American Journal of Molecular Biology
Vol.3 No.3(2013), Article ID:34682,9 pages DOI:10.4236/ajmb.2013.33018
Generation of pig primary fibroblast cells harboring defective MC4R genes by N-ethyl-N-nitrosourea mutagenesis: A gene-driven, nontransgenic approach to pig improvement
![]()
National Institute of Agrobiological Sciences, Tsukuba, Japan
Email: *yukari@affrc.go.jp
Copyright © 2013 Michiharu Sakurai et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 24 March 2013; revised 26 April 2013; accepted 1 June 2013
Keywords: pig; fibroblasts; N-ethyl-N-nitorosourea; mutagenesis
ABSTRACT
Transgenic pigs have been produced with the aim of further improving pigs in terms of economic and environmental traits, but these animals have not been allowed to enter the food chain. As an alternative approach to generating pigs with novel traits of economic importance that cannot be introduced by conventional breeding, we propose a strategy for combining in vitro mutagenesis of pig primary cells with N-ethyl-N-nitrosourea (ENU) and somatic-cell nuclear transfer (SCNT) technology. To explore the feasibility of this strategy, we treated pig primary fibroblast cells with ENU, estimated the per-base mutation frequency induced by the mutagen, clonally cultured about 4000 of the mutagenized cells, and screened them for mutation within the coding region of the melanocortin-4 receptor (MC4R) gene, a key gene in energy homeostasis. Through this screening, we obtained 14 cell clones, each harboring a heterozygous base change within the coding region for MC4R. Of the mutant cell clones, each of two contained a mutant allele encoding MC4R with greatly reduced receptor activity. By SCNT using these cell clones as donors, pigs harboring mutated MC4R alleles with reduced receptor activity can be produced. Our strategy for generating pigs with novel genetic traits likely will be more acceptable to the public than is the use of transgenic technology.
1. INTRODUCTION
Thus far, improvement of commercial pigs has been achieved only by conventional breeding. A number of transgenic pigs have been developed with enhanced growth and production traits, or yielding a product that has been altered to make it more environmentally safe, but none of these pigs has been approved to enter the human food chain [1]. In plants, mutagenesis with chemical mutagens or radiation followed by phenotypic screening has been used by breeders to generate many improved commercial crops, and recently mutagenesis followed by gene-driven reverse genetic screening has been proposed as an important tool in crop improvement [2].
Mutant mice derived from embryonic stem (ES) cells mutagenized with chemical mutagens such as N-ethylN-nitrosourea (ENU) in vitro have been produced [3-7]. Somatic cell nuclear transfer (SCNT) of pigs has been developed, and many gene-knockout pigs have been generated by exploiting this technology [1,8]. Thus, generation of pigs with novel genetic traits by combining SCNT technology and in vitro mutagenesis of pig fibroblast cells with ENU would be an intriguing strategy for improving commercial pigs without the legislative regulations imposed on and social opposition to genetically modified organisms (GMOs). ENU mutagenesis is a conventional and safe method of altering the genetic traits of organisms, and foods derived from livestock produced by the use of SCNT technology and their progeny have been approved as safe in many countries, including the United States and Japan [9].
The melanocortin-4 receptor (MC4R) is a G-proteincoupled receptor and is positively coupled to adenylate cyclase via stimulatory heteromeric G proteins [10]. MC4R is produced predominantly in the brain and is a key regulator of energy homeostasis. MC4R activation leads to decreased food intake and increased energy expenditure. When MC4R was inactivated through gene targeting, the knockout mice had severe obesity, together with increased body length [11]. Moreover, mice harboring a fully or partially defective MC4R have been produced through in vivo mutagenesis with ENU and have varying degrees of obesity [12,13]. Human genetic studies have confirmed that the MC4R is indispensable for human energy homeostasis. It is well accepted that mutation in human MC4R is the most common monogenic form of obesity, with more than 170 distinct mutations identified so far [14,15]. Two non-synonymous variants of pig MC4R with apparently normal receptor activity, R236H and D298N, have been reported, but the association between the presence of these variants and traits of economic importance such as growth and fatness remains unclear [16-18].
Here, we developed procedures that enabled the isolation of pig primary cell clones harboring ENU-induced mutations in MC4R gene. We obtained two cell clones, each harboring an allele encoding an MC4R mutant with greatly reduced receptor activity. Our results illustrate the feasibility of producing cloned pigs via SCNT of cells with mutations of interest induced by ENU mutagenesis.
2. MATERIALS AND METHODS
2.1. Fibroblast Cells
Pig fibroblast cells were prepared from a male fetus of Landrace ´ Large White at embryonic day 17, as previously described [8]. The sex of the fetus was verified according to a procedure previously described [19]. The cells were cultured at 38˚C in an atmosphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (Thermo Fischer Scientific, Waltham, MA, USA), 100 units/ml penicillin, 100 mg/ml streptomycin, 0.25 mg/ml amphotericin (Sigma-Aldrich, St. Louis, MI, USA), and 2 mM GlutaMAX (Life Technologies, Carlsbad, CA, USA). This medium will be referred to here as DME10. The cells were expanded to the third passage and stored at –190˚C in Cellbanker 1 medium (Nihon Zenyaku Kougyou, Kouriyama, Fukushima, Japan) until use. Cells of this strain were used throughout the study and will be referred to as M6 cells.
2.2. Mutagenesis of Pig Fibroblast Cells with ENU
Ten days before mutagen treatment, one vial of M6 cells was thawed and placed in DMEM containing 20% fetal calf serum, the antibiotics listed above, and 2 mM GlutaMAX. This medium will be referred to as DME20. In subsequent cultures, the M6 cells and their mutants were maintained in DME20 at 38˚C in an atmosphere of 5% CO2. The thawed M6 cells were subcultured (1:5) 8 days before mutagen treatment. The cells were confluent 3 days before mutagen treatment; they were refed with fresh medium on this day and the next day. One day before mutagenesis, the cells were replated in appropriate numbers of 10-cm dishes so that the number of cells per dish was 1.1 ´ 106. Sixteen hours after the replatings, the culture medium was removed, and the cells were incubated at 38˚C for 1 h with DMEM containing 0 mM, 0.5 mM, 1.0 mM, or 1.5 mM ENU (Sigma-Aldrich) and 10 mM HEPES (pH 7.4). The cells were then washed with PBS and cultured in DME20. The colony-forming ability of the cells in one dish for each dose of ENU was assayed immediately after the ENU treatment. Two days after ENU treatment, most of the cells treated with 1.0 mM or 1.5 mM ENU were trypsinized and frozen until use in clonal culture, as described below. The remaining cells for each dose of ENU were seeded in six 10-cm dishes (3 ´ 105 cells/dish) to examine mutation frequency. The next day, the culture medium was replaced with DME20 containing 0.3 μM ouabain (Sigma-Aldrich). Six days after the addition of ouabain, the cells were refed with DME containing fresh 0.3 μM ouabain. Thirteen days after the start of ouabain selection, the cells were stained with methylene blue to count the colonies. At the start of ouabain selection, a portion of the cells was diluted and plated at cloning density in medium lacking ouabain to determine the cloning efficiency at the time of selection. This value was used to determine the number of ouabain-resistant (Ouar) colonies per total number of clonable cells.
2.3. Isolation of Independent Mutant Cell Clones Resistant to Ouabain
M6 cells were treated with 1.5 mM ENU as described above, and, at the end of the treatment, the cells were washed with PBS, trypsinized, and seeded onto 24-well plates at a density of 5000 cells/well. They were then cultured so that each mutant clone was independently isolated. After 3 days, the medium was replaced with DME20 containing 0.3 μM ouabain. After another 6 days, the cells were refed with fresh DME20 containing 0.3 μM ouabain. 7 days later, colonies growing in the wells (no more than one colony per well) were separately replated into the wells of 24-well plates and maintained. In subsequent cultures, all isolated cell clones were maintained in the presence of 0.3 μM ouabain.
2.4. Nucleotide Sequence Analysis of Cell Clones Resistant to Ouabain
Total RNA was prepared from M6 cells and each of the Ouar cell clones described above by using an RNeasy Mini Kit (Qiagen, Venio, The Netherlands). cDNA was synthesized with a ReverTra Ace-α-cDNA synthesis kit (Toyobo, Osaka, Osaka, Japan) and used as a template in PCR with the primers GGGACACAGACCACCGCCAC TATG (forward) and GTCGTTCCCACAGACGGATGT TTCTC (reverse) to amplify the entire coding region of the pig Na+/K+-ATPase α1 subunit gene (ATP1A1) [20]. PCRs were performed in 50-μl reactions by using KOD FX DNA polymerase (Toyobo), with the following cycling parameters: 94˚C for 2 min; five cycles of 98˚C for 10 s and 74˚C for 3 min; five cycles of 98˚C for 10 s and 72˚C for 3 min; five cycles of 98˚C for 10 s and 70˚C for 3 min; 21 cycles of 98˚C for 10 s and 68˚C for 3 min; and 68˚C for 7 min. Amplified DNA was purified by using a Wizard SV Gel and PCR Clean-up System (Promega, Fitchburg, WI, USA), and the nucleotide sequence of the DNA was determined by using external and internal primers on a DNA sequencer (Applied Biosystems Model 3100; Life Technologies). Nucleotide sequences were analyzed with DNASIS software (Hitachi, Hitachi, Ibaraki, Japan).
2.5. Clonal Culture of Mutagenized Cells
A vial of cells treated with 1.0 mM or 1.5 mM ENU was thawed, seeded, and cultured in a 10-cm dish for 3 days until the cells became confluent. The cells were then trypsinized and suspended in 5 ml Hanks’s balanced salt solution (Life Technologies) containing 2% fetal calf serum, 20 mM HEPES (pH 7.4), and 1 mM isopropyl iodide (Sigma-Aldrich). The cells were delivered to forty 96-well plates (each containing 150 μl DME20) with a flow cytometer (EPICS ALTRA, Beckmann-Coulter, Brea, CA, USA), such that each well contained one living cell. The 40-plate set of mutagenized cells was allowed to grow for 1 week, after which the cells in each plate were replicated into two 96-well plates. The cells in these two sets of 96-well plates were allowed to grow for 2 weeks, during which time the cells were refed with DME20 once a week. Twenty-four to 26 days after the cell sorting, one set of 96-well plates was inspected under an inverted microscope to identify wells containing more than about 1 ´ 104 cells. The cells in these wells were then subjected to screening for mutation, as will be described below. Cells in another set of 96-well plates for use as master plates were cultured further until wells containing cell clones with mutations within MC4R gene were identified as will be described below, after which the mutant cell clones in the master plates were expanded and frozen until use.
2.6. Screening for Mutations Following Clonal Culture
Genomic DNA was prepared from the cells in the 96- well plates by using Proteinase K and DirectPCR cell reagent (45 μl/well) (Viagen-Biotech, Los Angels, CA, USA). An aliquot of the DNA solution (2 μl) was used as a template in PCR with the primers GGAGAAAAT CGCTGAGGCTA (forward) and TGTCCAGACTGTG TAAGGAGAA (reverse) to amplify the entire coding region for MC4R [18]. The PCRs were performed in 20- μl reactions using TaKaRa Ex Taq Hot Start Version DNA polymerase (Takara Bio, Ohtsu, Shiga, Japan), with the following cycling parameters: 98˚C for 10 s; 35 cycles of 94˚C for 30 s, 63˚C for 30 s, and 72˚C for 1 min; and 72˚C for 7 min. Amplified DNA was subjected to sequencing on a DNA sequencer (Applied Biosystems 3130 × 1 Genetic Analyzer, Life Technologies). The nucleotide sequences of the cell clones were compared with that of M6 cells to identify mutations within the coding region for MC4R.
2.7. Construction of Plasmids Encoding Myc-Tagged MC4R Mutants
A pcDNA3.1-based expression vector producing MC4R was constructed essentially as described previously [21], after PCR amplification of the coding region for MC4R by using genomic DNA from M6 cells as a template. For immunologic detection purposes, the construct was tagged with an Myc-derived epitope (EQKLISEEDL) downstream of the initiation codon by using PCR-based sitedirected mutagenesis and restriction fragment replacement strategy. Expression plasmids, each harboring a nucleotide change corresponding to that found in the mutagenized cell clones, were constructed by using a QuickChange site-directed mutagenesis kit (Stratagene, LaJolla, CA, USA). All PCR-derived constructs were verified by DNA sequencing.
2.8. Transfection and cAMP Assays
COS-7 cells were grown in DME10 at 38˚C in an atmosphere of 5% CO2. Cells were seeded into six-well plates (2 ´ 105 cells/well), and 24 h later they were transiently transfected with plasmids encoding wild-type and mutant MC4Rs (2.0 μg/well), along with pCBR-Control vector (0.5 μg/well) (Promega), by using Lipofectamine LTX reagent (Life Technologies). One day after transfection, the cells were seeded into a 96-well plate (4 ´ 104 cells/well) precoated with polyethyleneimine (Sigma) and cultured in Waymouth’s MB 752/1 medium (Life Technologies) containing 1% bovine serum albumin (Waymouth-BSA). The remaining cells were seeded into a 96-well plate for chemiluminescence measurement (GreinerBio One, Frickenhausen, Germany) and cultured in Waymouth-BSA at 38˚C in an atmosphere of 5% CO2. The next day, cyclic adenosine monophosphate (cAMP) assay and chemiluminescence measurement were performed. Cells were first washed once with Waymouth-BSA and then incubated with 0.5 mM isobutylmethylxanthine (IBMX) (Sigma-Aldrich) in Waymouth-BSA for 15 min at 38˚C. The medium was removed and replaced with Waymouth-BSA with 0.5 mM IBMX and increasing concentrations of alpha-melanocyte stimulating hormone (α- MSH) (Peptide Institute, Osaka, Osaka, Japan), and the cells were incubated at 38˚C for 1 h. Intracellular cAMP was measured with a cAMP-Screen assay kit (Life Technologies). Chemiluminescence from the cells was measured by using Steady-Glo luciferase assay substrate (Promega). cAMP values were normalized against luciferase data to correct for variations in transfection efficiency and were then analyzed with GraphPad Prism 5 software (Graphpad Software, San Diego, CA, USA).
3. RESULTS
3.1. ENU Mutagenesis and Estimation of Per-Base Mutation Frequency
To our knowledge, no previous report of mutagenesis of primary pig fibroblasts with chemical mutagens has been published. We first sought to determine appropriate conditions for ENU mutagenesis of primary pig fibroblasts, in which we exploited ouabain selection to monitor the frequency of induced mutations. Ouabain selection of human and rodent cells after mutagenesis has been used to monitor the mutation frequency in such cells [22-25]. Cellular resistance to ouabain results from specific missense mutations in the ATP1A1 gene ([26] and references therein).
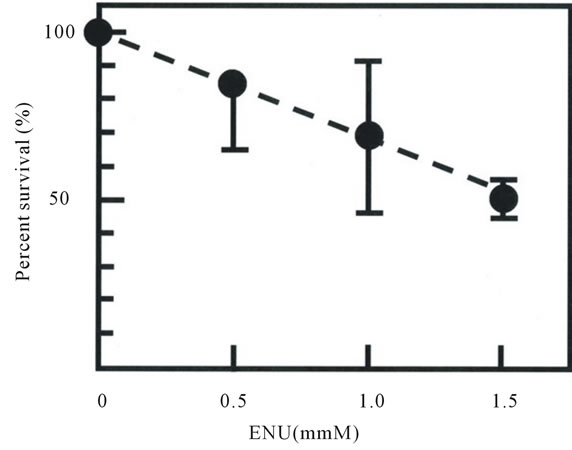
The percent survival and the frequency of mutation induced by ENU of M6 cells after ENU treatment were shown in Figure 1. The mutation frequency, expressed as the frequency of Ouar cells in the total number of clonable cells, increased with increasing ENU concentration, reaching 1.15 ± 0.16 ´ 10−4 at an ENU concentration of 1.5 mM (Figure 1(b)). The frequency of Ouar cells in mock-treated cells was less than 1 ´ 10−7 (not shown).
In studies of mutagenesis of mouse ES cells and subsequent generation of mutant mice, the mutation frequency in ES cells was monitored by exploiting the Xlinked hypoxanthine phosphoribosyl transferase (hprt) locus to determine appropriate concentrations of ENU for mutagenesis [3,5-7], and the relationship between the frequency of HPRT-deficient cells and the per-base mutation frequency in the mouse genome were inferred from sequence analysis of randomly isolated mutagenized cell clones [3,7]. However, for unknown reasons, we were not able to detect any pig fibroblast cells deficient in HPRT activity after treatment of the cells with ENU or ethyl methanesulfonate (data not shown). We needed to examine the relationship between the mutation frequency (determined as the frequency of Ouar cells) and the perbase mutation frequency in the swine genome. If the
 (a)
(a)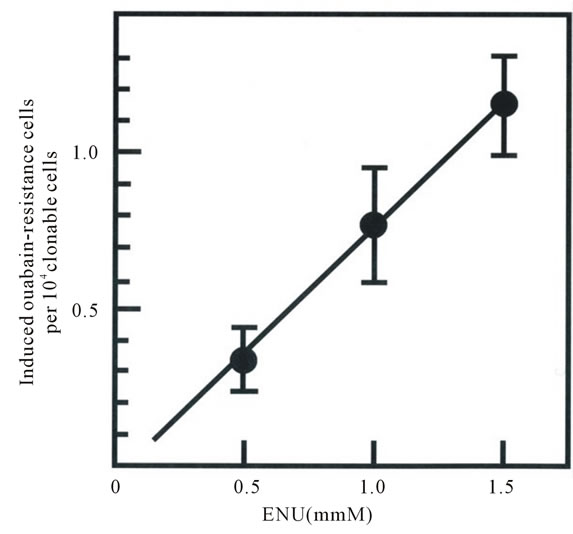 (b)
(b)
Figure 1. Cytotoxicity (a) and mutagenicity (b) induced by N-ethyl-N-nitrosourea (ENU) in pig fibroblasts. Percentage survival in panel A was calculated by dividing the cloning efficiency of cells treated with the indicated concentrations of ENU by that of mock-treated cells at the end of treatment. Cloning efficiency of the mock-treated cells ranged from 0.56 to 0.73. The frequency of induced Ouar cells (b) was calculated by dividing the number of Ouar colonies by the number of cells plated for selection, corrected by the cloning efficiency at the time of selection. Each point represents the mean of at least four experiments. Bars represent 1 SD.
per-base mutation frequency after mutagenesis were too low, we would have to screen too many clones of mutagenized cells to find one with a desired mutation, whereas if the frequency were too high, this high load of mutations in the genome would prevent the generation of fertile cloned animals after transfer of the nuclei of the mutant cells, just as has been pointed out in studies of mutant mice derived from mutagenized ES cells [3,7,27].
To examine the relationship between the frequency of Ouar cells after ENU treatment and the per-base mutation frequency induced by this treatment, we first isolated 18 independent Ouar cell clones after mutagenesis with 1.5 mM ENU, and we determined the nucleotide sequence of the entire coding region of the pig ATP1A1 gene in these cells. Comparison of the sequences in the Ouar clones with that in M6 cells (GenBank Accession No. AB703104.1) unequivocally identified a heterozygous point mutation in each clone (Table 1). All of these mutations were missense mutations. T-to-A transversions were found most frequently among the mutations (38%); this was in accordance with the results of sequence analysis of HPRT-deficient mutants after mutagenesis of HFW human fibroblast cells with ENU [28]. Amino-acid residues at a total of eight positions were substituted in the Ouar clones: these were C104, Q111, D121, N122, I315, F316, L793, and T797.
A series of extensive random-mutagenesis studies of the sheep ATP1A1 gene has identified single amino-acid substitutions, each of which renders the enzyme resistant to ouabain ([26] and references therein). These aminoacid changes resulting from missense mutations occur at a total of 16 positions: C104, Y108, Q111, P118, D121, N122, Y308, L330, A331, T338, F786, L793, T797,
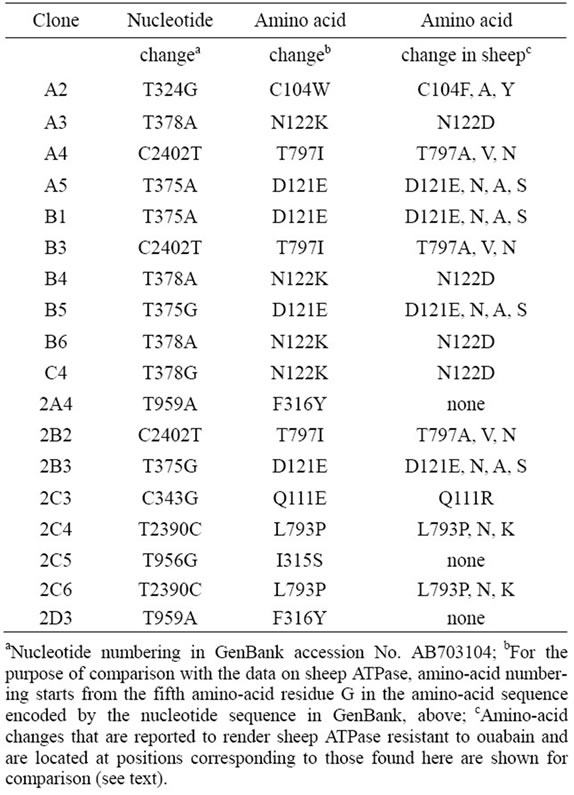
Table 1. Mutations found in 18 independent Ouar cell clones.
F863, R880, and F982. The amino-acid residues at these positions are assumed to be involved in binding between the enzyme and its inhibitor, ouabain; in fact, a recent X-ray crystallography study of a complex between pig ATP1A1 and ouabain has shown that all of the aminoacid residues mentioned above are included in the ouabain-binding pocket of the enzyme [29]. Notably, the amino-acid sequence is highly conserved between the sheep and pig ATP1A1 enzymes (99.3% identity), and all of the sheep enzyme amino-acid residues mentioned above are conserved at the same positions in the pig enzyme.
Of the eight positions where we found amino-acid substitutions in Ouar clones, six corresponded to the respective ones identified in the sheep enzyme as being involved in binding to ouabain, suggesting that each amino-acid substitution that we found altered the affinity between the enzyme and ouabain. The other two positions of amino-acid substitutions that we found—I315 and F316—had no counterparts in the positions identified in the sheep enzyme. These results suggest that not all of the amino-acid positions at which an amino-acid change makes the enzyme resistant to ouabain have been identified in the studies on the sheep enzyme; that the amino-acid substitutions at I315 and F316 have been missed in the studies of the sheep enzyme; and that I315 and F316 are in fact conserved in the sheep amino-acid sequence. We propose that the ratio between the number of amino-acid positions already identified in studies of the sheep enzyme and that of the positions not identified so far, including I315 and F316, can reasonably be estimated on the basis of our results as 6:2. This assumption gives a value of 21.3 (16 ´ [8/6]) for the estimated total number of amino-acid positions at each of which an amino-acid change results in resistance of the enzyme to ouabain.
Taking into account the fact that an amino-acid residue is coded for by three nucleotides and the Ouar phenotype is dominant [26] and assuming that 70% of random nucleotide substitutions induced by ENU are missense mutations [30], the total number of nucleotide positions at each of which a nucleotide substitution would lead to generation of Ouar enzyme, and thus Ouar cells, would be 89.5 (i.e., 21.3 ´ 3´ 0.7 ´ 2). This argument leads to an estimated per-base mutation frequency of (the frequency of Ouar cells/89.5). The frequency of Ouar cells induced by 1.5 mM ENU was 1.15 ´ 10−4 and that induced by 1.0 mM ENU was 0.79 ´ 10−4 (see Figure 1(b)); thus the estimated per-base mutation frequency was 1.3 ´ 10−6 (1 base change in 778 kilobases (kb)) with 1.5 mM ENU, and 0.9 ´ 10−6 (1 in 1132 kb) with 1.0 mM ENU. These results implied that, for example, one clone among 389 clones of the cells treated with 1.5 mM ENU would harbor a heterozygous base change in the coding region for MC4R.
3.2. Clonal Culture and Screening of Cell Clones
A total of 2100 clones of cells treated with 1.5 mM ENU and a total of 1893 clones of cells treated with 1.0 mM ENU were screened for the presence of mutation in eight separate clonal culture experiments (Table 2). The percentage of wells in 96-well plates that contained sufficient numbers of cells for DNA analysis at the time of inspection was 13.4%; this was lower than the cloning efficiency of the cells (0.56 - 0.73). Sorting of the cells on a flow cytometer at the beginning of the clonal culture experiments was indispensable, because once the cells had been grown at clonal densities and harvested, they could not grow when reseeded at clonal densities. For the screening, DNA including the entire coding region for MC4R was amplified by PCR using two primers, one just 5’ and the other just 3’ to the coding region, and its nucleotide sequence was determined by using the same primers: the coding region for pig MC4R is 999 base pairs long, and this region is continuous on the genome [18]. A total of 10 clones treated with 1.5 mM ENU and a total of four clones treated with 1.0 mM ENU were identified as each having a mutation within the coding region (Table 3 and Figure 2). The cells found to contain mutations were picked from the corresponding wells in the master plates, expanded, and stored in liquid nitrogen. The nucleotide sequences of the MC4R coding regions in the expanded cells were determined after PCR amplifica-
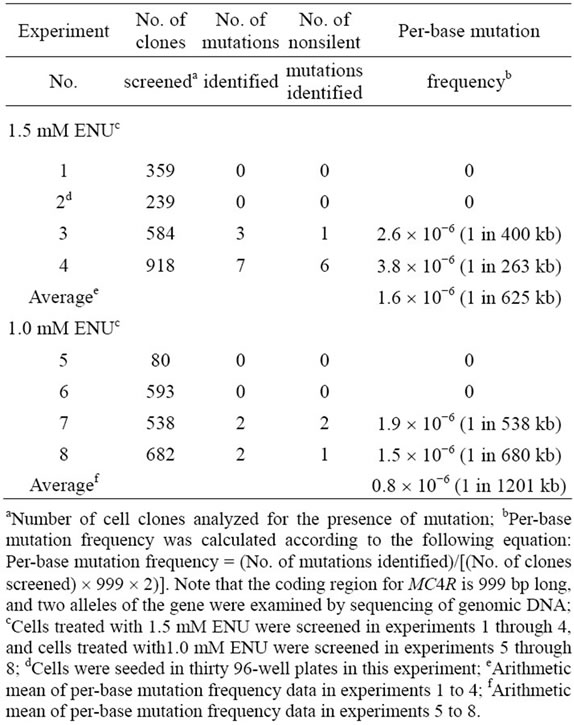
Table 2. Summary of clonal culture experiments.
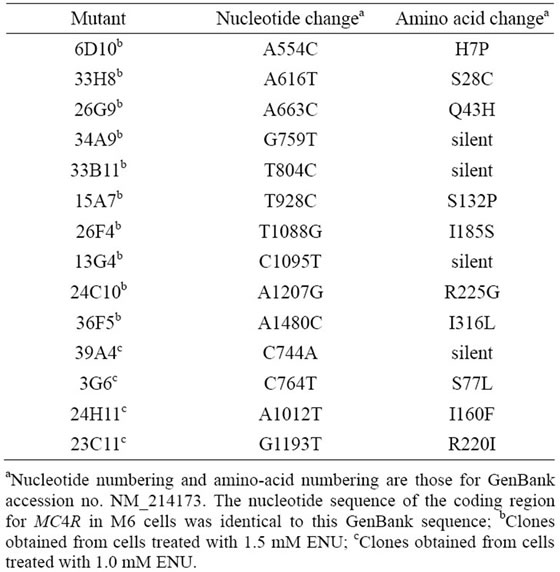
Table 3. Mutants obtained in clonal cultures of fibroblast cells treated with ENU.
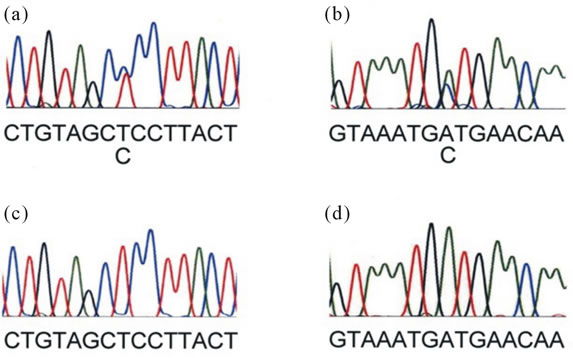
Figure 2. Sequencing traces of cell clones harboring MC4R mutants, S132P and I185S. Panel. (a) Sequencing trace (forward orientation) of the cell clone 15A7 harboring S132P. Panel; (b) Sequencing trace (reverse orientation) of the cell clone 26F4 harboring I185S. Panel; (c) Sequencing trace of wild-type cells which corresponds to panel A. Panel; (d) Sequencing trace of wild-type cells which corresponds to panel A. Nucleotide sequence is shown below each sequencing trace.
tion to verify the mutations.
All of the mutations were heterozygous base substitutions (Table 3). Ten were missense mutations, and the other four were silent. Average per-base mutation frequencies of cells treated with 1.5 mM ENU and 1.0 mM ENU, calculated on the basis of the sequence analysis, were 1.6 ´ 10−6 (1 in 625 kb) and 0.8 ´ 10−6 (1 in 1201 kb), respectively (Table 2). These average per-base mutation frequencies were in good agreement with the estimations of per-base mutation frequency based on the results of ouabain selection at the corresponding doses of ENU. Moreover, the average per-base mutation frequency when the cells were treated with 1.5 mM ENU was comparable to the per-base mutation frequency estimated on the basis of the sequence analysis of ENU-treated mouse ES cells (2.2 ´ 10−6), by which cells harboring mutations in Smad 2 and Smad 4 were obtained [7] This implies that pig fibroblast cells treated with ENU exhibited levels of mutability comparable to those reported for mouse ES cells.
3.3. Functional Analysis of Wild-Type and Mutant MC4Rs
α-MSH is considered to be an endogenous ligand of MC4R [10]. Wild-type pig MC4R and all the nonsynonymous mutants that we identified were examined for signal transduction activity upon stimulation with α-MSH in COS-7 cells transiently transfected with the receptor constructs. The responses of S132P and I185S were so greatly reduced that the maximum response (Rmax) and the half maximum effective concentration (EC50) values of these mutants could not determined (Table 4 and Figure 3). The Rmax values of S77L, R220I, R225G, and I316L were moderately reduced compared with that of the wild type, whereas H7P, S28C, Q43H, and I160F had Rmax levels similar to that of the wild type. The EC50 values of these active mutants were comparable to that of the wild type. Flow cytometry with an anti-myc antibody (Life Technologies) was used to analyze Cos-7 cells transiently transfected with plasmids encoding wild-type MC4R, I185S, and S132P and detected the expression of these encoded proteins on thesurfaces of the transfected cells at levels comparable to each other, suggesting that the apparent deficiency in receptor activity shown with I185S and S132P was due
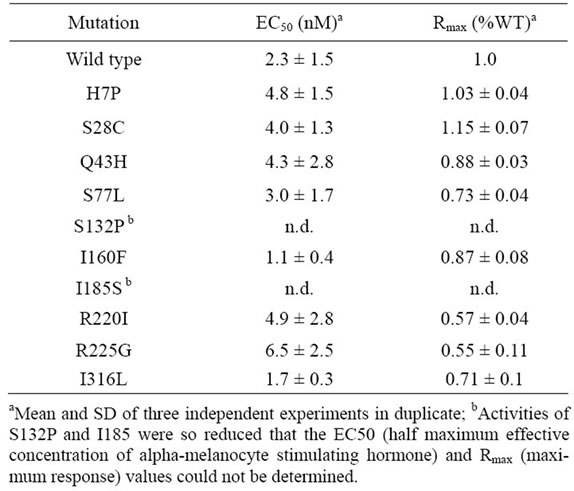
Table 4. cAMP-signaling activity of wild-type and mutant MC4Rsa.
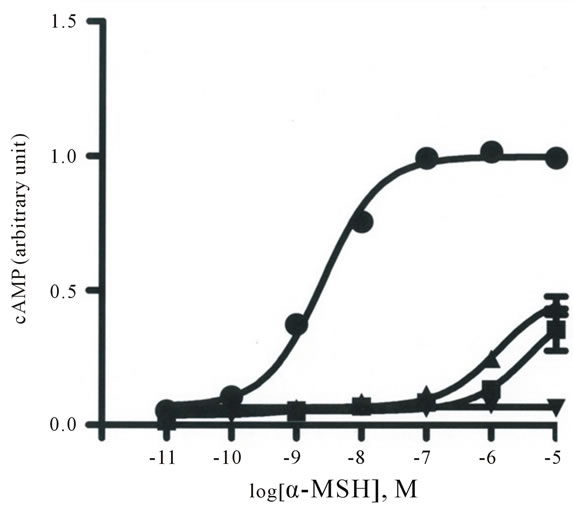
Figure 3. cAMP stimulation of wild-type (WT) MC4R and the mutants S132P and I185S in COS-7 cells with alpha-melanocyte stimulating hormone (α-MSH). The amounts of cAMP in the cells transfected with plasmids producing WT (●), S132P (▲), and I185S (■) after α-MSH stimulation were normalized against luciferase data in each experiment (see Materials and Methods). Each point represents the mean of three experiments in duplicate. Bars represent 1 SD.
to intrinsic defects in these mutants (data not shown).
4. DISCUSSION
The generation of cell clones harboring I185S or S132P provides proof-of-principle of the feasibility of generating pig primary fibroblast cells harboring mutations of functional importance in genes of interest. The relationships that have been shown in previous studies between the functional activity of MC4R mutants and the phenoltypes of humans or mice harboring MC4R mutants strongly suggest that cloned pigs produced via SCNT using cells harboring S132P or I185S as donors would have substantially increased appetite and decreased basal energy expenditure [11-14]. These pigs would be the founders of a generation of pigs with improved growth characteristics and would be valuable model animals for human obesity.
The estimated values for per-base mutation frequency based on ouabain selection were in good agreement with those based on the sequence analysis of randomly isolated mutagenized cell clones, suggesting that per-base mutation frequency in the genome can be reasonably well estimated by exploiting ouabain selection. Our results in this regard may also be applicable to large domestic animals, because all of the ouabain-binding sites in ATPase are conserved among mammals, with the exception of rodents [31]. It is pertinent to note here that the estimated values for per-base mutation frequency based on ouabain selection should be somewhat lower than actual per-base mutation frequency in a sense that mutations other than nucleotide changes (deletions or insertions) are not represented in the estimated values.
Vivian et al. [7] treated CT129 ES cells with 1.7 mM ENU for 2 h, and five clones of the mutagenized cells yielded germ-line chimeras among the 13 clones tested. The per-base mutation frequency in the study by Vivian et al. [7] was comparable to ours (see Results). Greber et al. [5] treated E14.1 ES cells with 1.5 mM ENU for 90 min; one out of two mutant cell clones gave rise to germ-line chimeras. Chick et al. [4] mutagenized c7.1 ES cells with 4.3 mM ENU for 2 h, and chimera formation using two of three mutagenized cell clones resulted in germ-line transmission. These results suggest that pig fibroblast cell clones harboring S132P or I185S would give rise to cloned pigs upon SCNT, because the per-base mutation frequency introduced into these pig cells in the present study seems no higher than that introduced into the mutant ES cells mentioned above.
The ENU-generated mutant alleles of genes of interest in cloned pigs can be introgressed into commercial pigs by serial backcrosses: Introgression of alleles controlling coat color, such as the MC1R * 2 allele, has been performed in pig breeding [32]. Marker-assisted selection of founder animals in each generation of the backcross would facilitate the elimination of bystander mutations induced by ENU [33]. Total-genome sequence data [34] and high-density SNP panels [35] of the pig can now greatly facilitate this process.
Recently, gene-knockout pigs have been produced by using zinc-finger-nuclease (ZFN) or transcription activator-like effecter nuclease (TALEN) [36,37]. In these studies, mutations were introduced into the genome of primary pig cells by use of ZFN or TALEN, and cells harboring the mutations were selected with the aid of marker genes and then subjected to SCNT. The resultant pigs are genetically modified owing to the presence of the marker genes, and they are therefore subject to the legislative regulations and social opposition surrounding GMOs. Moreover, licensing to produce and commercialize such pigs would be costly, because ZFN and TALEN are proprietary technologies [38,39], and the legislative question as to whether or not organisms engineered by the use of ZFN and TALEN—even when no exogenous DNA is stably integrated—are deemed non-GMO remains unsettled. Taking into consideration these uncertainties, we consider that our proposal for generating mutant pigs through in vitro ENU mutagenesis is currently the most feasible for generating commercial pigs with novel alleles of economical importance.
![]()
![]()
REFERENCES
- Prather, R.S., Shen, M. and Dai, Y. (2008) Genetically modified pigs for medicine and agriculture. Biotechnology and Genetic Engineering Reviews, 25, 245-266.
- Parry, M.A.J., Madgwick, P.J., Bayon, C., et al. (2009) Mutation discovery for crop improvement. Journal of Experimental Botany, 60, 2817-2825. doi:10.1093/jxb/erp189
- Chen, Y., Yee, D., Dains, K., et al. (2000) Genotypebased screen for ENU-induced mutations in the mouse embryonic stem cells. Nature Genetics, 24, 314-317. doi:10.1038/73557
- Chick, W.S., Drechsel, D.A., Hammond, W., et al. (2009) Transmission of mutant phenotypes from ES cells to adult mice. Mammalian Genome, 20, 734-740. doi:10.1007/s00335-009-9228-z
- Greber, B., Lehrach, H. and Himmelbauer, H. (2005) Mouse splice mutant generation from ENU-treated ES cells—A gene-driven approach. Genomics, 85, 557-562. doi:10.1016/j.ygeno.2005.01.011
- Greber, B., Tandara, H., Lehrach, H., et al. (2005) Comparison of PCR-based mutation detection methods and application for identification of mouse Sult1a1 mutant embryonic stem cell clones using pooled templates. Human Mutation, 25, 483-490. doi:10.1002/humu.20168
- Vivian, J.L., Chen, Y., Yee, D., et al. (2002) An allelic series of mutations in Smad2 and Sma4 identified in a genotype-based screen of N-ethyl-N-nitrosourea-mutagenized mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 99, 15542-15547. doi:10.1073/pnas.242474199
- Onishi, A., Iwamoto, M., Akita, T., et al. (2000) Pig cloning by microinjection of fetal fibroblast nuclei. Science, 289, 1188-1190. doi:10.1126/science.289.5482.1188
- Fox, J.L. (2008) Cloned animals deemed safe to eat, but labeling issues loom. Nature Biotechnology, 26, 249-250. doi:10.1038/nbt0308-249
- Tao, Y. (2010) The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocrine Reviews, 31, 506-543. doi:10.1210/er.2009-0037
- Huszar, D., Lynch, C.A., Fairchild-Huntress, V., et al. (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell, 88, 131-141. doi:10.1016/S0092-8674(00)81865-6
- Grosse, J. Tarnow, P., Roempler, H., et al. (2006) Nethyl-N-nitrosourea-based generation of mouse models for mutant G protein-coupled receptors. Physiological Genomics, 26, 209-217. doi:10.1152/physiolgenomics.00289.2005
- Meehan, T.P., Tabeta, K., Du, X., et al. (2006) Point mutations in the melanocortin-4 receptor cause variable obesity in mice. Mammalian Genome, 17, 1162-1171. doi:10.1007/s00335-006-0073-z
- Tao, Y. (2009) Mutations in melanocortin-4 receptor and human obesity. Progress of Molecular Biology and Transitional Science, 88, 173-204.
- Wang, Z. and Tao, Y. (2011) Functional studies on twenty novel naturally occurring melanocortin-4 receptor mutations. Biochimica et Biophysica Acta, 1812, 1190-1199. doi:10.1016/j.bbadis.2011.06.008
- Fan, Z., Sartin, J.L. and Tao, Y. (2008) Pharmacological analyses of two naturally occurring porcine melanocortin- 4 receptor mutations in domestic pigs. Domestic Animal Endocrinology, 34, 383-390. doi:10.1016/j.domaniend.2007.05.003
- Fan, B., Onteru, S.K., Plastow, G.S., et al. (2009) Detailed characterization of the porcine MC4R gene in relation to fatness and growth. Animal Genetics, 40, 401-409. doi:10.1111/j.1365-2052.2009.01853.x
- Kim, K.S., Larsen, N., Short, T., et al. (2000) A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and food intake traits. Mammalian Genome, 11, 131-135. doi:10.1007/s003350010025
- Sembon, S., Suzuki, S., Fuchimoto, D., et al. (2008) Sex identification of pigs using polymerase chain reaction amplification of the amelogenin gene. Zygote, 16, 327- 332. doi:10.1017/S0967199408004826
- Ovchinnikov, Y.A., Modyanov, N.N., Broude, N.E., et al. (1986) Pig kidney Na+, K+-ATPase. Primary structure and spacial organization. FEBS Letters, 201, 237-245. doi:10.1016/0014-5793(86)80616-0
- Tao, Y. and Segaloff, D.L. (2003) Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology, 144, 4544-4551. doi:10.1210/en.2003-0524
- Baker, R.M., Brunette, D.M., Mankovitz, R., et al. (1974) Ouabain-resistant mutants of mouse and hamster cells in culture. Cell, 1, 9-21. doi:10.1016/0092-8674(74)90149-4
- Balachandra Dass, S., Heflich, R.H. and Casciano, D.A. (1997) The mutagenic response at the ouabain resistance locus in T cells of mice exposed to N-ethyl-N-nitrosourea parallels the response at the Hprt locus and correlates with mutation target size. Carcinogenesis, 18, 2233-2237. doi:10.1093/carcin/18.11.2233
- Eldridge, S.R. and Gould, M.N. (1991) Specific locus mutagenesis of human mammary epithelial cells by ultraviolet radiation. International Journal of Radiation Biology, 59, 807-814. doi:10.1080/09553009114550701
- Mankovitz, R., Buchwald, M. and Baker, R.M. (1974) Isolation of ouabain-resistant human diploid cells. Cell, 3, 221-226. doi:10.1016/0092-8674(74)90135-4
- Croyle, M.L., Woo, A.L. and Lingrel, J.B. (1997) Extensive random mutagenesis analysis of the Na+/K+-ATPase subunit identifies known and previously unidentified amino acid residues that alter ouabain sensitivity—Implications for ouabain binding. European Journal of Biochemistry, 248, 488-495. doi:10.1111/j.1432-1033.1997.00488.x
- Monroe, R.J., Bergstrom, R.A., Zheng, et al. (2000) Mouse mutants from chemically mutagenized embryonic stem cells. Nature Genetics, 24, 318-321. doi:10.1038/73563
- Yang, J., Lee, P., Lin, S., et al. (1994) Comparison of mutation spectra induced by N-ethyl-N-nitrosourea in the hprt gene of Mer+ and Mer− diploid human fibroblasts. Carcinogenesis, 15, 959-945. doi:10.1093/carcin/15.5.939
- Yatime, L., Laursen, M., Morth, J.P., et al. (2011) Structural insights into the high affinity binding of cardiotonic steroids to the Na+, K+-ATPase. Journal of Structural Biology, 174, 296-306. doi:10.1016/j.jsb.2010.12.004
- Takahasi, K.R., Sakuraba, Y. and Gondo, Y. (2007) Mutational pattern and frequency of induced nucleotide changes in mouse ENU mutagenesis. BMC Molecular Biology, 8, 52-61. doi:10.1186/1471-2199-8-52
- Lingrel, J.B. (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na, KATPase. Annual Reviews of Physiology, 72, 395-412. doi:10.1146/annurev-physiol-021909-135725
- Giuffra, E., Kijas, J.M.H., Amarger, V., et al. (2000) The origin of the domestic pig: Independent domestication and subsequent introgression. Genetics, 154, 1785-1791.
- Markel, P., Shu, P., Ebeling, C., et al. (1997) Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nature Genetics, 17, 280-284. doi:10.1038/ng1197-280
- Groenen, M.A.M., Archibald, A.L., Uenishi, H., et al. (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature, 491, 393-398. doi:10.1038/nature11622
- Ramos, A.M., Crooijmans, R.P.M.A., Affara, N.A., et al. (2011) Design of high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE, 4, e6524. http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0006524 doi:10.1371/journal.pone.0006524
- Carlson, D.F., Tan, W., Lillico, S.G., et al. (2012) Efficient TALEN-mediated gene knockout in livestock. Proceedings of the National Academy of Sciences of the United States of America, 109, 17382-17387. doi:10.1073/pnas.1211446109
- Yang, D., Yang, H., Li, W., et al. (2011) Generation of PPAR mono-allelic knockout pigs via zinc-finger nucleases and nuclear transfer cloning. Cell Research, 21, 979- 982. doi:10.1038/cr.2011.70
- Chandrasekharan, S., Kumar, S., Valley, C.M., et al. (2009) Proprietary science, open science and the role of patent disclosure: The case of zinc-finger proteins. Nature Biotechnology, 27, 140-144. doi:10.1038/nbt0209-140
- DeFrancesco, L. (2011) More over ZFNs. Nature Biotechnology, 29, 681-684. doi:10.1038/nbt.1935
NOTES
*Corresponding author.