American Journal of Molecular Biology
Vol.3 No.1(2013), Article ID:26992,9 pages DOI:10.4236/ajmb.2013.31001
Effect of 4% glycerol and low aeration on result of expression in Escherichia coli of Cin3 and three Venturia inaequalis EST’s recombinant proteins
![]()
1The University of Samarrai, College of Medical Technology, Samarra, Iraq
2The Plant and Food Research Institute of New Zealand Ltd., CRI, Palmerston North, New Zealand
3The Plant and Food Research Institute of New Zealand Ltd., CRI, Auckland, New Zealand
Email: tahaalsamarrai@gmail.com
Received 2 November 2012; revised 5 December 2012; accepted 25 December 2012
Keywords: Venturia inaequalis; Expressed Sequence Tag (ESTs); Phytopathogenic Fungus; Appressorium; A Stroma
ABSTRACT
The phytopathogenic fungus Venturia inaequalis causes scab of apple. Once this fungus penetrates the plant surface, it forms a specialized body called a stroma between the inner cuticle surface and the epidermal cell wall. A novel Venturia inaequalis 5704 (Cin3) and three expressed sequence tags (ESTs): 38, 6987, and 4010 are strongly up-regulated in the early stages of infection. The CIN3 and three ESTs using two vectors pMAL-c2 and pET 21 were expressed in Escherichia coli. Recombinant proteins expression, solubility and yields were analyzed. 38, 5704 (Cin3) and 6987 recombinant proteins were expressed in soluble form and while 4010 was expressed in inclusion bodies. Resolution on native-PAGE, the recombinant proteins; 38, 5704 (Cin3), 6987 were shown to be present in dimmer, tetramer and polymer. A method was developed, consisting of induction of expression at various temperatures, and using enriched broth with 4% glycerol together with slow shaking, led to a decrease in concentration of nascent polypeptide and production of soluble recombinant proteins of 38, 5704 (Cin3), 6987 and 4010. Resolution on nativePAGE, the recombinant proteins were shown to be present as monomer.
1. INTRODUCTION
Ventura inaequalis (Cooke) Wint. causes black spot or scab of apple (Maluspumila (Mill.) Henry) [1]. V. inaequalis germinates on the apple leaf surface and forms an appressorium-like swelling prior to penetration of the cuticle. Once the cuticle has been breached, the fungal hyphae grow between the inner cuticle surface and the epidermal cell wall. At this time the hyphae differentiate, producing a specialized body called a stroma that is made up of single or multiple layers of pseudoparenchyma, a laterally dividing cell-type which is distinct from the usual tubular form of hyphal growth [1,2]. During the apple growing season, conidiophores are produced from the stromata through the perforated cuticle and are associated with the development of scab lesions.
Differentiation of sub-cuticular hyphae and stromata can be simulated by growing V. inaequalis in vitro on cellophane discs [2]. V. inaequalis penetrates and forms stromata within the cellophane. A differential cDNA screen was performed on a library made from mycelia grown on cellophane. Two novel genes, cin1 (EU189192) and cin3 (EU189193), were found to be differentially expressed in the cellophane library. Full-length versions of these genes were found in an expressed sequence tag library from apple leaves infected with V. inaequalis, and quantitative real-time PCR (qRT-PCR) indicated that these genes were up-regulated over 1000-fold during the early stages of infection compared with in vitro growth [2]. The genes encode putative secreted proteins with imperfect repeated domains. Three ESTs: 38(7006), 6987 and 4010 were selected since qRT-PCR indicated that these genes were up-regulated during the early stages of infection compared with in vitro growth (Gene accession number EB135294).
A reliable and efficient protein expression system was needed to make V. inaequalis proteins in their “native” form for protein crystallization and for production of monoclonal antibodies that would provide powerful tools for characterization of V. inaequalis genes function and protein interactions in signal transduction. In this report, we describe a strategy for expression and purification of the four recombinant proteins of V. Inaequalis: 38, 5704 (Cin3), 6987 and 4010.
2. MATERIALS AND METHODS
2.1. Materials
pMAL protein expression and purification system was purchased from New England BioLabs. Bacterial strain BL21 (DE3) was purchased from Novagen (Madison, WI, USA.). DNA manipulations were carried out by using standard procedures [3]. Plasmids were isolated with a Qiaprep spin plasmid kit (Qiagen).
2.2. Bacterial Strains and Plasmids Construction
The pETM plasmids from EMBL have been used as the back bone to construct the suitable expression vectors for our E. coli expression system. All of the vectors are IPTG inducible and Kanamycin resistant with a general formula of X-Y-rTEV-MCS-Z (see Figure 1), where X is either the fusion tag His (His-tag) or R (R-tag); Y is maltose-binding protein (MBP), thioredoxin A (TrxA), or glutathione Stransferase (GST) and Z is either a GlyGlyGlyCys tag (GGGC), His (His-tag), rTEV-R-tag, or blank (Nil). rTEV represents the cleavage site of recombinant Tobacco Etch Virus protease [4]. The EMBL vectors pETM-41 (MBP fusion), pETM-30 (GST fusion) and pETM-20 (TrxA fusion) were used as initial vectors for modification (EMBL vector data:
http://www.emblhamburg.de/~geerlof/webPP/genetoprotein/clo_ vector/our_Ec_vectors.html). The modifications of these vectors were carried out using the method reported previously previously [5]. A second rTEV site
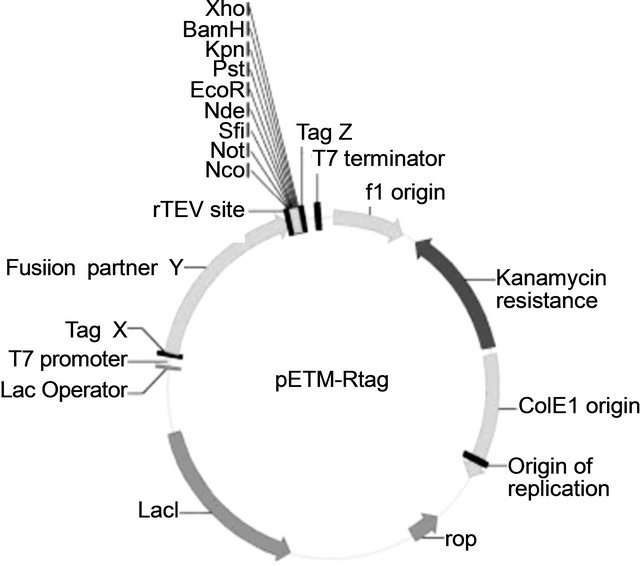
Figure 1. Vectors map for expression of Venturia inaequalis EST’s proteins in E. coli. The size of vectors varies from 5734 to 6517 bp.), where X is either the fusion tag His (His-tag) or R (R-tag); Y is maltose-binding protein (MBP), thioredoxin A (TrxA), Z is either a GlyGlyGlyCys tag (GGGC), His (His-tag), rTEV-R-tag, or blank (Nil). rTEV represents the cleavage site of recombinant Tobacco Etch Virus protease [4].
followed by R-tag at the 30 of the new MCS was incurporated using PCR with primers 5’CATGCTCGAGGAAAACCTGTACTTTCAGGGTCCGGATCAGTATGAATACAAATATCCGTAGTGAGATCCGGCTAACAAAGC-3’ and 5’-GCGATCCCCGGGAAAACAGCAT-3’. An in vitro biotinylation site, GlyGlyGlyCys peptide followed by two stop codons, was further incorporated into TrxA fusion vector at 3’ of the MCS using primers 5’CCGCTCGAGGGCGTGGCTGCTAGTGAGTCCGGCTGCTAAC-3’ and 5’-GCGATCCCCGGGAAAACAGCAT-3’.
The sequence of 38, 6987, 5704 and 4010 genes are: MSRPGTDPNQDPAYVPAAGDGSYTVCTPYDMPGICKRYKKDGTATKEVAKCRSASQCWVNG NGCVMVGNGFANCSG,MILEPKAGLGEIRSLEQRPAFSDLKASLSPKSACDKEDESDCTAFCKANDQTATCTAAGNKITCSCKGGKSESNCEERCLLCMPGKSQLEEASLLFKQGGSKLFKEDL, DKQEGSDAVNTRYLAKRQSDIPTYHLWDEEKESGVKAAYKVVDGQEVKGQVEKRQSDAPYYHKLWDEENGAVAKAAYRAVDGQKVKGQVEKRQSDAPYYHKLWDEENGAAAKAAYRAVD, and SQAQAPLQQPITQSTDLSPRILPVVTSNELLSLHR KLIEIESISGNEKPVGEWLKGYLEAKNLTVELQEVEEGRYNVFAYPGTERKTKVLVSSHIDTVPPYWSYERKTTDGVDEIWGRGSVDAKACVASQIIAVLDLLESGKHNLPSDALSLLFVIGEEVGGEGMRFFSDRKPTNYSAIVFGEPTEGKLVAGHKGMIGVKLNITGKAAHSGYPWLGISANNVLVQALSIVLALEKDDLPGSKKFGKTTVNIGRVSGGLAANVVAESSKADIAIRIAGGSPEEINKIITKALQPLKEETEKVGGIFELQWSKRAYGTVDIDTDVEGFDTITVNYGTDVPNIEGDHKRYLYGPGSIFVAHSDHEHLAVSELEQSVLDFQKIILAQF respectively.
2.3. Method of Expression of Soluble Recombinant Proteins of Venturia inaequalis
E. coli BL21 (DE3) clones were cultivated at 37˚C in Luria-Bertani (LB) medium containing Kanamycin (50 μg/mL). When OD600 reached 0.8 - 1.2, isopropyl-β-Dthiogalactopyranoside (IPTG) was added to the medium at a concentration of 0.3 mM, and the cells were continuously cultivated from 3 to 24 h at 10˚C, 15˚C, 25˚C, or 37˚C. Samples (10 mL) were collected at specific time intervals after induction. Bacterial growth was determined by monitoring the OD600 spectrophotometrically The cells harvested from 10 mL of each of the cultures, were washed with 2 mL of 20 mM phosphate buffer (pH 7.5), and then were resuspended with 1ml of 20 mM phosphate buffer (pH 7.5). The suspensions were sonicated on ice-bath and then centrifuged at 20,000 × g for 15 min at 4˚C. The supernatants were taken as soluble fractions. Proteins in the precipitates were suspended in 1 ml of 20 mM phosphate buffer to obtain insoluble fractions. The soluble and insoluble fractions were analyzed by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were visualized by staining with Coomassie blue-R. Protein concentration was estimated using image densitometry software ImageJ (http://rsb.info.nih.gov/ij/). The appropriate condition for further production of soluble fusion protein was selected based on the highest concentration of fusion protein obtained.
For production of soluble recombinant proteins, 2 - l flask containing 1000 mL of rich YT medium (30 g Tryptone and 15 g Yeast extract) supplemented with Kanamycin (50 μg/mL) and glycerol (4% v/v) were inoculated with 10 mL of overnight culture of E. coli BL21 (DE3) containing the MBP or Trx A tag fusion proteins. The cells were grown at 37˚C, with shaking (250 rpm) to an absorbance (600 nm) of 0.7 - 1.2, and then the medium was cooled to appropriate temperature in a water bath. IPTG was added to the medium at a concentration of 0.3 mM and the cells were continuously cultivated from 3 to 24 h at proper temperature with slow shaking (60 RPM) or static condition. Cells were collected by centrifugation at 6000 rpm for 10 min (Sorval RC5B). The pellets were resuspended in 40 mL of TrisHCl buffer pH 7.8 (20 mM Tris-HCl, 1 mM EDTA, 1 mM PMSF). The cells were disrupted by sonication on ice bath (20 × 10 s). Cell lysates were centrifuged at 16,000 rpm for 20 min, the supernatants collected and placed on ice. For purification of the MBP-tag fusion proteins, the soluble fraction containing MBP-tag fusion protein was loaded on an amylose affinity column, washed with six column volumes of 20 mM Tris, pH 7.8, and 100 mM NaCl, then with three column volumes of 20 mM Tris, pH 7.8, and 10 mM NaCl. Elution was performed with the same buffer supplemented with 10 mM maltose. Eluted recombinant proteins were further purified by employing DEAE column chromatography. The eluent was loaded into a 5 mL HiTrap DEAE column (GE Healthcare Life Sciences) equilibrated with 20 mL of sample buffer, the column washed with 20 mL sample buffer, and the protein was eluted at 2 mL/min, on an AKTA Prime (Amersham Biosciences) with a gradient of 0 - 1 M Nacl. A flow through that contain more than 50% - 80% of un-bound recombinant proteins was concentrated into 200 µL by using Vivaspin 20 ml Centrifugal Concentrators (Cole-Parmer 625 East Bunker Court, Vernon Hills, Illinois, USA). The recombinant protein was denatured by addition of 2 ml of buffer A (8 M urea, 100 mM Tris-HCl, 5 mM EDTA, 10 mM dithiothreitol, 1.5 mM reduced glutathione, 0.2 mM oxidized glutathione, pH 8.4). The insoluble material was removed by centrifugation using micro-centrifuge (14,000 rpm for 10 min). The supernatant was collected and refolding of proteins was carried as described previously [6]. The refolded protein was purified by using DEAE column chromatography. Fraction containing the purified recombinant protein was concentrated into 1 - 2 mL and stored in 10% glycerol at −80˚C Ni-NTA chromatography was employed in purification of TrexA His-Tagged proteins. Culture expressing recombinant protein was sonicated as describe above. The centrifuged supernatant was loaded onto a 5-mL Ni-NTA column equilibrated with buffer B contain 5 mM imidazole. The column was washed with the same buffer containing 30 mM imidazole and 1 M NaCl. Then the column washed with buffer B containing 30 mM imidazole and 10 mM NaCl. The His-tagged recombinant protein was eluted from the column with buffer B containing 500 mM imidazole and 10 mM NaCl. Fractions containing the His-tagged recombinant protein were pooled and concentrated by ultra filtration as described above. Recombinant protein was concentrated and stored in 10% glycerol at −80˚C.
2.4. Determination of Solubility and Expression Level
All of the soluble proteins were confirmed by the native PAGE. Soluble aggregates stay on the top of the gel and do not enter the body of the gel whereas non-aggregated proteins enter the body of the gel.
2.5. Electrophoresis and Western Blot Analysis
Protein samples were analyzed for purity and checked for degradation using SDS and native polyacrylamide gels. Samples were resolved on a 7.5% denaturing and native gel. Protein bands were either stained with Coomassie blue R or electro-transferred from an unstained gel onto a nitrocellulose membrane (Trans-Blot Transfer, Bio-Rad Laboratories, CA, USA) for Western blot analysis. The membrane was blocked in 0.5% I-block (Tropix, Bedfod, MA, USA) in phosphate-buffered saline containing 0.1% Tween 20 (PBST) for 2 h at room temperature and incubated for 1 h in 1:4000 dilution of peroxidase-labeled Anti-His6 (Roche # 1 965 085). The membrane was washed three times in PBS and developed using the ECL system (NEN Western Lightning Plus).
2.6. Protein Determination
His-tag, MBP-tag or Trx A tag recombinant proteins were separated on SDS-polyacrylamide gels and stained with Coomassie blue R. Protein concentration was estimated using image densitometry software ImageJ (http://rsb.info.nih.gov/ij/). Briefly samples were run on an SDS-polyacrylamide gel, Coomassie-stained, imaged on an HP Scan Jet 4300C and analysed with ImageJ software. The blots were processed with the ImageJ program to quantify relative signal intensities of his-tagg positive areas of the membrane.
Protein concentration was also determined using the BCA assay (Pierce chemicals) using bovine serum albumin as standard.
3. RESULTS AND DISCUSSIONS
The objective of this work was to develop a protocol for production of pure soluble full-length of four V. inaequalis ESTs to be used for immunization and production of monoclonal antibodies, by insertion of the genes; 38, 6987, 5704 (Cin3), and 4010 into two vectors [(pETM-41 (MBP fusion), and pETM-20 (Trx A fusion)] followed by expression in E. coli. To obtain strong immunological responses with small antigens such as EST proteins, it was necessary that these antigens be expressed as soluble proteins with fusion tags that were strongly immunogenic. We adopted MBP as the fusion partner for this purpose. Other fusion tags that could be cleaved by specific proteases, together with a variety of peptide tags suitable for either purification or attachment to a solid surface for protein arraying were also examined. Fusion tags have been reported to protect their fusion partners from intracellular proteolysis [7,8] and to have the ability to enhance the solubility of their fusion partners [9]. Results from our immunization experiments have shown MBP to be strongly immunogenic and suitable as a carrier for the V. inaequalis ESTs proteins. Trx A is another fusion partner that has been shown to be effective in enhancing proper folding of recombinant partner proteins and therefore their solubility. To avoid picking up antibodies against MBP, Trx A fused EST’s proteins will be used for cell fusion to prepare hybridoma cells and in ELISA for screening of cultures for monoclonal antibody.
The C-terminal GlyGlyGlyCys tag constructed together with N-terminal Trx A has proven to be excellent for biotinylation of V. inaequalis ESTs fusion proteins for use in cell fusion and screening experiments. Furthermore for detection and purification, use of a small peptide R-tag has proven extremely useful. R-tag does not occur in plant, E. coli or mammalian expression hosts and its use has minimized cross-reactions. We have previously reported the use of R-tag and an immuno-affinity matrix coupled with its monoclonal antibody for purification [5]. This tag has also been cloned into our expression vectors for V. inaequalis ESTs R-tag fusion.
All of these constructs incorporate different combinations of tags for different affinity purification procedures, and allow for flexibility during purification as well as applications for immunization, hybridoma production and screening for monoclonal antibody.
3.1. Efficiency of rTEV Cleavage
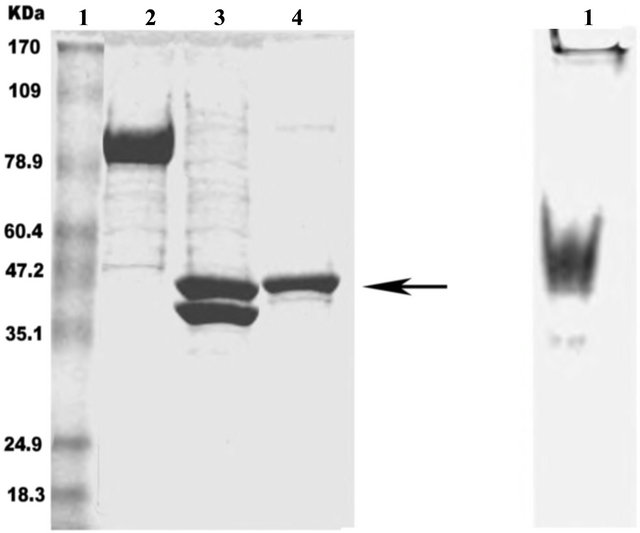
Proteolytic cleavage sites were introduced between the fusion tags and target protein in order to remove the fusion tags when this was needed. It has been reported that some commercial proteases used for removal of fusion tags, such as enterokinase and Factor Xa, cleave fusion proteins at non-canonical sites and often result in the degradation of the target proteins [10,11]. The highly site-specific rTEV protease [4] was used in our expression system for removing fusion tags at later stage of the protein purification. All of our cleavage experiments have indicated that the rTEV leads to specific and efficient cleavage and an example is shown in Figure 2.
3.2. Production of Soluble Proteins
For production of soluble proteins, six flask of 200 mL (numbered from 1 - 4) each contain 50 mL YT medium containing Kanamycin (50 μg/mL) (see Materials and Methods) were inoculated with E. coli BL21 (DE3) clone and cultivated at 37˚C. When OD600 reached 0.8 - 1.2, the culture media (1 - 4) were cooled to 10˚C, 15˚C, 25˚C, and 30˚C in water bath, respectively, then isopropyl-β-D-thiogalactopyranoside (IPTG) were added to the media at a concentration of 0.3 mM. The cells were continuously cultivated from 3 to 24 h at 14˚C, 25˚C, 30˚C, or 37˚C. Expression at various temperatures, using an optimal concentration of IPTG was tested for each recombinant protein. The test of solubility was carried out
 (a) (b)
(a) (b)
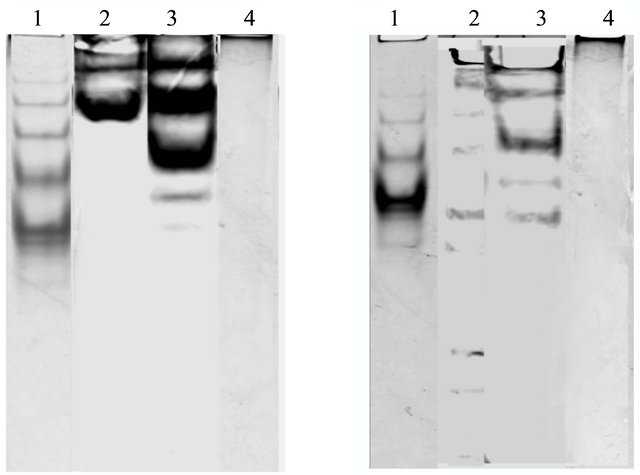
Figure 2. rTEV proteolytic cleavage of MBP fused N-terminal 4010 recombinant protein. Proteolysis was performed overnight at 4˚C. Proteins were separated by SDS-PAGE as described in Materials and methods. Lane 1a: protein ladder; lane 2a: 4010- MBP tagged before cleavage; lane 3a: 4010-MBP tagged after cleavage; lane 4a: purified 4010 fusion protein, 3b1: purified 4010 fusion protein loaded into Native PAGE as described in Materials and methods.
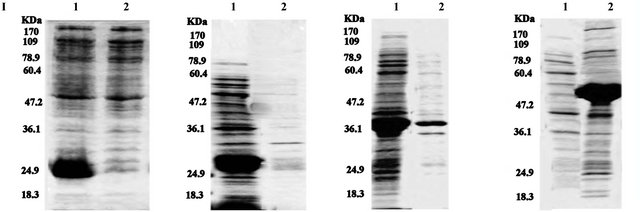
by centrifugation of the lysates followed by resolution by SDS-PAGE. The recombinant proteins; 4010 (TrexA-tagged or MBP-tagged) at 37˚C were expressed in the inclusion body while the recombinant proteins 38, 6987 and 5704 (Cin3) present in the supernatants as resolved on SDS gels to give a protein of the correct size (Figure 3), the proteins enter the body of a 7.5% native PAGE showing monomer, dimer and tetramer (Figure 4).
Expression at 30˚C showed that 38, 6987 and 5704 (Cin3) present in the supernatant as resolved on SDS gels to give a proteins of the correct size. The recombinant protein 38 enter the body of a 7.5% native PAGE and shown to be present as monomers, while the recombinant protein 6987 and 5704 (Cin3) protein enter the body of a 7.5% native PAGE present as monomer, dimer and tetramer (results not shown but similar to Figure 4).
Induction at 24˚C resulted in production of soluble 6987 and 5704 (Cin3) recombinant proteins and shown to be present as monomers (results not shown) and induction at 14˚C resulted in production of approximately 30% of soluble recombinant protein of 4010. Separation of soluble protein fraction of 4010 from soluble microaggregate, was carried out by employing DEAE column chromatography. The eluent from amylose affinity column was loaded into DEAE column. Approximately 30% of the recombinant protein was bind into the column and the rest of the protein appeared in the flow through. The unbound protein was collected and concentrated into 200 µL by using Vivaspin centrifugal concentrator. The recombinant protein was denatured and refolded as de scribed previously [6]. Resolution on native-PAGE followed by Western blotting and probing with antibodies to
 (a)
(a)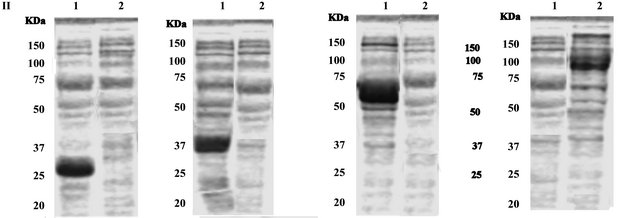 (b)
(b)
Figure 3. SDS-PAGE analysis of expression of Venturia inaequalis EST’s proteins in E. coli. Upper panel I, Lane 1a, supernatant of the cell lysate of 38-TrxA after induction; and lane 2a, pellet of the cell lysate of 38-TrxA after induction; Lane 1b, supernatant of the cell lysate of 6987-TrxA after induction; and lane 2b, pellet of the cell lysate of 6987-TrxA; Lane 1c, supernatant of the cell lysate of 5704 TrxA after induction; and lane 2c, pellet of the cell lysate of 5704-TrxA after induction; Lane 1d, supernatant of the cell lysate of 4010-TrxA after induction; and lane 2d, pellet of the cell lysate of 4010-TrxA after induction. Lowe panel II, Lane 1a, supernatant of the cell lysate of 38-MBP after induction; and lane 2a, pellet of the cell lysate of 38-MBP after induction; Lane 1b, supernatant of the cell lysate of 6987-MBP after induction; and lane 2b, pellet of the cell lysate of 6987-MBP; Lane 1c, supernatant of the cell lysate of 5704 MBP after induction; and lane 2c, pellet of the cell lysate of 5704-MBP after induction; Lane 1d, supernatant of the cell lysate of 4010-MBP after induction; and lane 2d, pellet of the cell lysate of 4010-MBP after induction.
 (a) (b)
(a) (b)
Figure 4. Purified fusion protein loaded into Native PAGE as described in Materials and methods. (a) Lane 1: 38-MBP, lane 2: 6987-MBP, lane 3: 5704-MBP, lane 4: 4010-MBP. (b) Lane 1: 38-TrexA, lane 2: 6987-TrexA, lane 3: 5704-TrexA, lane 4: 4010-TrexA.
the His-tag or rab-tag showed one major bands of the fusion protein (Figure 4(b)) correspondingto molecular weights of monomeric EST’s.
Production of insoluble fusion protein can be attributed to overproduction of the fusion protein itself. It has been observed that the increase in concentration of nascent polypeptide chain is sufficient to induce the formation of inactive aggregates even upon overexpression of homologous cytosolic proteins [11]. A well-known technique to limit the in vivo aggregation of recombinant proteins is to express protein at reduced temperatures [12]. This strategy has proven effective in improving the solubility of a number of difficult proteins [13]. Other workers have shown that the growth of transformed cells under a decreased growth temperature resulted in an increase in the yield of native, soluble protein [14,15]. It has been reported that cold shock can result in the production of CspA, the major cold-shock protein of E. coli, which is an RNA chaperone, preventing RNA from forming stable secondary structures for efficient translation of mRNAs at low temperature [13,16]. Also a direct consequence of temperature reduction is the partial elimination of heat-shock proteases that are induced under overexpression conditions [17-22].
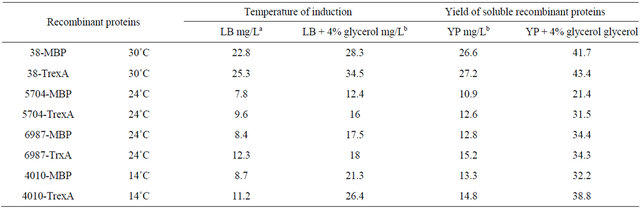
Also folding failures of about 60% recombinant proteins produced in E. coli is generally attributed to a limitation in the cell concentration of folding supporter elements, which cannot process the newly synthesized aggregation prone polypeptides. This assumption is physiologically supported by the overexpression of chaperone genes, in particular of chaperone genes from the heatshock protein family, in response to recombinant protein overproduction [23-25]. We therefore developed a method that involved induction of expression of recombinant proteins under changing temperature conditions and using rich broth that contain glycerol (4% v/v) with low aeration (see material and methods). Also, before induction, the cultures were cooled to required temperature followed by induction and incubation of cultures at low temperature with slow shaking (10 - 40 RPM) to decrease aeration. Andersen and Meyenburg [26] suggest that aerobic growth of E. coli in batch cultures is limited by the rate of respiration and the concomitant rate of ATP generation through oxidative phosphorylation [26]. The addition of glycerol (4% v/v) led to a decrease in concentration of nascent polypeptide. We found that expression of 38 recombinant protein MBP or TrxA tagged at 37˚C in presence of 4% glycerol resulted in the production of soluble momomeric recombinants protein as shown by native PAGE analysis. As for 5704 (Cin3) and 6987 MBP or TrxA tagged expression at 30˚C resulted in increase in the production of soluble monomer recombinants proteinas shown by native PAGE analysis. Although expression of 4010 of TrexAand MBP-tagged at 30˚C, showed that 30% of proteins were present in the supernatants as resolved on SDS gels to give a protein of the correct size, the protein did not enter the body of a 7.5% native PAGE suggestive of the “soluble” microaggregate. Expression at 14˚C with slow shaking (10 - 20 RPM) in presence of 4% glycerol showed that approximately 90% of 4010 with MBP or TrxA tagged recombinant proteins were present in supernatants as a soluble protein as shown by native PAGE analysis and the protein was shown to be present as monomer. The yields of soluble products for the four EST’s recombinant proteins were relatively higher from using rich YT medium in the presence of 4% glycerol in comparison to growth in LB medium in the presence of 4% glycerol. The yields of 38-MPB fusion protein in; LB medium, LB + 4% glycerol medium, YP medium and YP + 4% glycerol medium were; 22.8 mg/L, 28.3 mg/L, 26.6 mg/L and 41.7 mg/L respectively. The yields of 38-TrexA fusion protein in LB medium, LB + 4% glycerol medium, YP medium and YP + 4% glycerol medium were; 25.3 mg/L, 34.5 mg/L, and 43.4 mg/L respectively. Same findings were observed for; 5704-MBP, 5704-TrexA, 6987-MBP, 6987-TrxA, 4010- MBP and 4010-TrexA fusion proteins (see Table 1 for details).
Du Xiu-bol et al. [27] found that, the addition of glycerol (4% v/v), to the culture medium, enhanced the expression level of soluble scFv2F3. They proposed that, the chemical chaperones (glycerol) provide carbon source and protecting cells from IPTG’s toxicity. Hansen andEriksen [28] found that, the fraction of GST that was expressed as an active protein was 2.4 times higher in cultures grown on glycerol compared to cultures grown on glucose. They proposed that, the activity of GST is particularly significant when GST is used as a fusion partner since activity assays and affinity chromatographic methods for quantification and purification of the fusion proteins depend on the GST activity [28]. Our experimental work showed that, the final cell densities of cultures of rich YT medium (no NaCl) supplemented with 4% glycerol were lower than that of the rich YT medium grown cells (for example in the final OD600 nm of 2.82 ± 0.12, and 0.86 ± 0.02 for rich YT and YT with glycerol, respectively). Similar results were obtained from using LB and LB with 4% glycerol, though the final cell densities of cultures were lower than with the rich YP medium and the rich YP medium with 4% glycerol (final OD600 nm of 1.98 ± 0.11, and 1.742 ± 0.7, for LB medium and LB medium with 4% glycerol respectively) (Figure 5).
Table 1. The yield of soluble V. inaequalis EST’s fusion proteins expressed in E. coli grown in LB and YP media without or with 4% glycerol (growth condition presented in Materials and Methods).
aDetermined by using a BCA assay; bDetermined by image densitometry software (ImageJ: http://rsb.info.nih.gov/ij/) of SDS-polyacrylamide gel (data not shown).
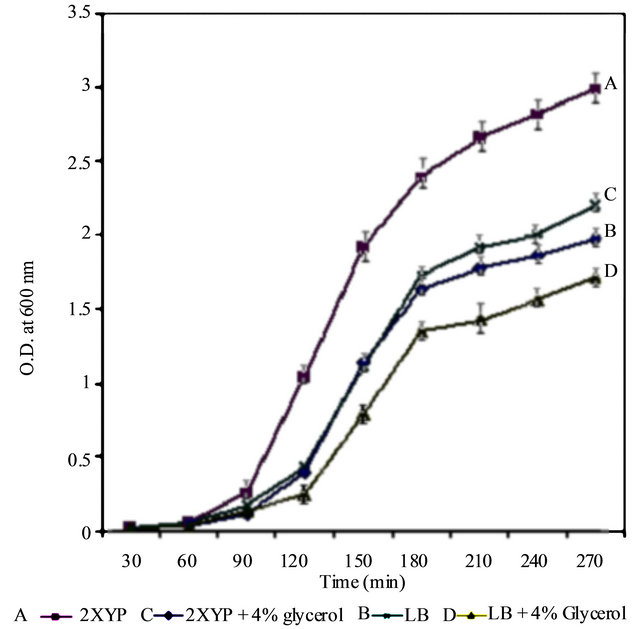
Figure 5. Growth of E. coli BL21 on LB [ ], LB with 4% glycerol [ ], Bacto-Tryptone + Yeasst Extract [ ], Bacto-Tryptone + Yeasst Extract with 4% glycerol [ ]. Data points are shown as averages with error bars representing the range of OD values for each time point. Each line represents a sigmoidal fit of the corresponding growth data.
4. ACKNOWLEDGEMENTS
We thank Christopher A. Kirk from Plant and Food Research Institute, Palmerston North, NZL for expert technical assistance.
REFERENCES
- MacHardy, W.E. (1996) Apple scab: Biology, epidemicology and management. APS Press, St. Paul.
- Kucheryava, N., Bowen, J.K., Sutherland, P.W., Connolly, J.J., Mesarich, C.H., Rikkerink, E.H.A., Kemen, E., Plummer, K.M., Hahn, M. and Templeton, M.D. (2008) Two novel Venturia inaequalis genes induced upon morphogenetic differentiation during infection and in vitro on cellophane. Fungal Genetics and Biology, 45, 1329-1339. doi:10.1016/j.fgb.2008.07.010
- Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular cloning: A laboratory manual. 2nd Edition, Cold Spring Harbor Press, New York, 47-59.
- Parks, T.D., Howard, E.D., Wolpert, T.J., Arp, D.J. and Dougherty, W.G. (1995) Expression and purification of a recombinant tobacco etch virus NIa proteinase: Biochemical analyses of the full-lengthand a naturally occurring truncated proteinase form. Virology, 210, 194-201. doi:10.1006/viro.1995.1331
- Jones, W.T., Harvey, D., Kirk, C., Rakonjac, J., Sun, X., Frearson, N. and Al-Samarrai, T.H. (2007) Novelpeptide tag for detection and purification of recombinant expressed proteins. Protein Expression and Purification, 53, 404-410. doi:10.1016/j.pep.2007.01.006
- Al-Samarrai, T.H., Kirk, C.A., Jones, W.T., Harvey, D. and Sun, X.L. (2007) Expression in Escherichia coli and in vitro refolding of the plant transcription factor Arabidopsis thaliana RGL3. Protein Expression and Purification, 53, 289-292. doi:10.1016/j.pep.2007.01.008
- Martinez, A., Knappskog, P.M., Olafsdottir, S., Døskeland, A.P., Eiken, H.G., Svebak, R.M., Bozzini, M., Apold, J. and Flatmark, T. (1995) Expression of recombinant human phenylalanine hydroxylase asfusion protein in Escherichia coli circumvents proteolytic degradation by host cell proteases, Isolation and characterization of the wild-type enzyme. Biochemical Journal, 306, 589- 597.
- Jacquet, A., Daminet, V., Haumont, M.L., Garcia, Chaudoir, S., Bollen, A. and Biemans, R. (1999) Expression of a recombinant Toxoplasma gondii ROP2 fragment as a fusion protein in bacteria circumvents insolubility and proteolytic degradation. Protein Expression and Purification, 17, 392-400. doi:10.1006/prep.1999.1150
- Kapust, R.B. and Waugh, D.S. (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science, 8, 1668-1674. doi:10.1110/ps.8.8.1668
- Stevens, R.C. (2000) Design of high-throughput methods of protein production for structural biology. Structure, 8, 177-185. doi:10.1016/S0969-2126(00)00193-3
- Georgiou, G. and Valax, P. (1996) Expression of correctly folded proteins in Escherichia coli. Current Opinion in Biotechnology, 7, 190-197. doi:10.1016/S0958-1669(96)80012-7
- Sørensen, H.P. and Mortensen, K.K. (2005) Advanced genetic strategies for recombinant expression in Escherichia coli. Journal of Biotechnology, 115, 113-128. doi:10.1016/j.jbiotec.2004.08.004
- Schein, C.H. (1989) Production of soluble recombinant proteins in bacteria. Biotechnology, 7, 1141-1148.
- Vasina, J.A. and Baneyx, F. (1996) Recombinant protein expression at low temperatures under the transcriptional control of the major Escherichia coli cold shock promoter cspA. Applied and Environmental Microbiology, 62, 1444-1447.
- Vanzi, F., Madan, B. and Sharp, K. (1998) Effect of the protein denaturants urea and guanidinium on water structure: A structural and thermodynamic study. Journal of the American Chemical Society, 120, 10748-10753. doi:10.1021/ja981529n
- Jess, A.V. and Baney, F. (1997) Expression of aggregation-prone recombinant proteins at low temperatures: A comparative study of the Escherichia coli cspA and tac promoter systems. Protein Expression and Purification, 9, 211-218. doi:10.1006/prep.1996.0678
- Chesshyre, J.A. and Hipkiss, A.R. (1989) Low temperatures stabilize interferon α-2 against proteolysis in Methylophilus methylotrophus and Escherichia coli. Applied Microbiology and Biotechnology, 31, 158-162. doi:10.1007/BF00262455
- Jiang, W., Hou, Y. and Inouye, M. (1997) CspA, the major cold-shock protein of Escherichia coli, is an RNA chaperone. The Journal of Biological Chemistry, 272, 196-202. doi:10.1074/jbc.272.1.196
- Arnold, U.R. and Ulbrich-Hofmann, R. (2001) Proteolytic degradation of ribonuclease A in the pretransition region of thermally and urea-induced unfolding. European Journal of Biochemistry, 268, 93-97. doi:10.1046/j.1432-1327.2001.01849.x
- Sachdev, D. and Chirgwin, J.M. (1998) Solubility of proteins isolated from inclusion bodies enhanced by fusion to maltose-binding protein or Thioredoxin. Protein Expression and Purification, 12, 122-132. doi:10.1006/prep.1997.0826
- Speed, M.A., Wang, D.I.C. and King, J. (1996) Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nature Biotechnology, 14, 1283. doi:10.1038/nbt1096-1283
- Emerick, A.W., Bertolani, B.L., Ben-Bassat, A., White T.J. and Konrad, M.W. (1984) Expression of a beta-lactamase preproinsulin fusion protein in Escherichia coli. BioTechnology, 2, 165-168. doi:10.1038/nbt0284-165
- Jurgen, B., Lin, H.Y., Riemschneider, S., Scharf, C., Neubauer, P., Schmid, R., Hecker, M. and Schweder, T. (2000) Monitoring of genes that respond to overproduction of an insoluble recombinant protein in Escherichia coli glucose-limited fed-batch fermentations. Biotechnology and Bioengineering, 70, 217-224. doi:10.1002/1097-0290(20001020)70:2<217::AID-BIT11>3.0.CO;2-W
- Lesley, S.A., Graziano, J., Cho, C.Y., Knuth, M.W. and Klock, H.E. (2002) Gene expression response to misfolded protein as a screen for soluble recombinant protein. Protein Engineering, 15, 153-160. doi:10.1093/protein/15.2.153
- Hoffmann, F. and Rinas, U. (2000) Kinetics of heat-shock response and inclusion body formation during temperature-induced production of basicfibroblast growth factor in high-cell-density cultures of recombinant Escherichia coli. Biotechnology Progress, 16, 1000-1007. doi:10.1021/bp0000959
- Andersen, K.B. and von Meyenburg, K. (1980) Aregrowth rates of Escherichia coli in batch cultures limited by respiration? Journal of Bacteriology, 144, 114-123.
- Xiu-bol, D.U., Sun, Y., Lin, F., Zheng, K.-Y., Wang, K.-W., Lin T.-T., Liu J.-Q., Shen, J.-C. and Luo G.-M. (2007) Chemical chaperones increasing expression level of soluble single-chain Fv antibody (scFv2F3). Chemical Research in Chinese Universities, 23, 69-75. doi:10.1016/S1005-9040(07)60014-2
- Hansen, R. and Eriksen, N.T. (2007) Activity of recombinant GST in Escherichia coli grown on glucose and glycerol. Process Biochemistry, 42, 1259-1263. doi:10.1016/j.procbio.2007.05.022