Open Journal of Physical Chemistry
Vol.06 No.01(2016), Article ID:61872,20 pages
10.4236/ojpc.2016.61001
Effect of Surface Site on the Spin State for the Interaction of NO with Pd2, Rh2 and PdRh Nanoparticles Supported at Regular and Defective MgO(001) Surfaces
S. Abdel Aal
Department of Chemistry, Faculty of Science, Benha University, Benha, Egypt

Copyright © 2016 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 18 July 2013; accepted 11 December 2015; published 14 December 2015

ABSTRACT
An attempt has been made to analyze the effect of surface site on the spin state for the interaction of NO with Pd2, Rh2 and PdRh nanoparticles that supported at regular and defective MgO(001) surfaces. The adsorption properties of NO on homonuclear, Pd2, Rh2, and heteronuclear transition metal dimers, PdRh, that deposited on MgO(001) surface have been studied by means of hybrid density functional theory calculations and embedded cluster model. The most stable NO chemisorption geometry is in a bridge position on Pd2 and a top configuration of Rh2 and PdRh with N-down oriented. NO prefers binding to Rh site when both Rh and Pd atoms co-exist in the PdRh. The natural bond orbital analysis (NBO) reveals that the electronic structure of the adsorbed metal represents a qualitative change with respect to that of the free metal. The adsorption properties of NO have been analyzed with reference to the NBO, charge transfer, band gaps, pairwise and non- pairwise additivity. The binding of NO precursor is dominated by the
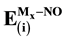 pairwise additive components and the role of the support was not restricted to supporting the metal. The adsorbed dimers on the MgO surface lose most of the metal-metal interaction due to the relatively strong bond with the substrate. Spin polarized calculations were performed and the results concern the systems in their more stable spin states. Spin quenching occurs for Rh atom, Pd2, Rh2 and PdRh complexes at the terrace and defective surfaces. The adsorption energies of the low spin states of spin quenched complexes are always greater than those of the high spin states. The metal-support and dimer-support interactions stabilize the low spin states of the adsorbed metals with respect to the isolated metals and dimers. Although the interaction of Pd, Rh, Pd2, Rh2 and PdRh particles with Fs sites is much stronger than the regular sites O2−, the adsorption of NO is stronger when the particular dimers are supported on an anionic site than on an Fs site of the MgO(001). The encountered variations in magnetic properties of the adsorbed species at MgO(001) surface are correlated with the energy gaps of the frontier orbitals. The results show that the spin state of adsorbed metal atoms on oxide supports and the role of precursor molecules on the magnetic and binding properties of complexes need to be explicitly taken into account.
pairwise additive components and the role of the support was not restricted to supporting the metal. The adsorbed dimers on the MgO surface lose most of the metal-metal interaction due to the relatively strong bond with the substrate. Spin polarized calculations were performed and the results concern the systems in their more stable spin states. Spin quenching occurs for Rh atom, Pd2, Rh2 and PdRh complexes at the terrace and defective surfaces. The adsorption energies of the low spin states of spin quenched complexes are always greater than those of the high spin states. The metal-support and dimer-support interactions stabilize the low spin states of the adsorbed metals with respect to the isolated metals and dimers. Although the interaction of Pd, Rh, Pd2, Rh2 and PdRh particles with Fs sites is much stronger than the regular sites O2−, the adsorption of NO is stronger when the particular dimers are supported on an anionic site than on an Fs site of the MgO(001). The encountered variations in magnetic properties of the adsorbed species at MgO(001) surface are correlated with the energy gaps of the frontier orbitals. The results show that the spin state of adsorbed metal atoms on oxide supports and the role of precursor molecules on the magnetic and binding properties of complexes need to be explicitly taken into account.
Keywords:
Surface Reactions, NO, Bimetallic Nanoparticles, Spin State and Charge Transfer

1. Introduction
Fundamental understanding of the electronic structure and activity of transition metal atoms and nanoclusters supported on metal-oxide surfaces is of great interest due to their broad applications in catalysis, coating for thermal applications, corrosion protection, and other technologically important fields [1] -[4] . Theoretical calculations have proved very helpful to gain insight into the mechanisms of growth of nanoclusters on oxide surfaces [5] [6] . It has been found that under typical conditions, formation of dimers constitutes the first step in the process of the growth of metal clusters on the oxide surface [7] . Even though in the gas phase there are dimers, trimers, etc., the cluster growth on the surface of the support is dominated by diffusion of adsorbed atoms and not by direct deposition of already existing gas phase clusters. It is observed that, diffusion is stopped at point defects, where the atoms are more strongly bound and nucleation takes place [8] . In addition, the properties of the deposited nanoclusters depend on the oxide substrate and in particular on the presence of point defects where the cluster can be stabilized. In general, there has been a consensus that defects not only can act as catalytic centers for chemisorption of small species but also as nucleation centers for growing metal clusters and can modify the catalytic activity of these adsorbed metal particles via the metal-support interaction at the interface [9] .
The strength of interaction between metal and substrate is due to metal-substrate covalent bonding that implies a polarization of the metal orbitals or redistribution of the atomic orbital population. The metal s-orbital combines with the oxygen p-orbital perpendicular to the surface of an oxide material resulting in a bonding (occupied) and antibonding (unoccupied) combinations. This leads to a decrease in the atomic population of the metal atom [9] - [11] . When the free metal atom electronic configuration is dns2, the resulting electronic configuration of the metal and atom may be expressed as dn+1s1 or even dn+2. The strength of the metal-oxide interaction varies with the resulting d-population. This change in the electronic configuration of the adsorbed metal may result in a concomitant spin quenching with respect to the ground state multiplicity of the isolated metal atom.
On the basis of the performance of different density functionals, Markovits et al. [12] reported that the electronic state of Ni with the oxygen regular and defective sites of MgO is the result of a balance between the tendency of Hund’s rule to preserve the atomic state and chemical covalent terms tending to form chemical bonds and hence to quench the atomic magnetic moment. Indeed, the stronger the interaction, the smaller the difference between the high and low spin states; in other words, the larger the interaction, the stronger the spin quenching.
Sousa et al. [13] calculated the low to high spin transition energy of Ni adsorbed on regular and defective sites of MgO and magnetic properties of first row transition metal oxides. The previous investigations suggest that the final spin state of an adsorbed metal can be different, when it interacts with an oxide support. However, the combined effect of oxide support and adsorbed species, such as NO on the final spin state is overlooked.
Bimetallic nanoparticles may create a synergistic catalytic effect that involves the change in local electronic properties of pure metal nanoparticles to modify the strength of the surface adsorption for oxygen reduction reactions [14] [15] . Although pure Pd and Rh clusters on MgO(001) and TiO2 have been widely studied [16] [17] , no reports are available on the geometrical and electronic structure of PdRh bimetallic that deposited on MgO surface. Efforts have been focused on the possibility of associating Pd with another noble metal, rhodium, to prepared bimetallic Pd-Rh/alumina catalysts and compared to reference Pd/alumina and Rh/alumina solids [18] . A. M. Ferrari [19] , Shinkarenko et al. [20] , Neyman and Illas [21] , Nasluzov et al. [22] , and Matveev et al. [23] have experimentally and theoretically studied the adsorption properties of different metal atoms and metal clusters deposited on the MgO (001) surface. Palladium and rhodium clusters of small size have been extensively studied at various semiempirical and ab initio levels of the theory by G. Berthier [24] . As the smallest cluster, homonuclear and heteronuclear transition metal dimers have been studied both experimentally and theoretically [25] [26] . For a systematic theoretical study, the homonuclear dimers of 4d transition metals were examined by use of diverse density functional methods [27] . The structures of AgPd clusters supported on MgO(001) are investigated via a combination of global optimization searches within an atom-atom potential model and density-functional calculations [28] . The reactions of H2 with the heteronuclear dimers PdCu, PdAg, PdAu have been studied by the hybrid density functional method B3LYP [29] . CO adsorption on monometallic and bimetallic Au-Pd nanoparticles deposited onto well-ordered thin films of Fe3O4(111), MgO(001), and CeO2(111) were studied by [30] .
It is frequently observed that a transition metal atom doped in a small cluster of other metal can strongly change the properties of the host cluster [31] [32] . Previous theoretical calculations have been devoted to the study of heteroatomic or impurity-doped as well as homoatomic metal clusters, which indicate that the impurity atoms can strongly influence geometric, electronic, and bonding properties of mixed clusters [33] . The first objective of this work is to generalize the possibility that electron-rich MgO surface can be used to determine how the substrate could affect the structural, energetic and electronic properties of small bimetallic Rh-Pd dimers that belonging to a completely different valence structure, i.e. Rh(4d8 5s1) with unfilled d and Pd(4d10 5s0) with complete d shell. For this purpose, the simplest bimetallic particle, PdRh, is considered and the results are compared with monometallic Pd2 and Rh2 dimers. Second, to clarify the roles of defects as nucleation centers for the formation of dimers and represent how these defects can induce modifications in the electronic, geometric and chemical properties of the supported dimers. Third, to identify the bonding mechanism of NO with Pd2, Rh2 and PdRh nanoparticles that supported on regular and defective sites of MgO(001). Finally, to induce qualitatively different changes in the electronic states of the supported particles and on the transition energy required to switch from low spin to high spin state.
The intriguing heterogeneous processes associated with nitric oxide, NO, observed at transition metal and metal-oxide surfaces, are a continuous topic for research. The molecule, which is one of the simplest and most stable radicals, is spontaneously formed in combustion processes at elevated temperatures. Being a major environmental hazard, it is of vital importance to remove NO from the exhaust gases. The reduction of NO by CO on palladium is of practical interest and experimental investigations show that nanosized palladium clusters have significant capacity to catalyze the CO + NO reaction at low temperatures [34] [35] . As one of the key factors to understand the catalytic mechanism, the adsorption behaviors of NO on Pd clusters have been extensively studied [36] [37] . Viñes et al. performed a combined experimental and theoretical study on the adsorption of NO on Pd nanoparticels, using infrared reflection adsorption spectroscopy (IRAS) and calculations based on density functional theory (DFT) [36] .
2. Computational Details and Surface Models
Hybrid density functional theory and embedded cluster models have been extensively employed in the description of the electronic and geometrical structures of Pd2, Rh2 and PdRh particles nucleated on regular and defect sites on the MgO(001) surface [5] [38] [39] . These models have demonstrated to be powerful in the description of the defective and non defective non polar oxide surface [40] . Sousa, et al. [13] used a cluster/periodic comparison within the same computational model (either DFT or HF) for the ionic systems (MnO, FeO, CoO, NiO, and CuO) to establish that embedded cluster models provide an adequate representation. They used a lattice parameter (421 pm) the same as was determined for the bulk, with no surface relaxation or rumpling in the defect- free system. The embedded cluster model considers a finite cluster embedded in the rest of the host crystal, by assuming that the electronic structure in this external region has remained the same as in the defect free system. This approach is adequate in principle, but is computationally demanding and requires an accurate analysis of the energy terms. Its flexibility is moderate and can describe the charged defects [41] .
To represent the substrate, the ionic clusters Mg9O14 and Mg9O13 Fs have been embedded in arrays of point charges. This was done by following an embedding procedure previously reported for alkaline earth oxides [42] . A finite ionic crystal of 292 point charges was first constructed. The Coulomb potentials along the X and Y axes of this crystal are zero by symmetry as in the host crystal. The ±2 charges on the outer shells were then modified, by using a fitting procedure; to make the Coulomb potential at the four central sites closely approximates the Madelung potential of the host crystal, and to make the Coulomb potential at the eight points with coordinates (0, ±R, ±R) and (±R, 0, ±R) where R is half the lattice distance, which for MgO is 2.105, equal to zero as it should be in the host crystal. With these charges, 0.818566 and 1.601818, the Coulomb potential in the region occupied by the central ions is very close to that in the unit cell of the host crystal. The Coulomb potential was calculated to be (1.748) at the four central sites (compared with 1.746 for a simple cubic ionic crystal) and (0.0) at the previously defined eight points (compared with 0.0 for the same crystal). All charged centers with cartesian coordinates (±X), (±Y) and (Z > 0) were then eliminated to generate the (001) surface of MgO with 176 charged centers occupying the three dimensional space (±X), (±Y) and (Z ≤ 0). The clusters were then embedded within the central region of the crystal surface, and the electrons of the embedded clusters were included in the Hamiltonians of the ab initio calculations. Other crystal sites entered the Hamiltonian either as full or partial point charges as demonstrated in [42] .
The density functional theory calculations were performed by using Becke’s three parameter exchange functional B3 with LYP correlation functional [43] [44] . The B3LYP hybrid functional has been used since it provides a rather accurate description of the metal/oxide interaction [45] . Moreover, for the magnetic systems it provides a reasonable albeit not perfect picture which lies midway between the HF and pure GCA descriptions [46] . Even if the DFT has well known problems with the description of magnetic properties, hybrid functionals such as B3LYP provides a fair indication of the relative energies. B3LYP correctly reproduce the thermochemistry of many compounds including transition metal atoms [47] and seems to be able to properly describe the band structure of insulators [12] . B3LYP ensures a correct description of the electronic ground state of first row transition metal atoms and a reasonable description of the energy difference between low lying electronic states with different spin multiplicity. Finally, B3LYP is able to describe magnetic coupling in systems with localized spins although the magnetic coupling constant is too large [48] .
The Stevens, Basch and Krauss Compact Effective Potential (CEP) basis sets [49] [50] were employed in the calculations. In the CEP basis sets, the double zeta calculations are referred to as CEP-31G, and similarly triple zeta calculations to as CEP-121G. It may be noted that there is only one CEP basis set defined beyond the second row, and the two basis sets are equivalent for these atoms. These basis sets have been used to calculate the equilibrium structure and spectroscopic properties of several molecules and the results are compared favorably with the corresponding all-electron calculations [51] . In the present calculations, the effective core potential of the cep-121g basis set was used for all atoms in the clusters.
The defect free surfaces exhibit very small relaxations only and therefore they have been kept fixed when studying deposition of metal atoms. A minimal energy search on a defect free surface does not usually include surface relaxation since this is experimentally very small, less than 5% [52] . Surface relaxation effects can be significant if discontinuities, like steps or point defects, are present [53] . Sousa et al. [13] focused in the problems when applying DFT methods to open-shell systems with particular emphasis on the consequences on the description of magnetic properties. They found for ionic systems with unfilled d- shells, such as the present Rh atom, that the resulting open-shell electrons are localized and hence it is possible to model these systems by means of embedded cluster models. All calculations are of spin unrestricted type and carried out by using Gaussian 98 system [54] . The figures were generated by using the corresponding Gauss View software.
The binding energy, Ea, of the Pd2, Rh2 and PdRh dimers at various sites of the metal oxide surface can be calculated as follows:
 (1)
(1)
Positive values of the binding energies mean that the formed dimers are stable.
The high to low spin transition energies were calculated from the relation
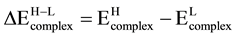 (2)
(2)
where Ecomplex is the total electronic energy of the complex.
The nucleation energy (Enucl), dimer formation energy, is an important parameter to study the atom-by-atom growth of a particle from atoms in the gas phase. It is defined as the energy associated with the formation of homonuclear dimers Rh2,Pd2, and heteronuclear dimers, PdRh, when an atom of the gaseous phase, Rh or Pd bonds with a pre-adsorbed metallic particle, Rh/MgO_site or Pd/MgO_site [54] , respectively:
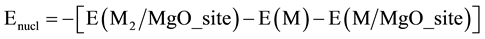 (3)
(3)
where MgO_site indicates the nucleation site. These two quantities, Ea (M2) and Enucl., measure the binding energy of gas phase Pd2, Rh2 and PdRh to a given MgO site [55] .
The dimer binding energy, Eb, measures the stability of the adsorbed dimer with respect to Pd and Rh adatoms, where one of which is bound on a five coordinated terrace anion, O5c. Eb is simply the difference between the adsorption energy of transition metal, TM, atom to the supported TM/MgO and the binding energy of the atom to the metal oxide terrace.
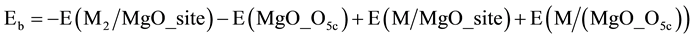 (4)
(4)
The trapping energy, Et, measures the energy gain when Pd and Rh atoms move from a terrace site to a strongly binding site, anion vacancy. The trapping energy is the difference in Ea between a regular and a defect site. Et can be quite large on some specific defects, indicating their strong tendency to capture metal atoms. Thus, metal atoms have a high probability to find a defect in the diffusion process and to stick to this defect [56] .
3. Results and Discussion
3.1. Adsorption of Single Pd and Rh Atoms
It was well established that small metal particles adsorb preferentially on sites where negative charge accumulates [6] [39] [57] [58] ; more specifically the O2− ionic sites for regular and the Fs centers for defective metal oxide. Experimentally, metal clusters are often formed on a surface by exposing it to a beam of gas-phase atoms. These atoms adsorb onto the surface and diffuse to the sites at O2− or Fs centers. From these single adsorbed atoms, clusters are formed by nucleation. For this reason, the adsorption of a single Pd and Rh atoms on both O2− and Fs of MgO is investigated as a first step in this study. The results provide a clear indication that the atomic Rh adsorbs more strongly on both sites than that of Pd. The interaction of Rh and Pd atom on F site is characterized by stronger binding energy (Eads = 3.263 eV, 3.186 eV) with shorter equilibrium adsorption distance (1.62 Ǻ, 1.54 Ǻ) than on the surface O2− site (Eads = 1.301 eV, 1.251 eV) with equilibrium distance (2.06Ǻ, 2.19 Ǻ), respectively. The presence of trapped electrons at the defect site results in a more efficient activation of the supported Pd and Rh atoms. These results are in agreement with [5] [59] [60] . In general, a good agreement is established between the geometrical parameters obtained in this work and the reported theoretical values for Rh/MgO(001) surface (2.09 Å [61] ) and for Pd/MgO(001) (2.15 Å [62] ) at the low coordinated surface.
In addition, it is not a trivial task to conclude a priory which one of the 4F (4d8s1) and 2D (d9) states of Rh determines the ground state energy of the unit cell of MgO(001) surface with the adsorbed Rh atom. Therefore, the effect of the substrate on the electronic states of the adsorbate and the energy required to switch from high-spin to low-spin state are analyzed. By using the B3LYP calculation, high- to low-spin transition energies of Rh atoms free,
 , and supported on O2− and Fs sites of MgO (001),
, and supported on O2− and Fs sites of MgO (001),
 , are summarized in Table 1. Since
, are summarized in Table 1. Since
 is negative value, the spin-polarized structure with one unpaired electron is the most stable state, in agreement with [62] . Consequently, the interaction of Rh at MgO (001) surface induces a quenching of the magnetic moment, which results in a doublet ground state, separated by 0.267 eV from the lowest quartet. Whereas, upon interaction with O2− and Fs surface sites, the high to low spin transition energies
is negative value, the spin-polarized structure with one unpaired electron is the most stable state, in agreement with [62] . Consequently, the interaction of Rh at MgO (001) surface induces a quenching of the magnetic moment, which results in a doublet ground state, separated by 0.267 eV from the lowest quartet. Whereas, upon interaction with O2− and Fs surface sites, the high to low spin transition energies
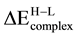 of Pd atom is positive indicating that the spin states are preserved and the low spin states are favored. Hence, the number of unpaired electrons in the Pd adatom tends to be the same as in the gas phase and the ground state of Pd?MgO is spin singlet. The interaction of Rh and Pd atoms with the FS center merits a separate discussion since results show that in almost all cases
of Pd atom is positive indicating that the spin states are preserved and the low spin states are favored. Hence, the number of unpaired electrons in the Pd adatom tends to be the same as in the gas phase and the ground state of Pd?MgO is spin singlet. The interaction of Rh and Pd atoms with the FS center merits a separate discussion since results show that in almost all cases
 exhibits the largest increase. Hence, it is clear that, the low-spin state is more favored because of the formation of a direct bond between the adsorbed species and the electronic levels corresponding to the oxygen vacancy electrons.
exhibits the largest increase. Hence, it is clear that, the low-spin state is more favored because of the formation of a direct bond between the adsorbed species and the electronic levels corresponding to the oxygen vacancy electrons.
Nevertheless, the important point here is the trend of the adsorbed atoms from one site to another. The analysis of these results clearly show that, there is a change in the transition energy required to switch from high spin to low spin,
 , which is induced by the MgO (001) surface. It is observed that the transition energy from the high-spin to the low-spin state increases when the oxide support is present. This is a clear indication that the metal-support interaction tends to stabilize the low-spin state with respect to the isolated atom. The difference in the equilibrium distance perpendicular to the surface was calculated. Notice that, as expected, there is an inverse correlation between adsorption energy and equilibrium distance, the larger the former the shorter the later, Table 1.
, which is induced by the MgO (001) surface. It is observed that the transition energy from the high-spin to the low-spin state increases when the oxide support is present. This is a clear indication that the metal-support interaction tends to stabilize the low-spin state with respect to the isolated atom. The difference in the equilibrium distance perpendicular to the surface was calculated. Notice that, as expected, there is an inverse correlation between adsorption energy and equilibrium distance, the larger the former the shorter the later, Table 1.

Table 1. Transition energy,
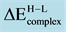 , required to excite adsorbed monomer, Pd and Rh from the high- to low-spin state. A positive sign indicates that the ground state is provided by the low-spin coupling.
, required to excite adsorbed monomer, Pd and Rh from the high- to low-spin state. A positive sign indicates that the ground state is provided by the low-spin coupling.
 are the change in the equilibrium distances of Pd and Rh atoms that supported to the regular (O2−) and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d is larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin. Ne: number of unpaired electrons.
are the change in the equilibrium distances of Pd and Rh atoms that supported to the regular (O2−) and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d is larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin. Ne: number of unpaired electrons.
There are some differences in the adsorption heights between the lowest spin state and the highest spin state of the adsorbed Rh and Pd atoms on both the O5c and oxygen vacancy sites. In particular, for the perfect surface, the adsorption height of 2.54Åand 2.39 Å for the quartet Rh and triplet Pd that is larger by 0.48 Å and 0.2Å than for the doublet Rh and singlet Pd, respectively. The similar phenomena for the vacancy surface are also observed. This observation may result from the larger overlaps between the highest occupied molecular orbital, HOMO, of MgO cluster and the lowest unoccupied molecular orbital, LUMO, of the adsorbed metal atoms in the doublet and singlet state than those in the quartet and triplet states of the adsorbed Rh and Pd atoms that have anti-bonding character as shown from Figure 1. Therefore, a short bond distance and a strong interaction between the O2− and Fs sites and the Rh and Pd atoms at the lowest spin state have been observed (Table 1).
The increase in the adsorption heights of supported Pd and Rh can contribute to the Pauli repulsion of the valence s orbital of the Pd and Rh that is almost empty with those in the p orbitals of the surface oxygen atoms. As well as, the HOMO for the oxygen vacancy has a large s-like character, which would also lead to repulsive interaction with the metal s orbital; this orbital is occupied by ~−0.8e due to the charge transfer. The adsorption heights of supported Pd and Rh increased at oxygen anion and oxygen vacancy sites also the energy gain of 1.90 eV and 2.0 eV due to the electron occupying this bonding orbital of the Pd and Rh atom, where the binding energy is calculated to be 3.192 eV and 3.263 eV for the adsorption complexes Pd/Mg9O13Fs and Rh/Mg9O13 Fs, respectively.
It is interesting to explore the electronic configuration for the interaction of Pd and Rh atoms with the regular site at MgO(001) surface. The only appreciable change with respect to the free atom is the hybridization between 5s and 4d orbitals with negligible contribution of the 5p subshell, 5s0.284d9.745p0.016p0.02 and 5s 0.43 4d 8.6 5p 0.03 5d 0.01 6p 0.01 for the supported Pd and Rh atoms and no appreciable charge transfer, −0.054e and −0.07e, respectively, These results are consistent with [63] . The increased adsorption energy of Pd and Rh atoms with FS centers is accompanied by notable changes in the electronic configuration of the adsorbed metal, which is progressively changed by a charge transfer, −0.799e and −0.807e, with an increasing participation of the 5p orbitals, for the supported Pd and Rh atoms, 5s0.794d9.865p0.14 and 5s 0.83 4d 8.815p 0.13, respectively, Table 1.
As it has been shown later, the interaction of NO with supported Pd and Rh depends strongly on the metal- oxide interaction and it is essential to dispose of an adequate substrate model for the subsequent NO adsorption [64] [65] . Hence, as a first step in our approach, Rh2/MgO(001), Pd2/MgO(001)and PdRh/MgO(001) systems were studied using the above detailed cluster models.
3.2. Rh2, Pd2 and PdRh Particles Deposited on MgO (001)
To underscore the most stable configuration of Rh2, Pd2 and Pd-Rh dimer on the MgO(001) surface, two confi-



 Pd(1)/MgO(O2−) site Pd(1)/MgO(Fs) site Pd(3)/MgO(O2−) site Pd(3)/MgO(Fs) site(a) (b)
Pd(1)/MgO(O2−) site Pd(1)/MgO(Fs) site Pd(3)/MgO(O2−) site Pd(3)/MgO(Fs) site(a) (b)


 Rh(2)/MgO(O2−) site Rh(2)/MgO(Fs) site Rh(4)/MgO(O2−) site Rh(4)/MgO(Fs) site(c) (d)
Rh(2)/MgO(O2−) site Rh(2)/MgO(Fs) site Rh(4)/MgO(O2−) site Rh(4)/MgO(Fs) site(c) (d)
Figure 1. Schematic representation of the highest occupied molecular orbitals (HOMOs) of Pd and Rh atoms with (a) low spin state and (b) high spin state deposited on MgO(O2−) and MgO(Fs) sites using the embedded cluster model.
gurations, parallel and perpendicular to the surface plane have been considered. The best optimized geometries of Rh2, Pd2 and PdRh supported particles anchored on terrace sites of MgO(001) are with the molecular axis almost parallel to the surface and the supported two atoms of the dimer nearly on the top of two O5c anions, Figure 2. The results are agreement with [19] [56] [66] .
Because of the spin polarization, the corresponding values of binding, nucleation, trapping and charges transfer for the deposition of the Rh2, Pd2 and PdRh particles on the regular oxygen site and Fs center have been summarized in Table 2 by using B3LYP/CEP-121G at various spin states in order to find the most stable spin state for each dimer. It is interesting to note that, the ground state of Rh2, Pd2 is singlet, at variance with gas-phase of Pd2 and Rh2 which has a triplet
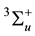 [67] [68] and quintet
[67] [68] and quintet
 [69] ground state, respectively, but in agreement with previous studies [5] [16] . Thus, the interaction with the substrate induces a change in the electronic structure of Pd2 and Rh2 dimers. The adsorption energies of Rh2, Pd2 and PdRh in the low spin states are stronger than those in the high spin states. Hence, the dimer-support interactions stabilize the low spin states of the adsorbed Rh2, Pd2 and PdRh dimers.
[69] ground state, respectively, but in agreement with previous studies [5] [16] . Thus, the interaction with the substrate induces a change in the electronic structure of Pd2 and Rh2 dimers. The adsorption energies of Rh2, Pd2 and PdRh in the low spin states are stronger than those in the high spin states. Hence, the dimer-support interactions stabilize the low spin states of the adsorbed Rh2, Pd2 and PdRh dimers.
From these results, it is observed that the interaction of Rh2, Pd2 and PdRh on Fs site is characterized by stronger binding energy with shorter equilibrium adsorption distance than on the surface O2− site. Although the binding energy is noticeably affected by the support, the nucleation energy is weakly affected; for both sites the values of Enucl are less significantly changed (0.001 - 0.246 eV). This can mean that (a) the regular metal oxide surface is always an appropriated place to form Rh2, Pd2 and Pd-Rh(b) the dimers formation is independent of the adsorption site (regular or diamagnetic Fs site). However the dimer formation will be favored on the Fs center due to the trapping energy, consistently with [3] .
On a terrace site, the addition of second TM atom leads to a nucleation energy Enucl. = 1.716 eV, 2.465 eV, and 2.354 eV that are 0.466 eV, 1.165 eV and 1.053 eV higher than the adsorption energies of the TM atom on a terrace site. Consequently, the dimer formation of Rh2, Pd2 and PdRh are preferred with respect to two isolated atoms adsorbed on O2− anions (Table 2), indicating that dimerization should be possible even on the MgO (001) terraces and the dimer nucleation is a thermodynamic favored process at O2− anions. Although this result is in contrast with the results reported by Bogicevic and Jennison [70] who reported almost no difference in stability between the dimer and two isolated atoms on the MgO (001) terraces, it is agreement with [5] [66] .
The elongation of the Pd-Pd, Rh-Rh and Pd-Rh distances with respect to the gas-phase is explained by the fact that the dimer is oriented towards two nearest neighbor O2− anions on the surface to maximize the bonding with the O2− anions. The Pd-Pd bond length becomes close to 2.98Å, is only 0.22 Å longer than in the free molecule, in agreement with [71] .
Concerning the bimetallic PdRh particle, the ground state geometry of the bimetallic is significantly modified

Figure 2. Frontier orbitals of regular surfaces Mg9O14, free NO molecule, Pd/Mg9O14, Rh/Mg9O14, Pd2/Mg9O14, Rh2/Mg9O14, PdRh/Mg9O14, NO/Pd2/Mg9O14, NO/Rh2/Mg9O14 and NO/PdRh/Mg9O14 at their high and low spin states.
Table 2. Geometrical parameters, binding, nucleation, trapping energies and atomic charges for the adsorption of Pd2, Rh2 and PdRh dimers with various spin multiplicities at regular (O2−) site and defect center (Fs) site of the MgO (001) surface. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units.
q(M1), q(M2): atomic charges at each metal of the dimer. d(M1-S), d(M2-S): optimal distances between adsorbed metals of the dimer and surface site of MgO.
after deposition. The electronic density of states analysis reveals that after deposition, Pd-Rh favors doublet spin multiplicity as the lowest energy configuration, Table 2. In consequence, the binding, nucleation and dimer binding energies for Rh-Rh and Pd-Rh are very close. So that, such a comparison is of interest as many recent experimental studies on the supported palladium model catalytic systems [39] address the question whether the palladium can be used as an alternative to the expensive rhodium in the reaction of reduction of NO [72] .
In this section, the stability trends of the Pd2, Rh2 and PdRh dimers for ground state structures are analyzed in terms of the energy gaps between HOMO and LUMO. A large HOMO-LUMO gap has been considered as an important requisite for the chemical stability of transition metal clusters [68] . The calculated HOMO-LUMO gaps for the ground states of Pd2, Rh2 and PdRh dimers are presented in Table 2. It can be seen from this table that, the calculated HOMO-LUMO gaps of Pd2, Rh2 and PdRh dimers follow the trend Pd2 < Rh2 < PdRh. Since a large energy gap corresponds to higher stability therefore, the PdRh dimers that deposited on the MgO (O2−) and MgO (Fs) sites have the highest chemical stabilities.
Indeed, the molecular orbital, MO, interaction is controlled by the level of the frontier orbitals. Therefore, the relations between spin quenching of supported Rh, Pd, Pd2, Rh2 and PdRh dimers at MgO surface and energy gaps between frontier orbitals are established. In Figure 3, the HOMOs and LUMOs of the supported metals and dimers at the defect free surface of MgO are presented. While spin preservation occurs for Pd complex, spin quenching occurs for Rh, Rh2, Pd2 and RhPd complexes; this is agreement with Tables 1-3. This is clearly correlated with frontier orbital energies where the HOMO energy levels of Rh, Rh2, Pd2 and Pd-Rh low spin complexes are lower than their high spin counterparts. This extra stability allows for stronger interaction with the surface hence, the interaction in this case is strong enough to quench the spin.

Figure 3. Schematic representation of the HOMO’s of Pd2/MgO, Rh2/MgO, PdRh/MgO, NO/Pd/MgO, NO/Rh/MgO, NO/Pd2/MgO, NO/Rh2/MgO and NO/PdRh/MgO deposited on MgO(O2−) and MgO(Fs) sites using the embedded cluster model.
Table 3. Transition energy,
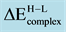 , required to excite the supported dimers, Pd2, Rh2 and PdRh, from the high- to low- spin states. A positive sign indicates that the ground state is provided by the low-spin coupling.
, required to excite the supported dimers, Pd2, Rh2 and PdRh, from the high- to low- spin states. A positive sign indicates that the ground state is provided by the low-spin coupling.
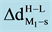 and
and
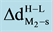 are the change in the equilibrium distance of each TM to the surface;
are the change in the equilibrium distance of each TM to the surface;
 is the change in charges at the dimers that supported to the regular (O2−) site and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d and q are larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin.
is the change in charges at the dimers that supported to the regular (O2−) site and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d and q are larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin.
*a: High spin multiplicity is (5) for Pd2 Rh2 and (6) for PdRh; *b: High spin multiplicity is (3) for Pd2 Rh2 and (4) for PdRh; Where low spin state is (1) for Pd2 Rh2 and (2) for PdRh
3.3. Interaction of NO with Supported Pd, Rh, Rh2, Pd2 and PdRh on MgO (001)
The binding energy for the interaction of NO molecule on different spin states of Pd, Rh, Rh2, Pd2 and PdRh that supported on MgO can be calculated as Ea(NO) = −[E(NO/Mx/MgO_site) − E(Mx/MgO_site) − E(NO)] where x = 1 or 2 [55] . In all cases, the positive values correspond to exothermic processes. It is observed that NO adsorbs much more strongly at the Pd with low spin state and Rh with different spin state that deposited on the MgO(O2−) site than on the MgO(Fs), Table 4. The reason of this behavior will be discussed later.
In Tables 5-7, the adsorption energies, optimal distances and charge transfer for the interaction of NO molecule on different spin states of Rh2, Pd2 and PdRh that supported at MgO (O2−) and MgO (Fs) through its N atom at various spin states have been calculated. Again, the NO molecule adsorbs much more strongly at the Pd2, with low spin state, Rh2 and PdRh with different two spin state that deposited on the MgO (O2−) site than on the MgO (Fs), Table 5. As well as, the NO has been adsorbed at the center of the Pd-Pd bond in a bridge position, which is energetically the most stable in agreement with previous works [37] [65] [73] . The Pd-Pd distance is about 2.98 Å - 3.05 Å, depending on the site considered, Table 5. The large Pd-Pd bond distance of adsorbed Pd2 is in close agreement with the STM measurements [20] . Globally, the binding energy calculated for the adsorption of NO on the palladium dimers,1.808 eV, is in the range of the experimental value that measured in thermal desorption by Ramsier et al. [74] .
At the high spin states a dramatic change is found when the NO which bind to the supported Pd and Pd2 where there is an increase in the binding energies at the MgO (Fs) site than on the MgO (O2−). The different behavior of Pd and Pd2 with high spin states that adsorbed on Fs centers towards NO can be explained as follows. At the low spin states of supported Pd and Pd2 the delocalization of the trapped electrons into the 5 s level leads to an increased Pauli repulsion with the NO molecule and in a strong weakening of the bond. On contrary, at the supported Pd and Pd2 with high spin states this effect is smaller because of the presence of an incomplete d shell. Figure 3 confirmed the results in Table 4 and Table 5 and the adsorption properties that discussed above.
Since the high occupied molecular orbital of NO is π* anti-bonding orbital with an unpaired electron therefore, the charge transferred from Pd2/MgO, Rh2/MgO and PdRh/MgO to the NO will occupy the π* orbital and weak the NO bond strength. The NO bond length is elongated after the adsorption of NO on the particular dimers, this
Table 4. Adsorption properties of NO interacting with regular (O2−) site and oxygen vacancy (Fs) site of the MgO (001) surface supported Pd and Rh atoms at the low and high spin state.
q(M): atomic charges at the adatom. q(NO): molecular charge at NO molecule. d(M-S): optimal distances between adatom and surface site of MgO. d (N-O): Equilibrium N-O distances. d(N-M): optimal distances between adatom and nitrogen atom.
Table 5. Adsorption properties of NO interacting with regular (O2−) site and oxygen vacancy (Fs) site of the MgO (001) surface supported Pd2 dimer at the low and high spin state.
Red spheres: O2−; yellow spheres: Mg2+; light blue spheres: Pd atom; dark blue sphere: N atom.
Table 6. Adsorption properties of NO interacting with regular (O2−) site and oxygen vacancy (Fs) site of the MgO (001) surface supported Rh2 dimer at the low and high spin state.
Red spheres: O2−; yellow spheres: Mg2+; blue spheres: Rh atom; dark blue sphere: N atom.
Table 7. Adsorption properties of NO interacting with regular (O2−) site and oxygen vacancy (Fs) site of the MgO (001) surface supported PdRh dimer at the low and high spin state.
Red spheres: O2−; yellow spheres: Mg2+; light blue spheres: Pd atom; blue spheres: Rh atom; dark blue sphere: N atom.
is consistent with the electron transfer direction [57] , Tables 5-7. The higher electron transfer from Pd2/MgO, Rh2/MgO and PdRh/MgOto NO can explain the larger stretching of the NO interatomic distance on the oxygen anion and oxygen vacancies sites.
The adsorption energy of NO on Rh2/MgO is larger than on Pd2/MgO (2.909 vs. 1.808 eV), possibly due the decrease of the d electrons on the Rh which can lead to a decrease of the σ-σ repulsion. The metal-nitrogen bond is shorter for Rh than Pd (1.74 vs. 2.01 Å), which also indicates a strong bonding between NO and Rhx/MgO. The larger M-N-O angle for Rh2 than Pd2 (180˚ vs. 132.2˚) indicates that the 5σ orbital is much more involved in the adsorption at Rh than Pd. Although, the charge of Rh2NO and Pd2NO supported on the (O2−) and (Fs) site is practically the same than that of the supported Rh2 and Pd2 at the same sites, the MgO(Fs) site acquires a much more significant negative charge, Tables 5-7. These results are confirmed by Figure 3.
Indeed, the different behavior of rhodium and palladium supported at MgO(001) towards NO can be explained as follows. On palladium the delocalization of the trapped electrons into the 5s level leads to an increased Pauli repulsion with the NO molecule and in a strong weakening of the bond, whereas on Rh this effect is smaller because of the presence of an incomplete d shell and the easier mixing of the 5s with the 4d orbitals to form new hybrid orbitals [62] . The high reactivity of Rh relative to Pd was also found by a periodic DFT calculation for NO adsorbed on (001) surfaces of Rh and Pd. Therefore, Rh appears to be more efficient than Pd for NO adsorption at Pd-Rhbimetallic and may justify its use as a catalyst in TWC. These observations agree well with previous theoretical calculations [75] . Therefore, it seems that NO prefers binding to Rh when both Rh and Pd sites co-exist in the bimetallic Pd-Rh. These results are confirmed by Table 7 and Figure 3.
The optimized geometry of admolecule NO with the N-end to the Rh atom of bimetallic PdRh and the molecular axis of NO normal to the surface plane is presented in Table 6. The N-O distance elongates from its value of 1.158 Ǻ for the free NO molecule to 1.207 Ǻ for Rh2 and PdRh at O2− and Fs, respectively. Therefore, the results show that the adsorption energies of NO at Rh-Rh and Pd-Rh, with low spin state, that deposited at O2− and Fs of MgO are very close, (2.909 vs. 2.666 eV) at oxygen anion and (2.971 vs. 2.872 eV) at Fs centers, Table 6 and Table 7. Figure 3 confirmed the results in Tables 2-7 with the adsorption properties discussed above.
Interestingly, the interaction is assumed to mainly be a HOMO-LUMO type [76] . The differences in the adsorption energies reported for the interaction of NO on supported PdRh at the O2− and Fs center can be due to the differences in energy between the HOMO of the surface and the LUMO of the NO molecule. The results show that, the formation of a vacancy on the MgO surface decreases the difference between the HOMO of PdRh supported at MgO (001) surface and LUMO of NO molecule by 0.367 eV. This result is in agreement with the greater strength of NO chemisorption on supported PdRh/MgO(Fs) in comparison with supported PdRh/MgO(O2−), HOMO-LUMO = 2.174 eV and 2.541 eV, respectively. As the interaction of NO with PdRh occurs through a charge transfer from the HOMO of the surface to the LUMO of the adsorbed NO molecule, the smaller the value of the HOMO?LUMO gap the easier the charge transfer and consequently, the larger the adsorption energy, this is agreement with [77] .
Recently, several authors were interested in studying the CO-induced modification of the metal-MgO interaction [78] [79] in the form of atoms and layers. For instance, it was reported that CO enhances the bonding between Pt or Pd atoms and the oxide, but for Au this effect is negligible [78] . To allow a similar analysis on NO, the adsorption of the MNO and M2NO complexes on MgO(001) was considered. The corresponding binding energy can be defined in a similar way as that for Mx on MgO, Ea(MxNO) = −[E(NO/ Mx/MgO_site) − E(MgO_site) − E(MxNO)] where x = 1 or 2. On terrace O5c sites the O5cRh2-NO and O5cPdRh-NO bonds are definitely stronger than the O5c-Rh2NO and O5c-PdRhNO. This means that an increase in temperature can lead to diffusion of the Rh2NO and PdRhNO complexes before NO desorption occurs. Whereas on neutral F centers, the bonding of the Pd2NO, Rh2NO and PdRhNO unit to the surface is so strong that no diffusion of this species occurs once the complex is trapped at an oxygen vacancy. An increase in temperature will result in the loss of NO and TM atoms filling the vacancy. An important result is that, on Fs sites, the Fs-Pd2NO, Fs-Rh2NO and Fs-PdRhNO bonding is stronger than that of the FsPd2-NO FsRh2-NO and FsPdRh-NO, Tables 5-7.
The spin transition energies,
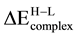 , of ON∙Pd2∙MgO, ON∙Rh2∙MgO and ON∙RhPd∙MgO complexes have been considered, Table 8. The trend emerging from the present model calculations indicates that the metal-support interaction tends to stabilize the low-spin state with respect to the isolated atom thus, the low-spin state becoming the ground state at regular and FS sites. Although, the combined effects of NO adsorbate and Pd2, Rh2 and RhPd supported at MgO were strong enough to quench the spin of Pd2, Rh2 and RhPd dimers (changes the sign of the spin transition energy), the low-spin states are preserved. Although, the interaction of ON.Rh2∙MgO (Fs)
, of ON∙Pd2∙MgO, ON∙Rh2∙MgO and ON∙RhPd∙MgO complexes have been considered, Table 8. The trend emerging from the present model calculations indicates that the metal-support interaction tends to stabilize the low-spin state with respect to the isolated atom thus, the low-spin state becoming the ground state at regular and FS sites. Although, the combined effects of NO adsorbate and Pd2, Rh2 and RhPd supported at MgO were strong enough to quench the spin of Pd2, Rh2 and RhPd dimers (changes the sign of the spin transition energy), the low-spin states are preserved. Although, the interaction of ON.Rh2∙MgO (Fs)
Table 8. Transition energy,
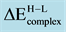 , required to excite ON・Pd2・MgO, ON・Rh2・MgO and ON・PdRh・MgO complexes from the high-to low-spin states. A positive sign indicates that the ground state is provided by the low-spin coupling.
, required to excite ON・Pd2・MgO, ON・Rh2・MgO and ON・PdRh・MgO complexes from the high-to low-spin states. A positive sign indicates that the ground state is provided by the low-spin coupling.
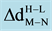 is the change in optimal distances between TM and nitrogen atom.
is the change in optimal distances between TM and nitrogen atom.
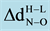 is the change in optimal distances between N and O atom of NO molecule.
is the change in optimal distances between N and O atom of NO molecule.
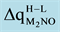 is the change in charges at the M2NO dimer that supported to the regular (O2−) site and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d is larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin.
is the change in charges at the M2NO dimer that supported to the regular (O2−) site and oxygen vacancy (Fs) site at MgO (001) going from high- to low-spin state. A positive value indicates that d is larger in the high-spin state. Energies are expressed in eV, the corresponding equilibrium distances (d) in (Å), and charges in electron units H: High spin. L: Low spin.
where low spin multiplicity is (2) for NOPd2, NORh2 and (1) for NOPdRh high spin multiplicity is (4) for NOPd2 , NORh2 and (3) for NOPdRh.
and ON∙RhPd∙MgO (Fs) shows that the transition energy exhibits the largest increase, the interaction of ON∙Pd2∙MgO (Fs)exhibits the largest decrease. Notice that, as expected, there is an inverse correlation between adsorption energy and equilibrium distance, the larger the former the shorter the later, Tables 5-8. In these cases, it is clear that the low-spin state is more favored because of the formation of a direct bond between the adsorbed transition metal dimer and the electronic levels corresponding to the oxygen vacancy electrons. The results show that the magnetic-spin states of transition metals atoms and clusters supported at metal oxide surface and the role of a precursor molecule on the considered magnetic and binding properties need to be explicitly taken into account.
3.4. Pairwise and Non-Pairwise Additivity
The concept of pairwise and non-pairwise additivity has been studied for atom clusters and insulators [80] -[82] . In studying a supported-metal catalyst system, it is very important to quantify the extent to which the support MgO (S) with regular and defective surface affects the interaction of the NO admolecule with the Pd, Rh, Pd2, Rh2 and PdRh particles. The interaction energy
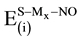 among three subsystems; the support (S), (Mx) where x = 1 for Pd, Rh atom and x = 2 forPd2, Rh2, PdRh dimer, and the adsorbate (NO) molecule can be defined as:
among three subsystems; the support (S), (Mx) where x = 1 for Pd, Rh atom and x = 2 forPd2, Rh2, PdRh dimer, and the adsorbate (NO) molecule can be defined as:
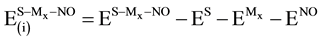 (5)
(5)
where every energy term on the right-hand side of Equation (5) is calculated using geometrical parameters corresponding to the equilibrium geometry of S-Mx-NO systems. The left-hand side represents the energy required to separate the three subsystems without altering any change in their geometrical parameters. Such energy can be divided into contributions from three-pairwise components and a non-additive term, εnadd, as follows:
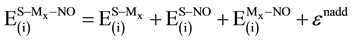 (6)
(6)
where εnadd is a measure of cooperative interactions among the three subsystems [38] [81] . The four energy terms on the right-hand side of Equation (6) are calculated from the relations:
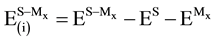 (7)
(7)
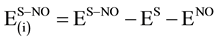 (8)
(8)
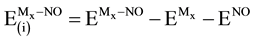 (9)
(9)
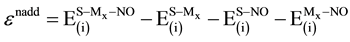 (10)
(10)
The total interaction energies, the pairwise energy components to the S-Mx-NO systems, and the non-additive energy term, εnadd, are presented in Table 9. As shown in this table, the total interaction energies of ON∙Pd∙Mg9O13O2−, ON∙Pd∙Mg9O13Fs, ON∙Rh∙Mg9O13O2−, ON∙Rh∙Mg9O13Fs, ON∙Pd2∙Mg9O13O2−, ON∙Pd2∙Mg9O13Fs, ON∙Rh2∙Mg9O13O2−, ON∙Rh2∙Mg9O13Fs, ON∙PdRh∙Mg9O13O2− and ON∙PdRh∙Mg9O13Fs complexes are dominated by the pairwise additive components
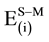 and
and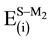 , respectively. On the other hand, the small values of
, respectively. On the other hand, the small values of
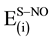 pairwise component that represent the interaction energy between support (S) and admolecule (NO) in the S-Mx-NO system may be attributed to the large separation between (S) and the NO admolecule. This result means that the binding of NO is mainly dominated by the
pairwise component that represent the interaction energy between support (S) and admolecule (NO) in the S-Mx-NO system may be attributed to the large separation between (S) and the NO admolecule. This result means that the binding of NO is mainly dominated by the
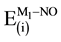 and
and
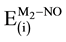 pairwise additive contributions of the considered complexes.
pairwise additive contributions of the considered complexes.
The non additivity term, εnadd, is a measure of cooperative interaction among the subsystems, decreases with surface defect-formation at ON∙Pd∙MgO, ON∙Rh∙MgO, ON∙Pd2∙MgO and ON∙Rh2∙MgO. Except at ON∙PdRh∙MgO complex, εnadd increases with surface defect-formation, Table 9. This suggests that the interaction of NO with Pd, Rh, Pd2, Rh2 and PdRh dimer is essentially affected by defect formation. This confirmed the adsorption properties of NO at supported Pd, Rh, Pd2, Rh2 and PdRh particles that discussed above. Finally, the role of metal oxide is not restricted only to supporting the metal, but influences the interaction of NO molecule with the Pd, Rh, Pd2, Rh2 and PdRh dimers.
4. Conclusions
An attempt has been made to understand the effect of surface site on the spin state for the interaction of NO with Pd2, Rh2 and PdRh nanoparticles that supported at regular and defective MgO (001) Surfaces. A spin-polarized treatment is considered to properly describe the ground-state electronic structure, adsorption energies and the low- to high-spin energy transition. The calculated results are compared with experimental data and previous theoretical studies as possible. The geometrical optimizations have been considered to represent the most stable structures for the adsorption of NO at the supported Pd2, Rh2 and PdRh and to investigate the changes induced by the oxide substrate in the chemisorption properties of the adsorbed particles.
Upon interaction with O2− and Fs surface sites, the high to low spin transition energies of Pd atom is positive indicating that the spin states are preserved, and the low spin states are favored. Hence, the number of unpaired electrons in the adatom tends to be the same as in the gas phase and the ground state of Pd-MgO is spin singlet. However, the main contributions to the Rh atom, Pd2, Rh2 and PdRh at MgO are the polarization of the metal electrons induced by the ionic substrate and the small mixing between the s and d orbitals of the transition metal with the 2p orbitals of the surface oxygen. Consequently, the interaction of Rh atom, Pd2, Rh2 and PdRh dimers at MgO (001) surface induces a quenching of the magnetic moment, which results in a doublet ground state for Rh atom and PdRh as well as a singlet ground state for Pd2 and Rh2 at MgO (001) surface. As a consequence, the formation of the dimer in its singlet state, Rh2 and Pd2, and doublet state, Pd-Rh deposited at MgO (001), is
Table 9. Interaction energies of ON.Pd.MgO, ON.Rh.MgO, ON.Pd2.MgO, ON.Rh2.MgO, ON.PdRh.MgO complexes with the most stable spin states at O2− and Fs sites, pairwise components and non additivity terms. All energies are given in eV.
favored with respect to the presence of two isolated atoms on the surface. Notice that, as expected, there is an inverse correlation between adsorption energy and equilibrium distance, the larger the former the shorter the later. In any case, the extent of metal-metal bonding in supported dimer has been increased compared with the gas-phase unit. This leads to a considerable elongation of the metal-metal bond to maximize the metal-O interaction. Notice that the dimer as a unit adsorbs much more strongly on the MgO (Fs) site than on the MgO (O2−) site. Moreover, the large enhancement in the activity of supported dimers is due mainly to the electron transfer from the cavity to the supported dimers. Theoretical calculations indicate that the formation of Rh2, Pd2 and PdRh dimer on an Fs center is favored by 0.466, 0.962, and 0.807 eV respectively with respect to a TM atom bound at the Fs center and other TM atom on a terrace site. The dimers deposited interact relatively strongly with the substrate oxide forming predominantly covalent bonds with the adsorbed sites. The interaction is not accompanied by a significant charge transfer at the interface. The PdRh bimetallic has larger HOMO-LUMO gap and is relatively more chemically stable than the Pd2 and Rh2 monometallic that deposited on the MgO (O2−). The transition energy,
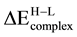 , for the interaction of Pd-Rh with the oxygen anion and FS center exhibits the largest increase, 2.223 eV and 2.222 eV respectively. In these cases, it is clear that the low-spin state is more favored because of the formation of a direct bond between the adsorbed bimetallic and the electronic levels corresponding to the oxygen anion and oxygen vacancy electrons. A molecular-scale understanding of the energetic and mechanisms for formation of such metal dimers on inert oxide surfaces can open new avenues to the design of catalysts with specific functions.
, for the interaction of Pd-Rh with the oxygen anion and FS center exhibits the largest increase, 2.223 eV and 2.222 eV respectively. In these cases, it is clear that the low-spin state is more favored because of the formation of a direct bond between the adsorbed bimetallic and the electronic levels corresponding to the oxygen anion and oxygen vacancy electrons. A molecular-scale understanding of the energetic and mechanisms for formation of such metal dimers on inert oxide surfaces can open new avenues to the design of catalysts with specific functions.
In summary, it seems that NO prefers to bound with Rh atoms when both Rh and Pd site co-exist in the Pd- Rh bimetallic. The electronic structures and N-O bond lengths of the chemisorbed systems are similar for NO∙Rh2∙MgO and NO∙PdRh∙MgO with the top geometries but show significant differences from bridge geometries, NO/Pd2/MgO. Bridge-site adsorption causes the N-O bond to lengthen and soften due essentially to increase an electrostatic repulsion between both N and O atoms. In addition, the NO adsorbs much more strongly at the PdRh that is deposited on the MgO (Fs) than on the MgO (O2−) site.
The transfer of electron charge density from such a defect to a dimer reinforces the metal-metal bonds. Therefore, color centers at the MgO surface not only reduce the diffusion of metal atoms and dimers, but also act as stabilizing agents for the whole structure. This point could be particularly important in the context of identifying methods to stabilize the support particles on an oxide substrate under chemical reaction conditions.
To summarize, the larger interaction of NO at bimetallic PdRh at oxygen anions and oxygen vacancies induces an enhancement of the energy required to switch from high spin to low spin 1.565 eV and 1.923 eV respectively. These results show that the spin state of adsorbed PdRh dimer on oxide supports tends to preserve the number of unpaired electrons as found in the case of the regular terrace sites.
Acknowledgements
My gratitude and deep thanks to Prof. Dr. A.S. Shalabi for his interest, and useful discussions.
Cite this paper
S. AbdelAal, (2016) Effect of Surface Site on the Spin State for the Interaction of NO with Pd2, Rh2 and PdRh Nanoparticles Supported at Regular and Defective MgO(001) Surfaces. Open Journal of Physical Chemistry,06,1-20. doi: 10.4236/ojpc.2016.61001
References
- 1. Piccolo, L. and Henry, C.R. (2000) Reactivity of Metal Nanoclusters: Nitric Oxide Adsorption and CO+NO Reaction on Pd/MgO Model Catalysts. Applied Surface Science, 162-163, 670-678.
http://dx.doi.org/10.1016/S0169-4332(00)00267-1 - 2. Xu, C., Oh, W.S., Liu, G., Kim, D.Y. and Goodman, D.W. (1997) Characterization of Metal Clusters (Pd and Au) Supported on Various Metal Oxide Surfaces (MgO and TiO2). Journal of Vacuum Science & Technology A, 15, 1261.
http://dx.doi.org/10.1116/1.580604 - 3. Florez, E., Mondragón, F., Truong, T.N. and Fuentealba, P. (2007) Density Functional Theory Characterization of the Formation of Copper Clusters on Fs and Centers on a MgO Surface. Surface Science, 601, 656-664.
http://dx.doi.org/10.1016/j.susc.2006.10.040 - 4. Wang, Y., Florez, E., Mondragón, F. and Truong, T.N. (2006) Effects of Metal-Support Interactions on the Electronic Structures of Metal Atoms Adsorbed on the Perfect and Defective MgO(100) Surfaces. Surface Science, 600, 1703-1713.
http://dx.doi.org/10.1016/j.susc.2005.12.062 - 5. Giordano, L., Di Valentin, C., Pacchioni, G. and Goniakowski, J. (2005) Formation of Pd Dimers at Regular and Defect Sites of the MgO(100) Surface: Cluster Model Calculations. The Journal of Chemical Physics, 309, 41-47.
- 6. Inntam, C., Moskaleva, L.A., Neyman, K.M. and Nasluzov, V.A. (2006) Adsorption of Dimers and Trimers of Cu, Ag, and Au on Regular Sites and Oxygen Vacancies of the MgO(001) Surface: A Density Functional study Using Embedded Cluster Models. Applied Physics A, 82, 181-189.
http://dx.doi.org/10.1007/s00339-005-3352-8 - 7. Brune, H. (1998) Microscopic View of Epitaxial Metal Growth: Nucleation and Aggregation. Surface Science Reports, 31, 125-229.
http://dx.doi.org/10.1016/S0167-5729(99)80001-6 - 8. Cinquini, F., Di Valentin, C., Finazzi, E., Giordano, L. and Pacchioni, G. (2007) Theory of Oxides Surfaces, Interfaces and Supported Nano-Clusters. Theoretical Chemistry Accounts, 117, 827-845.
http://dx.doi.org/10.1007/s00214-006-0204-3 - 9. Fernandez, S., Markovits, A. and Minot, C. (2008) Adsorption of the First Row of Transition Metals on the Perfect and Defective MgO(100) Surface. Chemical Physics Letters, 463, 106-111.
http://dx.doi.org/10.1016/j.cplett.2008.08.053 - 10. Markovits, A., Paniagua, J.C., Lopez, N., Minot, C. and Illas, F. (2003) Adsorption Energy and Spin State of First-Row Transition Metals Adsorbed on MgO(100). Physical Review B, 67, 115417.
http://dx.doi.org/10.1103/PhysRevB.67.115417 - 11. Neyman, K.M., Innatam, C., Nasluzov, V.A., Kosarev, R. and Rosch, N. (2004) Adsorption of d-Metal Atoms on the Regular MgO(001) Surface: Density Functional Study of Cluster Models Embedded in an Elastic Polarizable Environment. Applied Physics A, 78, 823-828.
http://dx.doi.org/10.1007/s00339-003-2437-5 - 12. Markovits, A., Skalli, M.K., Minot, C., Pacchioni, G., Lopez, N. and Illas, F. (2001) The Competition between Chemical Bonding and Magnetism in the Adsorption of Atomic Ni on MgO(100). The Journal of Chemical Physics, 115, 8172.
http://dx.doi.org/10.1063/1.1407824 - 13. Sousa, C., de Graaf, C., Lopez, N., Harrison, N.M. and Illas, F. (2004) Ab Initio Theory of Magnetic Interactions at Surfaces. Journal of Physics: Condensed Matter, 16, S2557-S2574.
http://dx.doi.org/10.1088/0953-8984/16/26/027 - 14. Paulus, U.A., Endruschat, U., Feldmeyer, G.J., Schimidt, T.J., Bonnemann, H. and Behm, R.J. (2000) New PtRu Alloy Colloids as Precursors for Fuel Cell Catalysts. Journal of Catalysis, 195, 383-393.
http://dx.doi.org/10.1006/jcat.2000.2998 - 15. Jalili, S., Isfahani, A.Z. and Habibpour, R. (2012) Atomic Oxygen Adsorption on Au (100) and Bimetallic Au/M (M = Pt and Cu) Surfaces. Computational and Theoretical Chemistry, 989, 18-26.
http://dx.doi.org/10.1016/j.comptc.2012.02.033 - 16. Sicolo, S. and Pacchioni, G. (2008) Charging and Stabilization of Pd Atoms and Clusters on an Electron-Rich MgO Surface. Surface Science, 602, 2801-2807.
http://dx.doi.org/10.1016/j.susc.2008.07.005 - 17. Kukovecz, á., Pótári, G., Oszkó, A., Kónya, Z., Erdöhelyi, A. and Kiss, J. (2011) Probing the Interaction of Au, Rh and Bimetallic Au-Rh Clusters with the TiO2 Nanowire and Nanotube Support. Surface Science, 605, 1048-1055.
http://dx.doi.org/10.1016/j.susc.2011.03.003 - 18. Rassoul, M., Gaillard, F., Garbowski, E. and Primet, M. (2001) Synthesis and Characterisation of Bimetallic Pd-Rh/ Alumina Combustion Catalysts. Journal of Catalysis, 203, 232-242.
http://dx.doi.org/10.1006/jcat.2001.3328 - 19. Ferrari, A.M. (1999) Pd and Ag Dimers and Tetramers Adsorbed at the MgO(001) Surface: A Density Functional Study. Physical Chemistry Chemical Physics, 1, 4655-4661.
http://dx.doi.org/10.1039/a904813h - 20. Shinkarenko, V.G., Anufrienko, V.F., Boreskov, G.K., Ione, K.G. and Yureva, T.M. (1975) Doklady Akademii Nauk SSSR, 223, 410.
- 21. Neyman, K.M. and Illas, F. (2005) Theoretical Aspects of Heterogeneous Catalysis: Applications of Density Functional Methods. Catalysis Today, 105, 2-16.
http://dx.doi.org/10.1016/j.cattod.2005.04.006 - 22. Nasluzov, V.A., Rivanenkov, V.V., Gordienko, A.B., Neyman, K.M., Birkenheuer, U. and Rösch, N. (2001) Cluster Embedding in an Elastic Polarizable Environment: Density Functional Study of Pd Atoms Adsorbed at Oxygen Vacancies of MgO(001). The Journal of Chemical Physics, 115, 8157.
http://dx.doi.org/10.1063/1.1407001 - 23. Matveev, A.V., Neyman, K.M., Yudanov, I.V. and Rösch, N. (1999) Adsorption of Transition Metal Atoms on Oxygen Vacancies and Regular Sites of the MgO(001) Surface. Surface Science, 426, 123-139.
http://dx.doi.org/10.1016/S0039-6028(99)00327-1 - 24. Berthier, G. (2001) Simulation of Ab Initio Results for Palladium and Rhodium Clusters by Tight-Binding Calculations. International Journal of Quantum Chemistry, 82, 26-33.
http://dx.doi.org/10.1002/1097-461X(2001)82:1<26::AID-QUA1018>3.0.CO;2-O - 25. Lombardi, J.R. and Davis, B. (2002) Periodic Properties of Force Constants of Small Transition-Metal and Lanthanide Clusters. Chemical Reviews, 102, 2431-2460.
http://dx.doi.org/10.1021/cr010425j - 26. Wu, Z.J. (2005) Theoretical Study of Transition Metal Dimer AuM (M = 3d, 4d, 5d Element). Chemical Physics Letters, 406, 24-28.
http://dx.doi.org/10.1016/j.cplett.2005.02.083 - 27. Wu, Z.J. (2004) Density Functional Study OF the Second Row Transition Metal Dimmers. Chemical Physics Letters, 383, 251-255.
http://dx.doi.org/10.1016/j.cplett.2003.11.023 - 28. Negreiros, F.R., Barcaro, G., Kuntová, Z., Rossi, G., Ferrando, R. and Fortunelli, A. (2011) Structures of AgPd Nanoclusters Adsorbed on MgO(100): A Computational Study. Surface Science, 605, 483-488.
http://dx.doi.org/10.1016/j.susc.2010.12.002 - 29. Wang, M.Y., Liu, X.J., Meng, J. and Wu, Z.J. (2007) Interaction of H2 with Transition Metal Homonuclear Dimers Cu2, Ag2, Au2 and Heteronuclear Dimers PdCu, PdAg and PdAu. Journal of Molecular Structure: THEOCHEM, 804, 47-55.
http://dx.doi.org/10.1016/j.theochem.2006.10.007 - 30. Gómez, T., Florez, E., Rodriguez, J.A. and Illas, F. (2010) Theoretical Analysis of the Adsorption of Late Transition-Metal Atoms on the (001) Surface of Early Transition-Metal Carbides. The Journal of Physical Chemistry C, 114, 1622-1626.
http://dx.doi.org/10.1021/jp910273z - 31. Die, D., Kuang, X.Y., Guo, J.J. and Zheng, B.X. (2009) First-Principle Study of AunFe (n = 1–7) Clusters. Journal of Molecular Structure: THEOCHEM, 902, 54-58.
http://dx.doi.org/10.1016/j.theochem.2009.02.009 - 32. Chin, Y.H., King, D.L., Roh, H.S., Wang, Y. and Heald, S.M. (2006) Structure and Reactivity Investigations on Supported Bimetallic Au-Ni Catalysts Used for Hydrocarbon Steam Reforming. Journal of Catalysis, 244, 153-162.
http://dx.doi.org/10.1016/j.jcat.2006.08.016 - 33. Liu, F.L., Zhao, Y.F., Li, X.Y. and Hao, F.Y. (2007) Ab Initio Study of the Structure and Stability of MnTln (M = Cu, Ag, Au; n = 1, 2) Clusters. Journal of Molecular Structure: THEOCHEM, 809, 189-194.
http://dx.doi.org/10.1016/j.theochem.2007.01.018 - 34. Vesecky, S.M., Rainer, D.R. and Goodman, D.W. (1996) Basis for the Structure Sensitivity of the CO+NO Reaction on Palladium. Journal of Vacuum Science & Technology A, 14, 1457.
http://dx.doi.org/10.1116/1.579969 - 35. Rainer, D.R., Vesecky, S.M., Koranne, M., Oh, W.S. and Goodman, D.W. (1997) The CO+NO Reaction over Pd: A Combined Study Using Single-Crystal, Planar-Model-Supported, and High-Surface-Area Pd/Al2O3Catalysts. Journal of Catalysis, 167, 234-241.
http://dx.doi.org/10.1006/jcat.1997.1571 - 36. Viñes, F., Desikusumastuti, A., Staudt, T., Gorling, A., Libuda, J. and Neyman, K.N. (2008) A Combined Density-Functional and IRAS Study on the Interaction of NO with Pd Nanoparticles: Identifying New Adsorption Sites with Novel Properties. The Journal of Physical Chemistry C, 112, 16539-16549.
http://dx.doi.org/10.1021/jp804315c - 37. Grybos, R., Benco, L., Bucko, T. and Hafner, J. (2009) Interaction of NO Molecules with Pd Clusters: Ab Initio Density-Functional Study. Journal of Computational Chemistry, 30, 1910-1922.
http://dx.doi.org/10.1002/jcc.21174 - 38. Abbet, S., Riedo, E., Brune, H., Heiz, U., Ferrari, A.-M., Giordano, L. and Pacchioni, G. (2001) Identification of Defect Sites on MgO(100) Thin Films by Decoration with Pd Atoms and Studying CO Adsorption Properties. Journal of the American Chemical Society, 123, 6172-6178.
http://dx.doi.org/10.1021/ja0157651 - 39. Mineva, T., Alexiev, V., Lacaze-Dufaure, C., Sicilia, E., Mijoule, C. and Russo, N. (2009) Periodic Density Functional Study of Rh and Pd Interaction with the (100)MgO Surface. Journal of Molecular Structure: THEOCHEM, 903, 59-66.
http://dx.doi.org/10.1016/j.theochem.2009.01.025 - 40. Lopez, N. and Illas, F. (1998) Ab Initio Modeling of the Metal-Support Interface: The Interaction of Ni, Pd, and Pt on MgO(100). The Journal of Physical Chemistry B, 102, 1430-1436.
http://dx.doi.org/10.1021/jp972626q - 41. D’Ercole, A., Giamello, E. and Pisani, C. (1999) Embedded-Cluster Study of Hydrogen Interaction with an Oxygen Vacancy at the Magnesium Oxide Surface. The Journal of Physical Chemistry B, 103, 3872-3876.
http://dx.doi.org/10.1021/jp990117d - 42. Abdel Halim, W.S., Abdel Aal, S. and Shalabi, A.S. (2008) CO Adsorption on Pd Atoms Deposited on MgO, CaO, SrO and BaO Surfaces: Density Functional Calculations. Thin Solid Films, 516, 4360-4365.
http://dx.doi.org/10.1016/j.tsf.2008.01.009 - 43. Becke, A.D. (1993) Density-Functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics, 98, 5648.
http://dx.doi.org/10.1063/1.464913 - 44. Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Physical Review B, 37, 785-789.
http://dx.doi.org/10.1103/PhysRevB.37.785 - 45. Lopez, N., Illas, F., Rösch, N. and Pacchioni, G. (1999) Adhesion Energy of Cu Atoms on the MgO(001) Surface. The Journal of Chemical Physics, 110, 4873.
http://dx.doi.org/10.1063/1.478373 - 46. Moreira, I.P.R., Illas, F. and Martin, R.L. (2002) Effect of Fock Exchange on the Electronic Structure and Magnetic Coupling in NiO. Physical Review B, 65, Article ID: 155102.
http://dx.doi.org/10.1103/PhysRevB.65.155102 - 47. Siegbahn, P.E. and Crabtree, R.H. (1997) Mechanism of C-H Activation by Diiron Methane Monooxygenases: Quantum Chemical Studies. Journal of the American Chemical Society, 119, 3103-3113.
http://dx.doi.org/10.1021/ja963939m - 48. Illas, F., Moreira, I.P.R., Graaf, C. and Barone, V. (2000) Magnetic Coupling in Biradicals, Binuclear Complexes and Wide-Gap Insulators: A Survey of Ab Initio Wave Function and Density Functional Theory Approaches. Theoretical Chemistry Accounts, 104, 265-272.
http://dx.doi.org/10.1007/s002140000133 - 49. Stevens, W., Krauss, M., Basch, H. and Jasien, P.G. (1992) Relativistic Compact Effective Potentials and Efficient, Shared-Exponent Basis Sets for the Third-, Fourth-, and Fifth-Row Atoms. Canadian Journal of Chemistry, 70, 612-630.
http://dx.doi.org/10.1139/v92-085 - 50. Cundari, T.R. and Stevens, W.J. (1993) Effective Core Potential Methods for the Lanthanides. The Journal of Chemical Physics, 98, 5555.
http://dx.doi.org/10.1063/1.464902 - 51. Larsen, G. (2000) A Performance Comparison between the CEP Effective Core Potential/Triple-Split Basis Set Approach and an All-Electron Computational Method with Emphasis on Small Ti and V Alkoxide Complexes. Canadian Journal of Chemistry, 78, 206-211.
http://dx.doi.org/10.1139/v99-225 - 52. Henrich, V.E. and Cox, P.A. (1994) The Surface Science of Metal Oxides. Cambridge University Press, Cambridge.
- 53. Grimes, R.W., Catlow, C.R.A. and Stoneham, A.M. (1989) Quantum-Mechanical Cluster Calculations and the Mott-Littleton Methodology. Journal of the Chemical Society, Faraday Transactions II: Molecular and Chemical Physics, 85, 485-495.
http://dx.doi.org/10.1039/f29898500485 - 54. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., et al. (1998) Gaussian 98. Gaussian Inc., Pittsburgh.
- 55. Fuente, S.A., Ferullo, R.M., Domancich, N.F. and Castellani, N.J. (2011) Interaction of NO with Au Nanoparticles Supported on (100) Terraces and Topological Defects of MgO. Surface Science, 605, 81-88.
http://dx.doi.org/10.1016/j.susc.2010.10.003 - 56. Giordano, L. and Pacchioni, G. (2005) Pd Nanoclusters at the MgO(100) Surface. Surface Science, 575, 197-209.
http://dx.doi.org/10.1016/j.susc.2004.11.024 - 57. Silvia, A., Patricia, G., Ferullo, M. and Castellani, J. (2008) Adsorption of NO on Au Atoms and Dimers Supported on MgO(100): DFT Studies. Surface Science, 602, 1669-1676.
http://dx.doi.org/10.1016/j.susc.2008.02.037 - 58. Yulikov, M., Sterrer, M., Heyde, M., Rust, H.-P., Risse, T., Freund, H.-J., Pacchioni, G. and Scagnelli, A. (2006) Binding of Single Gold Atoms on Thin MgO(001) Films. Physical Review Letters, 96, Article ID: 146804.
http://dx.doi.org/10.1103/PhysRevLett.96.146804 - 59. Moseler, M., Häkkinen, H. and Landman, U. (2002) Supported Magnetic Nanoclusters: Soft Landing of Pd Clusters on a MgO Surface. Physical Review Letters, 89, Article ID: 176103.
http://dx.doi.org/10.1103/PhysRevLett.89.176103 - 60. Xu, L., Henkelman, G., Campbell, C.T. and Jónsson, H. (2006) Pd Diffusion on MgO(100): The Role of Defects and Small Cluster Mobility. Surface Science, 600, 1351-1362.
http://dx.doi.org/10.1016/j.susc.2006.01.034 - 61. Stirling, A., Gunji, I., Endow, A., Oumi, Y., Kubo, M. and Miyamoto, A. (1995) Γ-Point Density Functional Calculations on Theadsorption of Rhodium and Palladium Particles on MgO(001) Surface and Their Reactivity. Journal of the Chemical Society, Faraday Transactions, 93, 1175-1178.
http://dx.doi.org/10.1039/a604388g - 62. Giordano, L., Vitto, A.D., Pacchioni, G. and Ferrari, A.M. (2003) CO Adsorption on Rh, Pd and Ag Atoms Deposited on the MgO Surface: A Comparative Ab Initio Study. Surface Science, 540, 63-75.
http://dx.doi.org/10.1016/S0039-6028(03)00737-4 - 63. Reed, A., Weinstock, R.B. and Weindhold, F. (1985) Natural Population Analysis. The Journal of Chemical Physics, 83, 735.
http://dx.doi.org/10.1063/1.449486 - 64. Zhao, S., Ren, Y., Ren, Y., Wang, J. and Yin, W. (2011) Density Functional Study of NO Binding on Small AgnPdm (n + m ≤ 5) Clusters. Computational and Theoretical Chemistry, 964, 298-303.
http://dx.doi.org/10.1016/j.comptc.2011.01.009 - 65. Dufaurea, C., Roques, J., Mijoule, C., Sicilia, E., Russo, N., Alexiev, V. and Mineva, T. (2011) A DFT Study of the NO Adsorption on Pdn (n = 1 - 4) Clusters. Journal of Molecular Catalysis A: Chemical, 341, 28-34.
http://dx.doi.org/10.1016/j.molcata.2011.03.020 - 66. Giordano, L., Valentin, C.D., Goniakowski, J. and Pacchioni, G. (2004) Nucleation of Pd Dimers at Defect Sites of the MgO(100) Surface. Physical Review Letters, 92, Article ID: 096105.
http://dx.doi.org/10.1103/PhysRevLett.92.096105 - 67. Zhang, W., Ge, Q. and Wang, L. (2003) Structure Effects on the Energetic, Electronic, and Magnetic Properties of Palladium Nanoparticles. The Journal of Chemical Physics, 118, 5793.
http://dx.doi.org/10.1063/1.1557179 - 68. Kumar, V. and Kawazoe, Y. (2002) Icosahedral Growth, Magnetic Behavior, and Adsorbate-Induced Metal-Nonmetal Transition in Palladium Clusters. Physical Review B, 66, Article ID: 144413.
http://dx.doi.org/10.1103/PhysRevB.66.144413 - 69. Yang, J.X., Cheng, F.W. and Guo, J.J. (2010) Density Functional Study of AunRh (n=1–8) Clusters. Physica B: Condensed Matter, 405, 4892-4896.
http://dx.doi.org/10.1016/j.physb.2010.09.029 - 70. Bogicevic, A. and Jennison, D.R. (2002) Effect of Oxide Vacancies on Metal Island Nucleation. Surface Science, 515, L481-L486.
http://dx.doi.org/10.1016/S0039-6028(02)02024-1 - 71. Efremenko, I. (2001) Implication of Palladium Geometric and Electronic Structures to Hydrogen Activation on Bulk Surfaces and Clusters. Journal of Molecular Catalysis A: Chemical, 173, 19-59.
http://dx.doi.org/10.1016/S1381-1169(01)00144-3 - 72. Piccolo, L. and Henry, C.R. (2001) NO-CO Reaction Kinetics on Pd/MgO Model Catalysts: Morphology and Support Effects. Journal of Molecular Catalysis A: Chemical, 167, 181-190.
http://dx.doi.org/10.1016/S1381-1169(00)00505-7 - 73. Yamaguchi, A. and Iglesia, E. (2010) Catalytic Activation and Reforming of Methane on Supported Palladium Clusters. Journal of Catalysis, 274, 52-63.
http://dx.doi.org/10.1016/j.jcat.2010.06.001 - 74. Ramsier, R.D., Gao, H.N.Q., Lee, K.W., Nooji, O.W., Lefferts, L. and Yates, J.T. (1994) NO Adsorption and Thermal Behavior on Pd Surfaces. A Detailed Comparative Study. Surface Science, 320, 209-237.
http://dx.doi.org/10.1016/0039-6028(94)90310-7 - 75. Tsai, M.H. and Hass, K.C. (1995) First-Principles Studies of NO Chemisorption on Rhodium, Palladium, and Platinum Surfaces. Physical Review B, 51, Article ID: 14616.
http://dx.doi.org/10.1103/PhysRevB.51.14616 - 76. Pacchioni, G. (1993) Physisorbed and Chemisorbed CO2 at Surface and Step Sites of the MgO(100) Surface. Surface Science, 281, 207-219.
http://dx.doi.org/10.1016/0039-6028(93)90869-L - 77. Florez, E., Fuentealba, P. and Mondragón, F. (2008) Chemical Reactivity of Oxygen Vacancies on the MgO Surface: Reactions with CO2, NO2 and Metals. Catalysis Today, 133, 216-222.
http://dx.doi.org/10.1016/j.cattod.2007.12.087 - 78. Sterrer, M., Yulikov, M., Risse, T., Freund, H.J., Carrasco, J., Illas, F., Valentin, C.D., Giordano, L., Pacchioni, G., Risse, T. and Freund, H.J. (2006) When the Reporter Induces the Effect: Unusual IR Spectra of CO on Au1/MgO(001)/ Mo(001). Angewandte Chemie International Edition, 45, 2633-2635.
http://dx.doi.org/10.1002/anie.200504473 - 79. Grönbeck, H. and Broqvist, P. (2003) CO-Induced Modification of the Metal/MgO(100) Interaction. The Journal of Physical Chemistry B, 107, 12239-12243.
- 80. Abbeta, S., Heizb, U., Ferraric, A.M., Giordanod, L., Valentin, C.D. and Pacchioni, G. (2001) Nano-Assembled Pd Catalysts on MgO Thin Films. Thin Solid Films, 400, 37-42.
http://dx.doi.org/10.1016/S0040-6090(01)01444-4 - 81. Abdel Halim, W.S., Assem, M.M., Shalabi, A.S. and Soliman, K.A. (2009) CO Adsorption on Ni, Pd, Cu and Ag Deposited on MgO, CaO, SrO and BaO: Density Functional Calculations. Applied Surface Science, 255, 7547-7555.
http://dx.doi.org/10.1016/j.apsusc.2009.04.026 - 82. Shalabi, A.S., Nour, E.M. and Abdel Halim, W.S. (2000) Characterization of van der Waals Interaction Potentials D4h and Td Configurations of He4. International Journal of Quantum Chemistry, 76, 10-22.
http://dx.doi.org/10.1002/(SICI)1097-461X(2000)76:1<10::AID-QUA2>3.0.CO;2-1