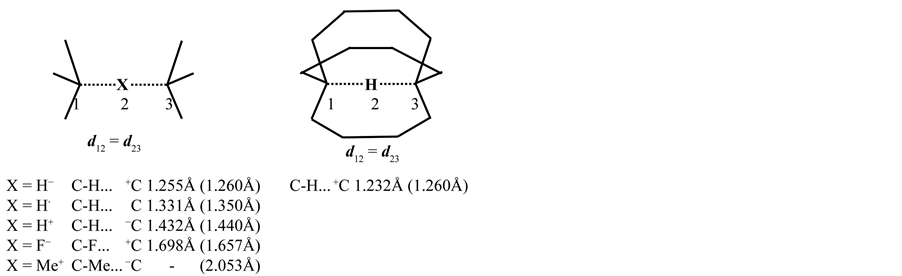
Figure 1. Three-center four-, three-, and two electron systems based on carbon-, hydrogen-, and halogen exchange at the MP2/6-31 + G(d, p) level of theory in combination with the model findings between parentheses. The transition distances in open models are slightly greater than in cage compounds, indicated as intra-annular pressure.
intermediate state is then d(XY) = R(p, δ) × d(XY) in which d (XY) is the conventional bonding. The ratio number corresponding with distances R(d) is expressed as R(d) = d(XY):d(XY), vide supra [5] . Following the dynamics from a tetrahedral configuration in a trigonal bipyramidal configuration, it can be derived that the pretransition state or intermediate illustrated for SN2 with [X−…CH3-X] represents the following distances in the principal reaction axis in combination with the angles XCH and HCH for methyl as cation, radical and anion i.e., 1.667d, d with 109.47˚, 109.47˚; 1.500d, d with 104.47˚, 113.98˚ and 1.334d, d with 99.61˚, 117.27˚, respectively [2] . Under Results and Discussion the aforementioned model reactions are fully analyzed in different sections: 1) Structure analysis and ab initio calculations of stabilized trivalent carbon cation and boron fixed between the apical positions of a trigonal bipyramid. A frozen transition state for pentavalent boron. 2) Dynamics in 2-norbornyl systems based on a three-center two-electron configuration with ab initio calculations, crystallographic data and model systems. 3) Hydrogen and fluor transitions as divalent atoms in a three-center configuration with experimental data and ab initio calculations.
2. Results and Discussion
2.1. Structure Analysis and ab Initio Calculations of Stabilized Trivalent Carbon Cation Fixed between the Apical Positions of a Trigonal Bipyramid. A Frozen Transition State for Pentavalent Boron
It will be clear that if the ratio number corresponding with C-X in the principal axis of a trigonal bipyramidal (TBP) configuration exceeds the value of 1.33, one is dealing with a stabilized (trivalent) carbenium ion or pentacoordinate carbon. Examples are given by Yamashita et al. [7] -[9] in their study on the synthesis and X-ray structural determination of ten-electron pentacoordinate carbon compounds bearing an anthracene skeleton with two apical oxygens (-OCH3) at the 1,8-positions linked with -C+(OCH3)2 at the 9-position. The X-ray crystallographic analysis showed a TBP geometry with relatively long almost identical apical distances of ca. 2.44Å resulting in a ratio number of 1.66 for a corresponding tetravalent value of 1.47Å as is shown in Figure 2.
With the X-ray results it was possible to find the borderline which is connected with the ratio value of 1.33. Introduction of the much stronger electrophile -C+(CF3)2 in comparison with -C+(OCH3)2 at the 9-position results in a tetrahedral configuration for the central carbon. For a pentavalent configuration in that case the apical distance should be 1.96Å, resulting in a strained conformation thereby cancelling the lone pair donating ability of O−(K+). The cation then relaxes into a pentacoordinate TBP structure in the absence of the ratio numbers necessary for the accommodation of a pentavalent state. On the other hand substitution of the central carbon by boron with two fluorines results in a ratio number of 1.36 (av 2.29Å:1.69Å) in consequence of the electron-withdrawing nature of the fluorine atoms. The tetrahedral value has been taken from a corresponding complex in which boron has two chlorines resulting in a short and long B-O distance of 1.69 and 2.79Å. Within the pentavalent TBP model the long B-O distance is then 2.82Å (= 1.66(7) × 1.69Å, see Figure 2) which is in good correspondence with the X-ray value. All the relevant results are summarized in Figure 2. The fact that the trivalent CH3 cation is isoelectronic with the corresponding neutral trivalent boron compound, BH3 (boron and carbon are consecutive second-period elements) demonstrates clearly the ability of boron to form a pentavalent TBP geometry conforms to the predicted ratio numbers. The latter configuration may be considered as a “frozen” transition state. Such a conclusion is based on the unique geometric construction of the organic frame in which the migration of the central group is restricted by the nearly fixed position of the apical sites. In fact this is a cage compound in which the “migrating” particle is already placed on the vital location of the principal reaction coordinate for the linear three-center four-electron transition state. This situation can be also realized using 1,8- dimethoxyanthracene as target for cationic insertion at the favoured 9-position under deprotonation. For this electrophilic addition, divalent methylene dications and boron monocations can be considered. It is tempting to introduce the expression molecular drone whose attack is controlled by explicit sensors on the organic frame (methoxyl groups) and the exclusive center (9-position) for accommodation of electrophilic attack. Focused on boron, BX2 monocations are suggested as a realistic substrate with X = F or Cl in combination with two amines as ligands in a tetrahederal configuration: 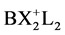 [10] -[12] , see Figure 3. This illustration for insertion is an excellent example for substrate-enzyme interactions.
[10] -[12] , see Figure 3. This illustration for insertion is an excellent example for substrate-enzyme interactions.
This modelling leads to an interesting spin-off conclusion. It can be concluded with a simple valence bond consideration that the locations of the methoxyl groups do not favour to a greater extent the electrophilic addition at the 9-position compared with the unsubstituted one. Replacing these substituents to the 2- and 7-positions then there is a considerable increase for this addition. However, the absence for coordination disfavours the formation of a possible pentavalent or pentacoordinate geometry. To the best of our knowledge this may be the first example in basic organic chemistry where the role of a strong electron-donating methoxyl group is used as coordination site via one of its lone pairs.
2.2. Dynamics in 2-Norbornyl Systems Based on a Three-Center Two-Electron Configuration with ab Initio Calculations, Crystallographic Data and Model Systems
Reactions based on a three-center two-electron construction mostly follows a nonlinear transition. In this alignment there will be a pursuit of cyclization for stabilization via homoaromaticity. In the case of this type of interaction then there is a preference for a homocyclopropenyl cation. The dynamics of such a process is discussed using recent X-ray crystallographic data of the symmetrically bridged, nonclassical geometry of the 2-norbornyl
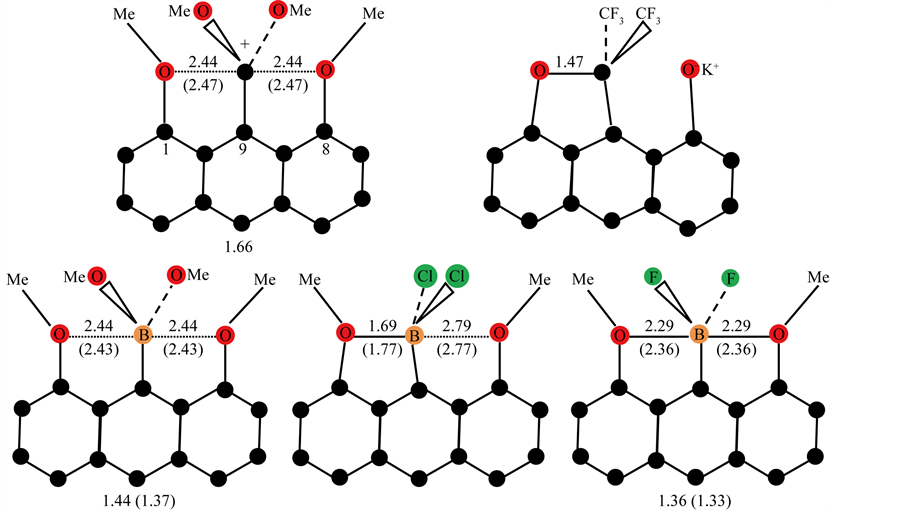
Figure 2. Structure (in Å) and ratio numbers of pentacoordinate 10-C and pentavalent 10-B compounds bearing an anthracene skeleton. The values in parentheses correspond with DFT calculations.
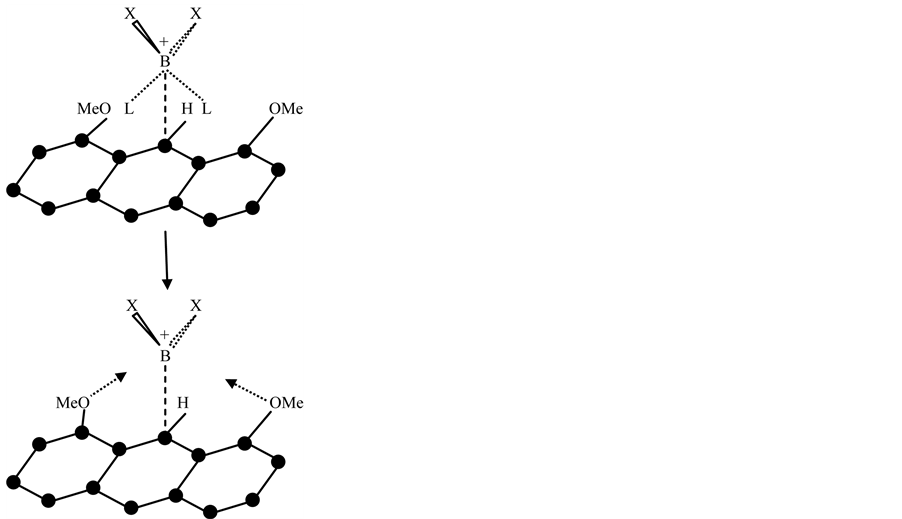
Figure 3. Initial state for the interaction of 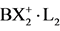 (X = F and Cl) with 1,8-dimethoxyanthracene. After loss of both ligands (L), coordination with the methoxyl groups starts ending with the in-plane pentavalent boron under deprotonation at the 9-position. Carbon numbers are given in Figure 2.
(X = F and Cl) with 1,8-dimethoxyanthracene. After loss of both ligands (L), coordination with the methoxyl groups starts ending with the in-plane pentavalent boron under deprotonation at the 9-position. Carbon numbers are given in Figure 2.
cation as has been published by Scholz et al. [13] . The molecular representation of the average bond distances is given in Figure 4. From the various data it appears that there is variation in the long bonds (C1C6 and C2C6) under the crystalline condition. The corresponding ab initio calculations at the MP2(FC)/def2-QZVPP level of theory differ to some extent from the experimental values. The attention was focused on the C2C1C6 arrangement in relation to more simple conversions as the 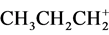 (1-propyl cation)-isomerization into protonated cyclopropane [14] .
(1-propyl cation)-isomerization into protonated cyclopropane [14] .
The study is directed on the reaction progress via an intermediate 2-norbornyl cation that can be described as a stationary point on the reaction coordinate resulting in its nonclassical structure. The ab initio optimized geometry of this transition intermediate was given by Schreiner et al. [15] . Of special interest are the C1C2, C1C6, and C1C7 bond lengths. The differences are striking in comparison with the parent saturated compound norbornane [16] . In most studies the focus was originated on the molecular C2C1C6 arrangement. However, the geometric results based on the ab initio results of the 2-norbornyl cation as transition structure, and on a more simple system as the 1-propyl cation, clearly demonstrate the exclusive information stored in the 2-norbornyl cation. It can be shown that the C1C7 bond in the intermediate state plays an explicit role in the reaction progress. Radom et al., performed ab initio geometries of nine  cations [14] . They suggested that the bond length of these cations will pertain to other nonclassical cations. Particular attention was given to the long C-C bonds (1.803Å) in corner-protonated cyclopropane. Although corner-protonated cyclopropane and the bridged 2-norbornyl cation have corresponding C-C bond lengths in the cyclic three-membered C1C2C3- and C1C2C6 structures respectively, the transition structure of the 2-norbornyl cation, compared with the various conformers of the 1-propyl cation, delivers additional geometric information. From an ab initio study of the former cation it appears that the partial double bond C1C2 (1.409Å) is interrelated with the C1C7 bond (1.671Å) and not, or to a small extent, with the other neighbour C1C6 bond (1.552Å) [15] . The 2-norbornyl cation then undergoes an intramolecular rearrangement via the 2-norpinyl cation [17] into the symmetrically bridged configuration. The results are summarized in Figure 5 with relevant bond distances.
cations [14] . They suggested that the bond length of these cations will pertain to other nonclassical cations. Particular attention was given to the long C-C bonds (1.803Å) in corner-protonated cyclopropane. Although corner-protonated cyclopropane and the bridged 2-norbornyl cation have corresponding C-C bond lengths in the cyclic three-membered C1C2C3- and C1C2C6 structures respectively, the transition structure of the 2-norbornyl cation, compared with the various conformers of the 1-propyl cation, delivers additional geometric information. From an ab initio study of the former cation it appears that the partial double bond C1C2 (1.409Å) is interrelated with the C1C7 bond (1.671Å) and not, or to a small extent, with the other neighbour C1C6 bond (1.552Å) [15] . The 2-norbornyl cation then undergoes an intramolecular rearrangement via the 2-norpinyl cation [17] into the symmetrically bridged configuration. The results are summarized in Figure 5 with relevant bond distances.

Figure 4. Molecular structure of the nonclassical 2-norbornyl carbonium ion. The average X-ray distances are C1C6 (C2C6) 1.802Å (1.825Å [13] , 1.829Å [15] ), C1C2 1.388Å (1.393Å [13] , 1.394Å [15] ), and C1C7 (C2C3) 1.495Å (1.506Å [15] ). The corresponding ab initio values are in parentheses.
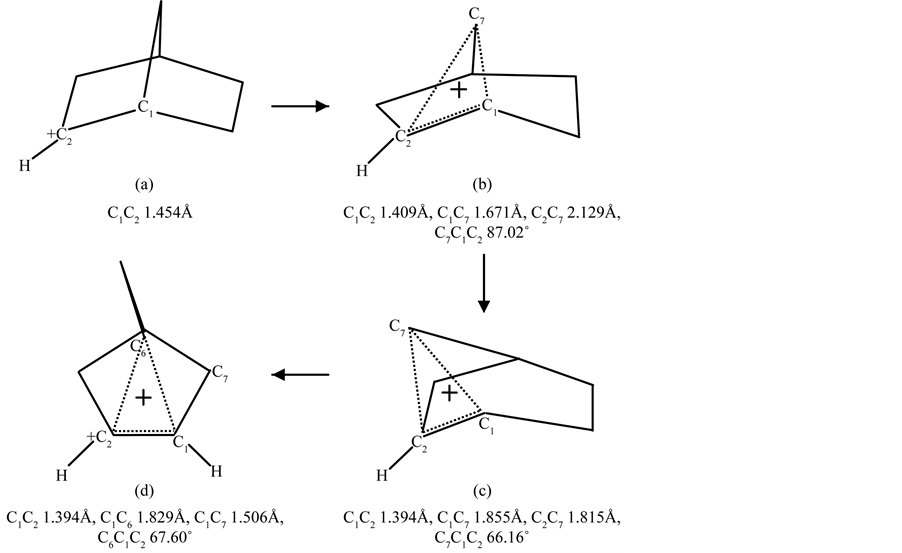
Figure 5. Rearrangement of the initial 2-norbornyl cation (a) via the transition structure (b) located as (classical) 2-norbornyl cation into the intermediate transition structure of the unsymmetrically bridged 2-norpinyl cation (c) resulting in the nonclassical symmetrically bridged, 2-norbornyl cation (d).
In the reaction sequence the relative energies proceed “downhill” from 21.80 (a), 13.64 (b), 12.39 (c) towards 0.00 kcal∙mol−1 (d), respectively [17] . A providing description for the formation of the symmetrically bridgednonclassical 2-norbornyl cation can be clearly visualized with the transition structure of the classical 2-norbornyl cation. The C1C6 bond electrons directed to the C1C2 bond restores this donation in the C1C7 (C2C3) bond (1.506Å [15] ; X-ray value 1.495Å [13] ). This exclusive geometry could be demonstrated by comparison with the dynamics of the 1-propyl cation into the corner-protonated cyclopropane, using a graph relation between the distances (d) of C2C3 and C1C2 for the methyl-staggered and methyl-eclipsed conformers of the 1-propyl cation, . This specific interaction between the electron-donating (dD) and electron-accepting bonds (dA) is extended with the corresponding bond distances C1C7 and C1C6 with C1C2 in the classical 2-norbornyl cation. It may be clear that this correlation is based on electron transfer via ϭ − π conversion and vice versa. The results for the various conformers concerning their interactive bond lengths of the 1-propyl cation are summarized in Table 1 in combination with the geometry of the classical 2-norbornyl cation. From a study of Chiavarino et al., [18] on the protonation of gaseous cyclopropane, a number of relevant
. This specific interaction between the electron-donating (dD) and electron-accepting bonds (dA) is extended with the corresponding bond distances C1C7 and C1C6 with C1C2 in the classical 2-norbornyl cation. It may be clear that this correlation is based on electron transfer via ϭ − π conversion and vice versa. The results for the various conformers concerning their interactive bond lengths of the 1-propyl cation are summarized in Table 1 in combination with the geometry of the classical 2-norbornyl cation. From a study of Chiavarino et al., [18] on the protonation of gaseous cyclopropane, a number of relevant 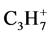 ions were calculated at the CCSD(T)/cc-pVDZ level. The optimized geometry of the unsymmetrical corner-protonated cyclopropane following the hydrogen transfer orientation via an unsymmetrical edge-protonated cyclopropane as transition state, shows correspondence with the methyl-eclipsed conformer of the 1-propyl cation through carbon (CH3) transfer. The geometric result of the unsymmetrical corner-protonated cyclopropane is also given in Table 1.
ions were calculated at the CCSD(T)/cc-pVDZ level. The optimized geometry of the unsymmetrical corner-protonated cyclopropane following the hydrogen transfer orientation via an unsymmetrical edge-protonated cyclopropane as transition state, shows correspondence with the methyl-eclipsed conformer of the 1-propyl cation through carbon (CH3) transfer. The geometric result of the unsymmetrical corner-protonated cyclopropane is also given in Table 1.
Using these results for dD and dA, a linear relationship between both distances could be established as is shown in Figure 6. The most fascinating result was the expression of the classical 2-norbornyl cation with the coordinate (1.409, 1.552). This pertinent deviation from the linear graph may be considered as support for the presented model description. There is no indication for a donor-acceptor interaction as follows from their d-values of the C1C6 and C1C2 bonds, respectively, whereas the corresponding interaction between C1C7 and C1C2 perfectly matches with the linear plot. To the best of our knowledge no attention has been given to this particular aspect in regard of the implication for the reaction dynamics caused by the geometry of the classical 2-norbornyl cation. The relationship also offers the possibility to find the extremes in the conformers of the 1-propyl cation with C-C bond values of 1.33Å and 1.54 Å, reflecting a double and saturated bond component, visualized as [H3C+]---CH2 = CH2 and H3C-CH2-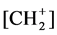 , respectively. It may be of interest to look more in detail to the calculations of Chiavarino et al. [18] . The coordinate (1.408, 1.716) of the unsymmetrical cornerprotonated cyclopropane matches to a certain extent with the suggested dD, dA linearity. The calculated geometries as the corner-protonated cyclopropane and the edge-protonated cyclopropane are in agreement with the results of Radom et al. [14] .
, respectively. It may be of interest to look more in detail to the calculations of Chiavarino et al. [18] . The coordinate (1.408, 1.716) of the unsymmetrical cornerprotonated cyclopropane matches to a certain extent with the suggested dD, dA linearity. The calculated geometries as the corner-protonated cyclopropane and the edge-protonated cyclopropane are in agreement with the results of Radom et al. [14] .
From the relationship in Figure 6, a number of interesting conclusions can be drawn. The coordinate (1.492, 1.541) of the methyl-staggered 1-propyl cation with a conformation in which the HC1H angle lies in the C1C2C3 plane, shows a match with the dD vs. dA linear plot. The evident absence of bond interaction in that case corresponds with the C2C3 saturated bond distance and the corresponding 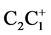 bond length with a positive charge localized at C1. The other extreme value of C2C3 corresponding with the C1C2 value of 1.330Å, amounts to 1.767Å. The latter value has been obtained by extrapolation of the linear plot. Calculations based on modelling for carbon transfer via a trigonal bipyramid results in 1.774Å [2] -[6] , vide supra. In this model the results are described for this specific complexation as a linear three-center system with two-, threeand four participating electrons. Adapted to the dD vs. dA linear plot, the coordinates are then (d, 1.334d), (d, 1.500d), and (d, 1.667d), respectively. The coordinate (d, 1.334d) corresponding with [H3C+]---CH2 = CH2 then results in (1.330, 1.774). The deviating coordinate (1.409, 1.552) of the intermediate classical 2-norbornyl cation is of particular interest for this study. It concerns the well-known neighbouring group participation C1C6 towards C1C2 in the favoured solvolytic pathway of 2-exo-norbornyl as opposed to 2-endo leaving groups [19] . In fact it looks like this specific coordinate may work counterproductive for the description of the solvolysis. The other coordinate that belongs to the classical 2-norbornyl cation (1.409, 1.671) represents the C1C7-C1C2 interaction. The bond length
bond length with a positive charge localized at C1. The other extreme value of C2C3 corresponding with the C1C2 value of 1.330Å, amounts to 1.767Å. The latter value has been obtained by extrapolation of the linear plot. Calculations based on modelling for carbon transfer via a trigonal bipyramid results in 1.774Å [2] -[6] , vide supra. In this model the results are described for this specific complexation as a linear three-center system with two-, threeand four participating electrons. Adapted to the dD vs. dA linear plot, the coordinates are then (d, 1.334d), (d, 1.500d), and (d, 1.667d), respectively. The coordinate (d, 1.334d) corresponding with [H3C+]---CH2 = CH2 then results in (1.330, 1.774). The deviating coordinate (1.409, 1.552) of the intermediate classical 2-norbornyl cation is of particular interest for this study. It concerns the well-known neighbouring group participation C1C6 towards C1C2 in the favoured solvolytic pathway of 2-exo-norbornyl as opposed to 2-endo leaving groups [19] . In fact it looks like this specific coordinate may work counterproductive for the description of the solvolysis. The other coordinate that belongs to the classical 2-norbornyl cation (1.409, 1.671) represents the C1C7-C1C2 interaction. The bond length

Table 1. Distances (d in Å) of electron-donating (dD) and electron-accepting (dA) bonds and angles (degrees) between the D and A bonds in the various conformers of the 1-propyl cation and the transition structure of the classical 2-norbornyl cation.
aThe bond distances and angles of methyl-eclipsed and methyl-staggered 1-propyl cation are from Ref. [14] . bThe bond distances and angle are from Ref. [18] ; in this reference the conformer is indicated as an unsymmetrical corner-protonated cyclopropane. cIn this conformation the HC1H angle lies in the C3C2C1 plane. dThe bond distances and angels in the classical 2-norbornyl cation are from Ref. [15] .
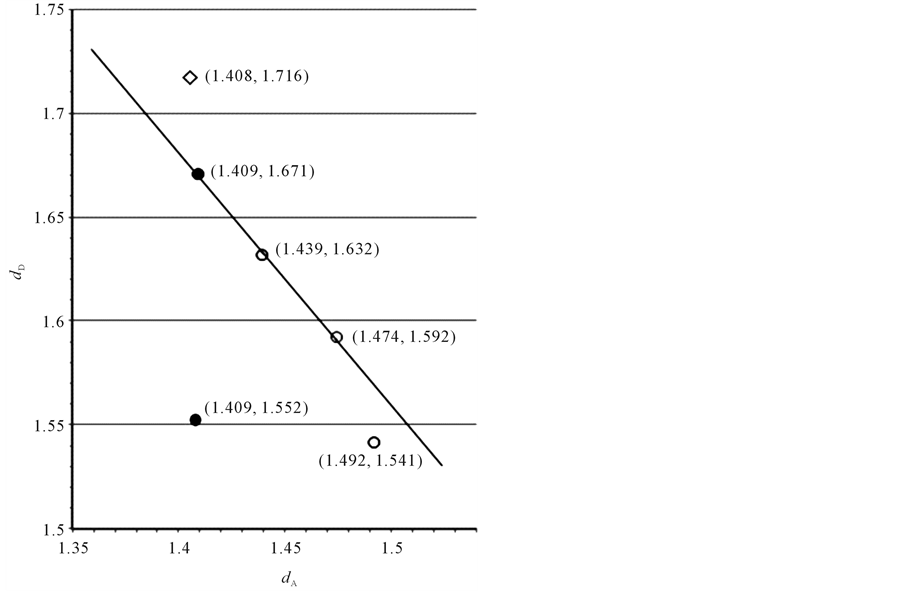
Figure 6. A linear relationship between the electron-donating (dD) and electron-accepting (dA) bond distances (Å) in the different conformers of the 1-propyl cation (○ [14] ), the unsymmetrical corner-protonated cyclopropane (◇ [18] ) and the classical 2-norbornyl cation (● [15] ). The coordinates correspond with the distances given in Table 1.
extension of C1C7 is accompanied with the C1C2 bond reduction, resulting in a partial double-bond character. This aspect correlated with the geometry of the classical 2-norbornyl cation, offers new insight in the mechanism for the formation of the symmetrically bridged 2-norbornyl cation. From the results available, it may be concluded that in the intermediate state of the 2-norbornyl cation, the C1C2 bond with its partial double-bond character is sufficiently activated for the neighbour C1C6 bond participation. An interesting observation can be demonstrated by a graph for dA/dD and the angle between donor and acceptor bond as shown in Figure 7. The result is a sigmoidal curve that shows an explicit demonstration for a pronounced intramolecular reaction profile between 80˚ - 120˚.
Summarizing, the ab initio study of the classical 2-norbornyl cation demonstrates the presence of an activated partial double bond C1C2 (1.409Å) through interaction with the C1C7 bond (1.671Å) and not or to a minor extent by the other neighbour C1C6 bond (1.552Å). Therefore the relaxation into the symmetrical bridged configuration takes place via the C1C6 bond electrons directed to an activated C1C2 bond. The activated C1C2 bond transfers this electron donation to the C1C7 (C2C3) bond (1.506Å [15] ; X-ray value 1.495Å [13] ) under increase of the C1C6 bond (1.825Å [13] , 1.829Å [15] ; X-ray value 1.802Å [13] ) and the straightforward reduction of the C2-C6 distance from 2.508Å to the relaxed value of 1.829Å.
2.3. Hydrogen and Fluor Transitions as Divalent Atoms in a Three-Center Configuration with Experimental Data and ab Initio Calculation
The hydrogen transfer reactions for linear three-center identity proton, hydrogen atom, and hydride anion (C-H…C) are also connected with the ratio numbers 1.333, 1.250, and 1.167, respectively. Generally proton exchange reactions occur in a linear mode. Deviations are found for identity hydride transfer going from isobutane to methane (CH3)3CH 1.156 (180˚), (CH3)2CHH 1.145 (165.8˚), CH3CH2H 1.141 (122.4˚), CH3H 1.135
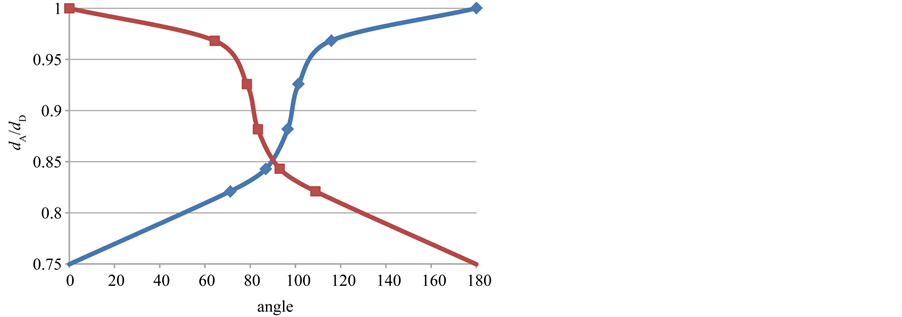
Figure 7. A sigmoidal curve for dA/dD versus the angle between donor and acceptor bond. The various bond lengths and angles are given in Table 1. The blue curve is anti clockwise and the red curve is clockwise. For a perfect sigmoidal curve we interchange the angles for dA/dD = 0.88(2).
(103.2˚) [2] . For the hydrogen atom transfer for methane the ratio number is 1.235, corresponding with a linear mode [2] . From a more general view it is clear that reduction of the number of electrons involved in the threecenter bonding may result in a geometric change of the CHC angle. Closely related to the hydride anion exchange are the halonium ions, in which the formal positively charged halogens are linked with carbons. Most of the halonium ions except for fluor has been prepared as stable dialkyl chloro-, bromo-, and iodohalonium ions as long-lived ions. Theoretically, it has been calculated for (CH3)2X+ that the C-C distances are 2.66Å (X = F), 2.94Å (X = Cl), 3.10Å (X = Br), and 3.33 Å (X = I) and the corresponding CXC angels are 120.2˚ (X = F), 105.0˚ (X = Cl), 101.4˚ (X = Br), and 97.7˚ (X = I) [20] [21] . In excellent correspondence with these results is the recent chemical and theoretical evidence for a symmetrical fluoronium ion in solution [22] . This ion can be described as a divalent fluorine within a C2v symmetric polycyclic cage compound with interlocking fiveand six-membered cyclic rings. For the three-center C-F-C bond it was calculated at the B3LYP/6-311++G level that the C-F and C-C bond lengths are 1.60Å and 2.70Å, respectively, with a CFC angle of 115.0˚. Although there is a perfect match with (CH3)2F cation, then the question arises what determines the exclusive stability of this fluoronium cation. The CFC angle in the artificial complex is somewhat smaller than in (CH3)2F+. Apparently, the decrease in CXC angle going from F to I favours this stability. Dissociation within the cage of the fluoronium ion into 7-norbornyl cation and the remaining C-F compound linked with each other, seems very unlikely. Since 7-norbornyl cation can be considered as a classical cation, the reactive center does not change its position [23] . In the case that we are dealing with a nonclassical ion then the C7 position moves away from the F-C bonding. It may be concluded that the structure can be described as a resonance contribution C+ F-C ↔ C-F +C with a F− exchange conform fluorine’s high electronegativity. As mentioned, this configuration is related the corresponding hydride transfer between carbenium ions. For that transfer a linear relation is found between R(d) and the CHC angle in the transition state: 103 × R(d) = 0.2276 × (CHC) + 1111.70 with R2 = 0.9967 [2] . Using for the tetrahedral C-F bond 1.406Å (MP2/6-31++G(d, p)), the R(d) value for the fluoronium ion is 1.138 that results in CHC angle of 115.50˚. The correspondence between both reactions seems a realistic point of view. Although the geometry of the fluoronium ion theoretically can be described as a real minimum, the C-F-C angle of 120.2˚ is apparently the borderline transition for dissociation in C+ and F-C. Suggesting an axis of symmetry by rotation of one of the C-F bonds around the other imaginary extended bond results in 59.8˚ [24] . The corresponding angles of the other halonium ions are much greater with an increase in the direction of the iodonium ion. This may be an interesting approach for the geometry of dissociative molecular complexes and their corresponding (in) stability.
3. Conclusions
Three-center four-, three-, and two-electron systems based on carbon-, boron-, hydrogen-, and halogen exchange were considered. For a linear three-center configuration, using the dynamics of a regular tetrahedron by changing its geometry in a trigonal bipyramidal transition complex, the bond lengths in the different states of the corresponding reaction profile can be predicted. All transfer or exchange reactions share the same ratio of the bond distances compared with its initial state. With X-ray data of model systems it was even possible to give the ratio numbers for a three-center four-electron configuration experimental evidence with additional ab initio data. Reactions based on a three-center two-electron configuration mostly follow a nonlinear transition. The molecular dynamics of such a process is discussed using recent X-ray crystallographic data of the symmetrically bridged, nonclassical geometry of the 2-norbornyl cation as has been published by Scholz et al. In the present paper the focus is aimed at the transition intermediate of the (classical) 2-norbornyl cation involved in the isomerization into the nonclassical geometry. This model description is compared with a simple molecular rearrangement of the 1-propyl cation into the corner-protonated cyclopropane using the ab initio data. The exclusivity of the former isomerization compared with the latter one could be demonstrated by the observation that the intramolecular electron shift can be expressed in a linear relationship between the concerned electron-donating and accepting bond lengths. Fluor transitions are described as divalent atoms in a three-center two-electron configuration. The role of fluor in comparison with the other halogens is striking. The attention was focused on an excellent correspondence between the recent chemical and theoretical evidence for a symmetrical fluoronium ion in solution.
Finally, the described molecular model inspired by Van’t Hoff has been tested on activated epigenetic systems in molecular biology and on (frozen) transition complexes in basic fundamental reactions in physical chemistry. The applied calculations are taken from the various references.
References
- Gunbas, G. and Mascal, M. (2013) Extraordinary Modes of Bonding Enabled by the Triquinane Framework. The Journal of Organic Chemistry, 78, 9579-9583. http://dx.doi.org/10.1021/jo401715s
- Buck, H.M. (2008) A Combined Experimental, Theoretical, and Van’t Hoff Model Study for Identity Methyl, Proton, Hydrogen Atom, and Hydride Exchange Reactions. Correlation with Three-Center Four-, Three-, and Two-Electron Systems. International Journal of Quantum Chemistry, 108, 1601-1614. http://dx.doi.org/10.1002/qua.21683
- Buck, H.M. (2010) A Linear Three-Center Four Electron Bonding Identity Nucleophilic Substitution at Carbon, Boron, and Phosphorus. A Theoretical Study in Combination with Van’t Hoff Modeling. International Journal of Quantum Chemistry, 110, 1412-1424. http://dx.doi.org/10.1002/qua.22252
- Buck, H.M. (2011) A Model Investigation of Ab Initio Geometries for Identity and Nonidentity Substitution with ThreeCenter Fourand Three-Electron Transition States. International Journal of Quantum Chemistry, 111, 2242-2250. http://dx.doi.org/10.1002/qua.22529
- Buck, H.M. (2012) Mechanistic Models for the Intramolecular Hydroxycarbene-Formaldehyde Conversion and Their Intermolecular Interactions: Theory and Chemistry of Radicals, Mono-, and Dications of Hydroxycarbene and Related Configurations. International Journal of Quantum Chemistry, 112, 3711-3719. http://dx.doi.org/10.1002/qua.24127
- Buck, H.M. (2013) An Adjusted Model for Simple 1,2-Dyotropic Reactions. Ab Initio MO and VB Considerations. Open Journal of Physical Chemistry, 3, 119-125. http://dx.doi.org/10.4236/ojpc.2013.33015
- Yamashita, M., Yamamoto, Y., Akiba, K., Hashizume, D., Iwasaki, F., Takagi, N. and Nagase, S. (2005) Syntheses and Structures of Hypervalent Pentacoordinate Carbon and Boron Compounds bearing an Anthracene Skeleton. Elucidation of Hypervalent Interaction Based on X-Ray Analysis and DFT Calculation. Journal of the American Chemical Society, 127, 4354-4371. http://dx.doi.org/10.1021/ja0438011
- Akiba, K., Moriyama, Y., Mizozoe, M., Inohara, H., Nishii, T., Yamamoto, Y., Minoura, M., Hashizume, D., Iwasaki, F., Takagi, N., Ishimura, K. and Nagase, S. (2005) Synthesis and Characterization of Stable Hypervalent Carbon Compounds (10-C-5) Bearing a 2,6-bis(p-Substituted Phenyloxymethyl)Benzene Ligand. Journal of the American Chemical Society, 127, 5893-5901. http://dx.doi.org/10.1021/ja043802t
- Kawachi, A., Tani, A., Shimada, J. and Yamamoto, Y. (2008) Synthesis of B/Si Bidentate Lewis Acids, o-(Fluorosilyl)(Dimesitylboryl)Benzenes, and Their Fluoride Ion Affinity. Journal of the American Chemical Society, 130, 4222- 4223. http://dx.doi.org/10.1021/ja710615r
- Hartman, J.S. and Shoemaker, J.A.W. (2001) Chelated Fluoroboron Cations. III. Spectroscopic Evidence for Ring Size and Steric Limitations to Chelate Formation by Amine Chelating Donors. Canadian Journal of Chemistry, 79, 426- 436. http://dx.doi.org/10.1139/v01-030
- Hartman, J.S., Shoemaker, J.A.W., Janzen, A.F., Ragogna, P.J. and Szerminski, W.R. (2003) The Coordination Chemistry of
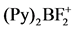 and Related Difluoroboron Cations. Journal of Fluorine Chemistry, 119, 125-139. http://dx.doi.org/10.1016/S0022-1139(02)00224-5
and Related Difluoroboron Cations. Journal of Fluorine Chemistry, 119, 125-139. http://dx.doi.org/10.1016/S0022-1139(02)00224-5 - Clark, E.R. and Ingleson, M.J. (2013) [(Acridine)BCl2]+: A Borenium Cation That Is a Strong Boronand Carbon-Based Lewis Acid. Organometallics, 32, 6712-6717. http://dx.doi.org/10.1021/om400463r
- Scholz, F., Himmel, D., Heinemann, F.W., Schleyer, P.V.R., Meyer, K. and Krossing, I. (2013) Crystal Structure Determination of the Nonclassical 2-Norbornyl Cation. Science, 341, 62-64. http://dx.doi.org/10.1126/science.1238849
- Radom, L., Pople, J.A., Buss, V. and Schleyer, P.V.R. (1972) Molecular Orbital Theory of the Electronic Structure of Organic Compounds. XI. Geometries and Energies of C3H7+ Cations. Journal of the American Chemical Society, 94, 311-321. http://dx.doi.org/10.1021/ja00757a001
- Schreiner, P.R., Severance, D.L., Jorgensen, W.L., Schleyer, P.V.R. and Schaefer III, H.F. (1995) Energy Difference between the Classical and the Nonclassical 2-Norbornyl Cation in Solution. A Combined Ab Initio-Monte Carlo Aqueous Solution Study. Journal of the American Chemical Society, 117, 2663-2664. http://dx.doi.org/10.1021/ja00114a037
- Schleyer, P.V.R. and Sieber, S. (1993) The Classical 2-Norbornyl Cation Rigorously Defined Ab Initio. AngewandteChemie International Edition, 32, 1606-1608. http://dx.doi.org/10.1002/anie.199316061
- Sieber, S., Schleyer, P.V.R., Vančik, H., Mesić, M. and Sunko, D.E. (1993) The Nature of the 7-Norbornyl Cation and Its Rearrangement into the 2-Norbornyl Cation. Angewandte Chemie International Edition, 32, 1604-1606. http://dx.doi.org/10.1002/anie.199316041
- Chiavarino, B., Crestoni, M.E., Fokin, A.A. and Fornarini, S. (2001) The Protonation of Gaseous Cyclopropane. Chemistry―A European Journal, 7, 2916-2921. http://dx.doi.org/10.1002/1521-3765(20010702)7:13<2916::AID-CHEM2916>3.0.CO;2-0
- Winstein, S. and Trifan, D.S. (1949) The Structure of the Bicyclo[2,2,1]2-Heptyl (Norbornyl) Carbonium Ion. Journal of the American Chemical Society, 71, 2953-2953. http://dx.doi.org/10.1021/ja01176a536
- Olah, G.A., Rasul, G., Hachoumy, M., Burrichter, A. and Surya Prakash, G.K. (2000) Diprotonated Hydrogen Halides (H3X2+) and Gitonic Protio Methyland Dimethylhalonium Dications (CH3XH22+ and (CH3)2XH2+): Theoretical and Hydrogen-Deuterium Exchange Studies. Journal of the American Chemical Society, 122, 2737-2741. http://dx.doi.org/10.1021/ja994044n
- Olah, G.A., Surya Prakash, G.K. and Rasul, G. (2013) Study of the Fluoroand Chlorodimethylbutyl Cations. Proceedings of the National Academy of Sciences of the United States of America, 110, 8427-8430. http://dx.doi.org/10.1073/pnas.1306252110
- Struble, M.D., Scerba, M.T., Siegler, M. and Lectka, T. (2013) Evidence for a Symmetrical Fluoronium ion in Solution. Science, 340, 57-60. http://dx.doi.org/10.1126/science.1231247
- Werstiuk, N.H. (2007) 7-Norbornyl Cation Fact or Fiction? A QTAIM-DI-VISAB Computational Study. Journal of Chemical Theory Computation, 3, 2258-2267. http://dx.doi.org/10.1021/ct700176d
- Buck, H.M. (2004) Quantum Chemical Study of Alternating N/B and N/C π Bonds in (Un)Charged Even-Membered 4n and 4n + 2 Cyclic Systems and Their Related Open Systems. International Journal of Quantum Chemistry, 101, 73- 83. http://dx.doi.org/10.1002/qua.20193