American Journal of Molecular Biology
Vol.2 No.4(2012), Article ID:23901,13 pages DOI:10.4236/ajmb.2012.24031
Anaplerosis in cancer: Another step beyond the Warburg effect
![]()
1Unidad de Genética de la Nutrición, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Mexico City, Mexico
2Facultad de Medicina, Programa de Posgrado en Ciencias Médicas, Odontológicas y de la Salud, Universidad Nacional Autónoma de México, Mexico City, Mexico
Email: rdruiz8@hotmail.com
Received 28 July 2012; revised 1 September 2012; accepted 10 September 2012
Keywords: Anaplerosis; Biosynthesis; Cancer; Glutaminase; Krebs Cycle; Metabolism; Mitochondria; Pyruvate Carboxylase; Warburg Effect
ABSTRACT
Biosynthesis is up-regulated in tumors and thus the demand for anabolic intermediates is increased. The metabolic routes providing the building blocks for macromolecules are thus a very attractive target as they are not normally up-regulated in a normal quiescent cell. Some routes for glycolysis-derived intermediates production have been identified, but these do not constitute the whole pool of biosynthetic molecules in the cell, as many of these derive from mitochondria in the Krebs cycle. Indeed, this metabolic pathway is considered a “biosynthetic hub” from which anabolism is fed. If a metabolite efflux is indeed occurring, anaplerotic reactions must keep a steady supply of substrates. In spite of this obvious relevance of anaplerosis, it has been poorly characterized in the malignant cell context. Glutaminolysis and and pyruvate carboxylation are two pathways that function in an anaplerotic fashion. In spite of the increasing evidence implicating these two processes in cancer metabolism their role as intermediate providers is overlooked. In this review we analyze the implications of an active anaplerosis in cancer and we discuss experimental evidence showing the relevance of these metabolic routes in tumor physiology.
1. INTRODUCTION
Cancer is a group of diseases considered as a public health issue because of their increasing incidence, the lack of effective treatments for the vast majority of the diagnosed cases, and the high mortality rate of cancer patients. These diseases can affect virtually every organ of the human body and they are characterized by the formation of malignant tumors, which in some cases can spread out to distant sites in the body (metastasis). During the last decades, the research on cancer has intensified and some important discoveries have been made in the field, such as the identification of oncogenes, tumor suppressor genes and the development of targeted antitumor drugs. In spite of these advances and some paradigm-shifting breakthroughs, the clinical progress is minimal and it is possible that additional and more specific therapeutic targets remain to be discovered. Still, one of the main challenges for cancer therapy is to discriminate between malignant and normal cells.
Almost 80 years ago Otto Warburg identified that cancer cells display a different metabolic program regarding their normal counterparts. He observed that tumors have a decreased respiration and an enhanced lactate production in presence of oxygen [Reviewed in 1]. The term coined for this phenomenon was “aerobic glycolysis”, nowadays known as “the Warburg effect”. Notwithstanding the relevance of these findings, the study of cancer metabolism did not receive much attention in the next decades after its discovery, as most of the work done in the cancer field was focused on genetics and the identification of deregulated signal transduction pathways. Nonetheless, the importance of aerobic glycolysis was demonstrated through the implementation of 18fluoro-deoxyglucose-based positron emission tomography (FDG-PET) [2]. Based on the high glycolytic capacity of tumors and their enhanced glucose uptake, this technique allowed the visualization of tumors in vivo resulting from the accumulation of the non-metabolizable probe by malignant cells.
However, a decade ago, the aerobic glycolysis phenomenon was rediscovered. Molecular evidence was gathered linking bona fide oncogenes and tumor suppressors with the metabolic alterations previously identified by Warburg (Table 1). Some of these findings were in accordance to Warburg’s original hypothesis: “in cancer cells an irreversible damage to mitochondria must

Table 1. Metabolic enzymes evaluated as essential for the maintenance of the glycolytic phenotype in tumors (see text for details and references).
exist concomitantly with the enhancement of the glycolytic flux” (see Tables 1 and 2) [3]. For instance, a mitochondrial damage was identified in phaeochromocytomas and paragangliomas. These tumors carry a mutation in different subunits of succinate dehydrogenase (the mitochondrial complex II), encoded by the genes SDHB, SDHC and SDHD [4]. Indeed, it has been demonstrated that these tumors display the Warburg effect [5]. Similarly, a biallelic inactivation of the FH gene, coding for mitochondrial fumarase, is inactivated in leyomiomas [4]. More evidence came from studies with the tumor suppressor p53, which is commonly mutated in most cancer types. The absence of p53 impairs the induction of SCO2 which is essential for the assembly of cytochrome oxidase subunit II (COX II), thus rendering the cells less dependent on oxidative phosphorylation [6].
The enhancement of glycolysis was explained by the overexpression of all the glycolytic enzymes, especially those ones considered as flux-limiting in the metabolic pathway: hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK) [Reviewed in 7]. The embryonic isoforms, are preferentially expressed in tumors, as they allow a higher metabolic flux compared to those expressed in differentiated tissues [7]. Moreover, some of these enzymes are presumably involved in nonconventional roles by endowing tumor cells with the ability to proliferate and overcome programmed cell death mechanisms. For instance, HK seem to dampen the apoptosis-induction mechanism of Bax by interacting with mitochondria [8]; The M2 isoform of PK appears to be essential for sustaining proliferation in conjunction with the epigenetic machinery and its activity may itself constitute a proliferation signal [9,10]. Table 1 lists some of the enzymes evaluated as essential for the maintenance of the glycolytic phenotype in tumors.
Furthermore, some oncogenes commonly activated in a number of tumors, such as Akt and MYC, are directly involved in the metabolic switch to aerobic glycolysis (Table 2) [11]. It was discovered that one of the main drivers of the Warburg effect is hypoxia-induced factor 1-α (HIF-1α), which is a transcription factor that activates the expression of all glycolysis enzymes, it triggers the translocation of glucose transporters to the plasma membrane, and it limits the substrate influx into the Krebs cycle by allowing the expression of pyruvate dehydrogenase kinase (PDK1) [12,13]. HIF-1α is stable and active in hypoxic conditions allowing the cells to switch to an anaerobic metabolism. However, in several tumors this transcription factor is active even in normoxic conditions by diverse mechanisms.
The relationship between metabolism and proliferation signals in cancer is being unveiled as other signaling molecules which are involved in the regulation of proliferation and differentiation (e.g. LKB1, Cyclin D1 and Notch) [14-16] are inextricably linked to energy metabolism as well. Table 2 lists some of these, along with their reported function for the maintenance of the glycolytic phenotype. The detailed discussion about oncogenes and tumor suppressors and their specific role in tumor metabolism is out of the scope of this review and the reader is referred to excellent reviews in the literature [11,17].
Based on these discoveries, it seemed that cancer cell metabolism could be regarded as a promising specific target. Several therapeutic approaches have been proposed, such as the inhibition of specific glycolysis enzymes (hexokinase, pyruvate kinase and lactate dehydrogenase) [18-20], the reactivation of mitochondrial metabolism by stimulating pyruvate oxidation [20], refeeding oxidative substrates, and the inhibition of the hypoxic metabolic program by interrupting the expression and/or activity of HIF-1α [21]. Encouraging results have been obtained in vitro for some of these strategies, but their effectiveness in cancer patients is still unknown.

Table 2. Signals commonly deregulated in cancer and their possible role for the aerobic glycolysis maintenance.
During the last five years it has been also demonstrated that cancer cell metabolism goes beyond a simple interplay between glycolysis and mitochondria, in contrast with Otto Warburg’s original proposal. A considerable body of evidence shows that some specific tumors rely mainly on oxidative phosphorylation [Reviewed in 22]. Other previously overlooked metabolic pathways are shown to be crucial for tumor survival: glutamine metabolism is highly important for energy obtaining and as a biosynthetic pathway [23], NADP+-dependent isocitrate dehydrogenases are directly implicated in tumorigenesis through the production of a putative oncometabolite (2-oxoglutarate) [24], and de novo fatty acid synthesis is overactive in malignant cells [25]. These findings highlight the need to evaluate cancer cell metabolism in a more exhaustive fashion as many other metabolic pathways might contribute to cancer cell survival and be susceptible of pharmacological inhibition for cancer therapy.
Aerobic glycolysis probably endows cancer cells with the ability to generate biosynthetic intermediates from the glycolytic pathway, thus enabling tumors with the ability to proliferate faster and outnumber their normal counterparts [26]. This current “biosynthesis model” envisions an overall predominance of anabolic metabolism, i.e. intermediates for protein, fatty acids and nucleic acid biosynthesis must be supplied at sufficiently high rates. Some of these can be supplied by glycolysis and the pentose phosphate shunt, and in theory, the rest of them should be obtained from the Krebs cycle. The latter constitutes one of the inconsistencies of this model, as Krebs cycle functioning is driven by the mitochondrial respiratory chain activity, which is assumed to be impaired or down-regulated in cancer. However, recent findings evidence the presence of functional mitochondria in tumor cells and show their significant contribution for anabolic flux in malignant cells (see Section 3.1). These discoveries challenge the “Warburg effect” hypothesis and unveil a complex scenario where energy metabolism in cancer goes beyond a simple interplay between glycolysis and mitochondria. Hence, if the Krebs cycle is indeed func-tioning as a provider for anabolism, the net metabolite efflux must be considerable, given the high demand for macromolecule building blocks during fast-rate proliferation. In order to keep the functioning of the pathway, the efflux rate must be equal to the influx rate. Thus, the substrate provision rate for Krebs cycle must be consequently increased. A constant pyruvate supply to mitochondria (provided by an accelerated glycolysis) is probably not sufficient to compensate for metabolite efflux, and hence some mechanism assuring a steady intermediate replenishment must be operating. Anaplerotic reactions fulfill such function, as in normal cells their physiological role is to ensure a constant supply of intermediates for the continuous metabolic activity of the Krebs cycle. In the context of a tumor cell with an increased demand for biosynthesis intermediates, anaplerosis reactions might be crucial. Although these metabolic pathways are overlooked in cancer metabolism studies, in this review we highlight their possible role in tumor physiology. We also discuss their potential as therapeutic targets in cancer.
2. ANAPLEROSIS
The Krebs cycle is a central pathway in energy metabolism. This metabolic route provides reducing equivalents that serve as substrate for the mitochondrial respiratory chain, thus contributing to energy (ATP) production in the cell. In addition, it is the source of anabolic precursors, such as aminoacids for protein biosynthesis, of citrate used for de novo fatty acid synthesis, of succinylCoA required for heme group synthesis, and oxaloacetate, which is an essential intermediate for gluconeogenesis [27]. Thus, there is a constant efflux of intermediates from the Krebs cycle, which must be replenished in order to maintain the carbon flux throughout the oxidative metabolism pathway (Figure 1). The anaplerotic reactions are those who regenerate these intermediates used for biosynthetic metabolism. Although there are several enzymes that might fulfill this role (see below), two of them are well characterized: pyruvate carboxylase (PC) and glutaminase (GLS) (Figure 1) [28].

Figure 1. Overview of anaplerotic reactions occurring in mitochondria. Dotted lines indicate the reactions participating in Krebs cycle intermediate replenishment. Pyruvate carboxylase (1) regenerates the oxaloacetate used for citrate synthesis; Aspartate aminotranferase can also produce oxaloacetate, which is the result from the transamination of aspartate (2); Propionyl-CoA carboxylase in the odd-chain fatty acid degradation pathway yield succinyl-CoA (3); Glutaminase deaminates glutamine producing glutamate (4); and the latter serve as the substrate of Glutamate dehydrogenase producing α-ketoglutarate. Alanine aminotransferase also produces α-ketoglutarate from the transamination of glutamate with pyruvate.
Pyruvate carboxylase (PC) is considered the main anaplerotic enzyme and it catalyzes the ATP-dependent conversion of pyruvate to oxaloacetate (Figure 1) in a process dependent on biotin as a coenzyme. PC is expressed in a wide variety if tissues, but it is of special relevance in liver, brain, adipose tissue, kidney and pancreatic islets. This enzyme has a relevant role for gluconeogenesis, glycerol synthesis and the down-regulation of fatty acid synthesis [29] (See below). Although there is only one gene coding for PC, there are several variants resulting from alternative splicing. These different forms might be expressed depending on the physiological state of the cell and their specific functionality remains unclear [30].
PC anaplerotic function is essential for insulin secretion since it maintains the pool of Krebs cycle intermediates in order to allow the complete oxidation of glucose to CO2 and H2O. This is important, as it is widely known that β-pancreatic cells must oxidize glucose from the blood stream in order to increase the ATP/ADP ratio that triggers insulin secretion [31]. Besides its direct anaplerotic role, PC is also important for the generation of a NADPH pool that might serve as substrate for anabolic pathways [30].
De novo glucose synthesis (gluconeogenesis) is the most studied function of PC in liver. This biosynthetic process is stimulated by glucagon, which enhances pyruvate conversion to oxaloacetate. In a second step, phosphoenol pyruvate carboxykinase (PEPCK) uses the latter to produce phosphoenolpyruvate, which is required for the gluconeogenic pathway [32]. Nonetheless, PC possibly has an anaplerotic role in liver as demonstrated in a NMR analysis in which it was shown that pyruvate carboxylase is the major pathway for pyruvate influx into the Krebs cycle. This study measured an anaplerotic flux seven-fold higher in comparison with pyruvate oxidation [33].
In central nervous system, anaplerotic reactions are essential for neurotransmitter synthesis, such as glutamate and γ-aminobutyric acid (GABA). Both of them can be produced from α-ketoglutarate, which is normally consumed during Krebs cycle functioning. PC role is predominant in astrocytes, where it compensates the constant efflux of intermediates because of glutamate and GABA synthesis [34]. The glutamate synthesized by astrocytes must reach the neurons and it is released in the form of glutamine, which is reconverted to glutamate by the latter in a global process called “the glutamate/glutamine cycle”. It has been demonstrated that the glutamine flux is dependent on the activity of astrocyte PC [35]. Thus, normal brain functioning rely on PC activity.
Glutaminase (GLS) is a mitochondrial enzyme that catalyzes the deamination of glutamine to produce ammonium ions and glutamate (Figure 1). There are two glutaminase isoforms: the K-type, the main isoform detected in kidney, and the L-type, which is predominant in liver. The former is the ubiquitous and detected in non-hepatic tissues. In spite of the long-time consideration that the K-form was the only one present in brain, both isoforms can be found in this organ and they might play a differential metabolic role [36]. In liver, this enzyme can be part of an anaplerotic route to produce α-ketoglutarate in conjunction with aspartate-alanine aminotransferase (ALT), which catalyzes its transamination with pyruvate to yield α-ketoglutarate and alanine. In brain, a similar pathway has been identified as part of the catabolism of the neurotransmitter glutamate, which can also be transaminated in a similar manner by astrocytes [37].
Some other metabolic pathways can provide for Krebs cycle intermediates and thus could be classified as anaplerotic. Such is the case for the reaction catalyzed by adenylosuccinate synthetase in skeletal muscle that synthesizes fumarate and AMP from adenylosuccinate in the purine nucleotide cycle. Other pathway for oxaloacetate synthesis is the transamination of aspartate catalyzed by aspartate aminotransferase (AST), which is of special importance in liver and skeletal muscle. The catabolism of odd-chain fatty acids can derive in the production of succinyl-CoA in a reaction catalyzed by the biotin-dependent enzyme propionyl-CoA carboxylase.
3. ANAPLEROSIS IN CANCER
3.1. The Existence of Functional Mitochondria in Cancer?
According to Warburg’s original hypothesis, mitochondria ought to be irreversibly damaged [3]. Although this seems to be the case for some malignancies such as phaeochromocytomas and leiomyomas (see Section 1), it has been well documented that several cell lines derived from a wide variety of tumors rely mainly on oxidative phosphorylation (Reviewed in [19]). For instance, breast cancer cells obtain their energy mainly from mitochondria [38], and this seems to be the case for other epithelial cancers as well, for which the activity of cytochrome oxidase (complex IV) is up-regulated causing a hyperactivation of mitochondrial metabolism [39]. A similar feature has been observed in liver and brain cancer, where a fully functional oxidative phosphorylation has been characterized [40,41]. Mitochondrial oxidative metabolism probably prevails in brain tumors in vivo, as it has been demonstrated that glioblastomas display a preference for glucose oxidation when implanted in mouse brain [42]. Further evidence comes from the study of lipolysis in cancer, as the hydrolysis of triglycerides is essential for cancer-associated cachexia induced by lung carcinoma and melanoma implantation in mice [43]. This clearly points out that cancer cells can use fatty acids (FA) as fuels in vivo. A similar mechanism occurs in ovarian cancer, as adipocytes provide malignant cells with lipids as substrates in order to support metastasis and invasion [44]. Of note, FA metabolism is carried out through the β-oxidation pathway in mitochondria, which is a process highly dependent on a functional oxidative phosphorylation.
Furthermore, some tumors seem to display metabolic flexibility, i.e. they can switch between aerobic glycolysis and oxidative metabolism depending on their microenvironment [45,46]. These cells trigger adaptive mechanisms (e.g. mitochondrial biogenesis and the formation of an extensive mitochondrial network) in order to optimize their oxidative phosphorylation according to the substrate supply and their energy demand [47]. In the vast majority of the in vitro studies, functional mitochondria and metabolic adaptation mechanisms could have been masked because of the cell culture conditions, in which a defined metabolic program is imposed on the cells as a function of the media composition and the optimal growth conditions. However, it has been shown that tumor microenvironments are highly heterogeneous due to glucose and oxygen gradients [47]. In the in vivo scenario, the metabolic transitions could be crucial for tumor physiology. For instance, experimental evidence shows a “lactate shuttling” between glycolytic and oxidative cells in lung carcinoma and colorectal adenocarcinoma [48]. This might facilitate a symbiosis between different populations within a tumor: the glycolytic cells would function as the “substrate providers” for those who are able to synthesize ATP through oxidative phosphorylation. A similar phenomenon, termed “the reverse Warburg effect”, has been characterized between stromal fibroblasts and breast cancer cells where the former provide lactate, ketones, glutamine and free fatty acids (being the four of them substrates for the mitochondrial respiratory chain) to the oxidative malignant cells [49].
Originally, Warburg’s observations posed a conundrum for researchers in the cancer field: how come a highly proliferative cell would support its proliferation using mainly glycolysis, a relatively inefficient pathway in terms of energy yield? One possible explanation is that, even if glycolytic ATP production is lower, it might be accelerated in order to meet the energy demand imposed by the fast proliferation rate of tumors [50]. Another non-exclusive possibility is that glycolytic flux enhancement brings as consequence the accumulation of metabolites subsequently used as building blocks for biosynthetic processes [26]. Some experimental evidence supports this theory. For instance, there is an alternative glycolytic pathway that uncouples ATP production at the pyruvate kinase step in order to promote metabolite accumulation for biosynthetic purposes [51]. In this regarda possible source of intermediates for serine and glycine biosynthesis might be at the phosphoglycerate dehydrogenase step, which function as a shunt from glycolysis and whose gene it is commonly amplified in tumors [52]. Moreover, the overall glycolytic intermediate accumulation may be linked to the tyrosine kinase deregulated signaling pathways commonly observed in tumors [53].
Nevertheless, in order to cope with the increased demand for biosynthesis intermediates, those ones derived solely from glycolysis are most probably insufficient to sustain a high anabolic flux. In normal cells the overall anabolic flux is heavily dependent on mitochondria and the Krebs cycle functioning (see Section 2). In support of this hypothesis, it is known that some molecules previously identified as essential for cancer cell proliferation, such as citrate [25] and heme prosthetic groups [54], are exclusively derived from Krebs cycle intermediates. Although anaplerosis in cancer remains ill-defined, recent findings suggest that mitochondria-localized metabolism is of special relevance for cancer. Some of these processes are linked to anaplerotic pathways and their relevance is beginning to be elucidated.
3.2 Glutaminolysis
Based on FDG-PET scan observations it was previously considered that tumors avidly consumed only glucose. This observation was supported by the detected overexpression of glucose transporters in several cancer cell lines [55]. Nevertheless, glucose is not the only highly consumed substrate in tumors, as in vivo studies demonstrated a considerable glutamine uptake by malignant overgrowths [56]. Although glutamine per se can serve as a biosynthetic intermediate (for a review see [57]), glutaminolysis is nowadays a well-characterized process inherent of several tumors. In accordance to this, both Land K-type glutaminase isoforms were found to be simultaneously expressed at high levels in leukemia cells, although the predominance of the latter was evident [58]. The relevance of the K isoform was further verified in lymphomas and on colorectal adenomas and carcinomas [59]. Based on these findings, it has been suggested that K-glutaminase has a defined role for tumor cell proliferation, although more studies need to be conducted in this regard.
The overexpression of glutaminase is observed in other malignancies as well, such as liver, brain and breast tumors [60-62]. Of note, these tumors possess an active mitochondrial metabolism in vitro as characterized by other studies (see Section 3.1).
Glutamine uptake and metabolism have been detected in proliferating non-tumor lymphocytes and in HeLa cells. In these studies it was discovered that glutaminase expression and activity is regulated according to the cell cycle stage, being most active during the replication (S)
phase [63,64]. Interestingly, during this same cell cycle phase, mitochondrial biogenesis and oxidative phosphorylation are up-regulated [65]. Furthermore, it has been demonstrated that glutaminase expression depends on MYC, which is a commonly activated oncogene in a wide variety of tumors [66]. It is noteworthy the fact that MYC also promotes mitochondrial biogenesis [67]. This experimental evidence highlights the dependence of glutamine metabolism on functional mitochondria. Moreover, it points out the possible existence of a specific program in which MYC, mitochondrial biogenesis and anaplerosis are coordinated as central elements of energy metabolism in some malignancies (see below).
As pointed out earlier, glutaminase serves to replenish α-ketoglutarate to the Krebs cycle. It has been proposed as the only anaplerotic route in tumors [68]. Although there are no studies focused to elucidate the anaplerotic function of glutaminolysis in cancer, recent reports indirectly demonstrate its implication in metabolic processes in some malignant cells. For instance, glutamine consumption feeds the Krebs cycle yielding fumarate, malate and citrate in an overall process coordinated by MYC in Burkitt lymphoma cells [69]. Citrate can be exported to cytosol where it serves as substrate of ATP citrate lyase (ACL) producing cytosolic acetyl-CoA, which is required as an intermediate for de novo fatty acid synthesis. ACL is found to be essential for tumor cell survival as acetyl groups provider for histone acetylation in brain cancer [25], and our own findings demonstrate that ACL is activated in primary glioblastomas but not in normal human astrocytes (Díaz-Ruiz, R. Unpublished results). The involvement of anaplerosis as a compensating mechanism of mitochondrial citrate production in brain cancer is an issue that will be subject of further research.
In this regard, another report proposed a “reductive” metabolic pathway in which the α-ketoglutarate derived from glutamine metabolism is used in order to produce cytosolic acetyl-CoA for lipid synthesis in a process dependent on the activity of the cytosolic form of isocitrate dehydrogenase (IDH1) [23]. There are some inconsistencies with this model, as they propose IDH1 as a central element of this reductive pathway. However, glutamine needs to be processed in mitochondria in order to produce α-ketoglutarate, whether if it is being supplied through the oxidation of the glutamate produced by glutaminase activity, or through glutamate transamination by alanine aminotransferase (ALT). And even if α-ketoglutarate is being carboxylated to isocitrate, the latter is not used as lipogenic precursor, being citrate the one used for such purposes. Moreover, citrate efflux from mitochondria is mandatory for cytosolic acetyl-CoA production and this feature has been clearly identified in hepatomas [70]. An alternative explanation is that glutaminolysis is functioning in an anaplerotic fashion in order to compensate mitochondrial citrate efflux and IDH1 may be supplying the NADPH cofactors required by two of the six steps in de novo fatty acid biosynthesis (Figure 2).
According to the reductive carboxylation hypothesis, it is proposed that a reversed Krebs cycle is the source of citrate and acetyl-CoA for biosynthesis [71]. The drawback in this proposal is of thermodynamic nature, as the reversal of the Krebs cycle implies a build-up of intramitochondrial NADH that would inhibit the pyruvate dehydrogenase complex thus inducing the dampening of pyruvate supply to mitochondria thereby inhibiting the Krebs cycle. It has been demonstrated by other reports that exogenous glutamine yields citrate [69], therefore it is more plausible that cytosolic citrate and acetyl-CoA are derived from the normal functioning of the Krebs cycle supported by an enhanced anaplerosis from glutamine (Figure 2).
In spite of the caveats mentioned above, it is clear that glutamine metabolism is being carried out in order to contribute with intermediates for biosynthesis in cancer. A more detailed study focusing on its anaplerotic role might clarify the inconsistencies of the reductive carboxylation hypothesis and it could offer some insight into the overall tumor energy metabolism. Anaplerotic glutaminolysis could be a therapeutic target as well. Several in vitro studies show that glutaminase inhibition affect tumor cell viability [72,73]. Moreover, by interfering with the signaling routes linked with glutaminase it is possible to selectively eliminate malignant cell such as lymphomas and breast cancer cells [74]. Although anaplerosis assessments in these conditions are lacking, most probably they are being affected as well. In order to confirm this, studies on the impact of anaplerosis inhibition on tumor cell viability need to be addressed.
3.3 Pyruvate Carboxylase
As mentioned in Section 2, pyruvate carboxylase (PC) is the main anaplerotic enzyme in normal cells. Early reports proposed that it has no participation in tumor cell metabolism as its activity is low in gliomas [75] and also in hepatomas [76]. However, an up-regulation of biotin-binding enzymes have been found in pancreatic tumors, particularly in ductal adenocarcinomas [77], increased levels of PC have been identified in several tumor cell lines [78], and perfused livers of tumor-bearing
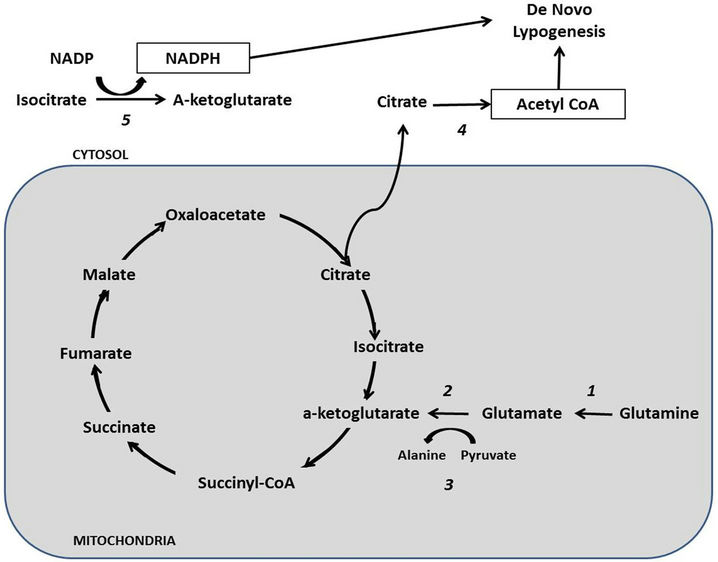
Figure 2. Proposed model for glutamine reductive metabolism for fatty acids biosynthesis based on anaplerotic reactions. Glutaminase (1) is the entry point of this pathway by catalyzing the deamination of glutamine to glutamate, which subsequently enters the Krebs cycle as α-ketoglutarate after either being oxidized by Glutamate dehydrogenase (2); or after being transaminated by Alanine aminotransferase (3); Citrate is generated after substrate influx. The latter can be exported to cytosol where it serve as the substrate of ATP Citrate lyase (4); producing the acetyl-CoA required for de novo fatty acid synthesis. Isocitrate dehydrogenase 1 (5) generates NADPH, the other substrate needed for fatty acid biosynthesis.
rats displayed a high alanine flux arising from PC activity [79]. These inconsistencies might arise from the use of different cell culture/incubation conditions. As pointed out earlier (see 3.1), brain and liver tumors are sensitive to the composition of the cell culture media, particularly to glucose levels. High glucose levels tend to repress oxidative metabolism in a reversible fashion and this does not necessarily implies a mitochondrial impairment [80]. Thus, it is possible that the contribution of PC and other anaplerotic enzymes was masked if these experiments were conducted using standard commercial media containing high supra-physiological glucose concentrations.
The relevance of PC in cancer is highlighted by recent findings as it was discovered a correlation between the Wnt signaling (which is commonly activated in breast and colon cancer) and anaplerosis in tumors. In this study it was reported that the activation of Wnt repressed mitochondrial cytochrome oxidase (COX) expression concomitantly with the stimulation of lactate production. Surprisingly, PC expression was enhanced, suggesting a possible activation of the anaplerotic pathways [81]. These results seem contradictory as they imply the induction of the aerobic glycolysis, while anaplerosis is indicative of a high Krebs cycle activity. A more detailed characterization of energy metabolism and anaplerosis is needed in order to draw some conclusions. However, there are interesting implications arising from this study as Wnt is an important trigger for the epithelial-to-mesenchymal transition (EMT). The latter is an expression program that confers to the cell with the ability to detach from its niche in order to migrate. Although this was a feature identified in embryogenesis, it has been found operating in metastatic tumors as well. Thus, a possible link between metastasis and anaplerosis is envisaged. Given the fact that a transition to oxidative metabolism induce and support cancer cell invasiveness in harsh microenvironments [82], it is plausible that anaplerosis serves as a mechanism preserving the functionality of oxidative metabolism in these conditions. More experimental work should be addressed to this particular issue, as anaplerosis if overactive, can be a very attractive therapeutic target to treat invasive tumors.
A further link between Wnt and pyruvate carboxylase is also suggested elsewhere, as intranuclear biotin-enriched particles containing PC and propionyl-CoA carboxylase (PCC) were found in gallbladder adenocarcinomas [83]. These accumulations could be dependent on β-catenin activation, which is the main downstream effector of the Wnt signaling activation. It is not quite clear if these biotin-containing fractions are actually engaged in active anaplerosis because of their nuclear localization, neither its role on energy metabolism functioning. It remains to be shown if in gallbladder carcinomas, as well as in other tumors, PC and PCC are overexpressed and/or overactive, and the mechanism by which they are connected to the Wnt/β-catenin axis.
Pyruvate carboxylation might have a more prominent role than it was thought, as PC can compensate the lack of glutaminase in some tumors. Moreover, these malignant cells were resistant to glutamine deprivation in a PC-dependent fashion. Additional results suggest that even if these cells are grown in a complete medium (nutrient replete conditions), they might depend on both PC activity and other anaplerotic reactions [84]. Additional evidence came from metabolomics studies in human lung tumors in situ. It was shown that PC-mediated anaplerotic flux is overactive in tumors regarding normal tissue [85].
PC most obvious role would be the sustaining of the constant efflux of biosynthetic intermediates. However, as suggested above, more defined roles are probably implied such as cell migration and metastasis. It remains to be defined the specific roles of PC in this scenario and also the elucidation of the advantages conferred by PC to the overall tumor physiology.
In this regard, given the complexity and the branched nature of the Krebs cycle along with its interrelationship with anaplerosis, the implementation of metabolomic analyses will serve to clarify this issue. Up to this date, metabolomics has been proposed as a diagnostic tool for biomarker screening in plasma and urine from cancer patients. Nonetheless, its implementation is being done in order to explore the particular features and the mechanisms governing the metabolic phenotypes in cancer [86]. Metabolomic studies need to be conducted in tumors arising from highly anaplerotic tissues in contrast to their respective malignant counterparts. As liver, glia and pancreatic islets display active anaplerosis, their metabolomic characterization will give an insight of the biosynthetic activities in malignancy perhaps revealing other metabolic deregulations at this level. It is worth to mention that the global metabolite pattern will offer even more therapeutic targets that will be discovered when integrated with data from genetic expression and metabolic fluxes assessment.
4. CONCLUSIONS
Our understanding of tumor cell metabolism is beginning to be broadened. With recent experimental evidence, the conceptual framework set by Otto Warburg early observations is being modified. The paradigm of the “aerobic glycolysis” has even been challenged by the finding of “oxidative” cancer cells and the “reverse Warburg effect”. These concepts are further reinforced by the discovery of glutaminolysis as one of the new hallmarks of cancer cell metabolism. Previously overlooked enzymes turned out to be pivotal, not only for tumor metabolism, but to the tumorigenic process as well (e.g. IDH1 and IDH2). All these discoveries force us to adopt more rigorous approaches in order to clearly identify potential “druggable” targets that might be used in future therapies.
The current biosynthesis model suggests anabolic processes as an attractive therapeutic target in cancer, but we just recently began to understand the anabolic deregulations in malignancies. The steady supply of metabolic intermediates is mandatory in order to synthesize de novo macromolecules. In this scenario, anaplerosis is crucial by keeping the availability of anabolic precursors. Based on recent discoveries, it is becoming clear that both glutaminolysis and pyruvate carboxylation can fulfill this role in tumors. Furthermore, anaplerosis might be implicated in important aspects of tumor physiology such as metastasis. Based on this and given the therapeutic implications, anaplerotic pathways in cancer deserve further study.
5. ACKNOWLEDGEMENTS
We would like to thank Dr. Antonio Velázquez for his invaluable help and advises during the preparation of the manuscript.
REFERENCES
- Koppenol, W.H., Bounds P.L. and Dang, C.V. (2011) Otto Warburg’s contribution to current concepts of cancer metabolism. Nature Reviews on Cancer, 11, 325-337. doi:10.1038/nrc3038
- Jones, T. and Price, P. (2012) Development and experimental medicine applications of PET in oncology. The Lancet Oncology, 13, e116-e125. doi:10.1016/S1470-2045(11)70183-8
- Warburg, O. (1956) On the origin of cancer cells. Science, 123, 309-314. doi:10.1126/science.123.3191.309
- Eng, C., Kiuru, M., Fernandez, M.J. and Aaltonen, M.A. (2003) A role for mitochondrial enzymes in inherited neoplasia and beyond. Nature Reviews on Cancer, 3, 193-202. doi:10.1038/nrc1013
- Pollard, P.J., Brière, J.J., Alam, N.A., Barwell, J., Barclay, E., Wortham, N.C., Hunt, T., Mitchell, M., Olpin, S., Moat, S.J., Hargreaves, I.P., Heales, S.J., Chung, Y.L., Griffiths, J.R., Dalgeish, A., McGrath, J.A., Gleeson, M.J., Hodgson, S.V., Poulsom, R., Rustin, P. and Tomlinson, I.P. (2005) Accumulation of Krebs cycle intermediates and overexpression on HIF1alpha in tumours which result from germline FH and SDH mutations. Human Molecular Genetics, 14, 2231-2239. doi:10.1093/hmg/ddi227
- Matoba, S., Kang, J.G., Patino, W.D., Wragg, A., Boehm, M., Gavrilova, O., Hurley, P.J., Bunz, F. and Hwang, P.M. (2006) p53 regulates mitochondrial respiration. Science, 312, 1650-1653. doi:10.1126/science.1126863
- Diaz-Ruiz, R., Uribe-Carvajal, S., Devin, A. and Rigoulet, M. (2009) Tumor cell energy metabolism and its common features with yeast metabolism. Biochimica et Biophysica Acta—Reviews on Cancer, 1796, 252-265.
- Pastorino, J.G., Shulga, N. and Hoek, J.B. (2002) Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. The Journal of Biological Chemistry, 277, 7610-7618. doi:10.1074/jbc.M109950200
- Yang, W., Xia, Y., Ji, H., Zheng. Y., Liang, J., Hwang, W., Gao, X., Aldape, K. and Luz, Z. (2012) Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature, 480, 118-122.
- Yang, W., Xia, Y., Hawke, D., Li, X., Liang, J., Xing, D., Aldape, K., Hunter, T., Alfred Yung, W.K. and Lu, Z. (2012) PKM2 phosphorylates histone H2 and promotes gene transcription and tumorigenesis. Cell, 150, 685-696. doi:10.1016/j.cell.2012.07.018
- Cairns, R.A., Harris, I.S. and Mak, T.W. (2011) Regulation of cancer cell metabolism. Nature Reviews on Cancer, 11, 85-95. doi:10.1038/nrc2981
- Semenza, G.L., Roth, P.H., Fang, H.M. and Wang, G.L. (1994) Transcriptional regulations of gene encoding glycolytic enzymes by hypoxya-inducible factor 1. Journal of Biological Chemistry, 269, 23757-23763.
- Kim, J.W., Tchernyshyov, I., Semenza, G.L. and Dang, C.V. (2006) HIF-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism, 3, 177-185. doi:10.1016/j.cmet.2006.02.002
- Lee, K.H., Hsu S.C., Guh, J.H., Yang, H.C., Wang, D., Kulp, S.K., Shapiro, C.L. and Chen, C.S. (2011) Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator, The Journal of Biological Chemistry, 286, 39247-39258. doi:10.1074/jbc.M111.264598
- Sakamaki, T., Casimiro, M.C., Ju, X., Quong, A.A., Katiyar, S., Liu, M., Jiao, X., Li, A., Zhang, X., Lu, Y., Wang, C., Byers, S., Nicholson, R., Link, T., Shemluck, M., Yang, J., Fricke, S.T., Novikoff, P.M., Papanikolaou, A., Arnold, A., Albanese, C. and Pestell, R. (2006) Cyclin D1 determines mitochondrial function in vivo. Molecular and Cellular Biology, 26, 5449-5469. doi:10.1128/MCB.02074-05
- Landor, S.K., Mutvei, A.P., Mamaeva, V., Jin, S., Busk, M., Borra, R., Grönroos, T.J., Kronqvist, P., Lendahl, U. and Sahlgren, C.M. (2011) Hypoand hyperactivated notch signaling induce a glycolytic switch through distinct mechanisms. Proceedings of the National Academy of Sciences USA, 108, 18814-18819.
- Jones, R.G. and Thompson, C.B. (2009) Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes & Development, 23, 537-548. doi:10.1101/gad.1756509
- Ko, Y.H., Pedersen, P.L. and Geschwind, J.F. (2001) Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Letters, 173, 83-91. doi:10.1016/S0304-3835(01)00667-X
- Vander Heiden, M.G., Christofk, H.R., Schuman, E., Subtenly, A.O., Sharfi, H., Harlow, E.E., Xian, J. and Cantley, L.C. (2010) Identification of small molecule inhibitors of pyruvate kinase M2. Biochemical Pharmacology, 79, 1118-1124. doi:10.1016/j.bcp.2009.12.003
- Bonnet, S., Archer, S.L., Allalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C.T., Lopaschuk, G.D., Puttagunta, L., Bonnet, S., Harry, G., Hashimoto, K., Porter, C.J., Andrade, A., Thebaud, B. and Michelakis, L.D. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell, 11, 37-51. doi:10.1016/j.ccr.2006.10.020
- Semenza, G.L. (2012) Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends in Pharmacological Sciences, 33, 207-214. doi:10.1016/j.tips.2012.01.005
- Moreno-Sánchez, R., Rodríguez-Enríquez, S., MarínHernández, A. and Saavedra, E. (2007) Energy metabolism in tumor cells. The FASEB Journal, 274, 1393-1418.
- Metallo, C., Gameiro, P.A., Bell. E.L., Mattiani, K.R., Yang, J., Hiller, K., Jewell, C.M., Johnson, Z.R., Irvine, D.J., Guarente, L., Kelleher, J.K., Vander Heiden, M.G., Iliopuolos, O. and Stephanopoulos, G. (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature, 481, 380-384.
- Lu, C., Ward, P.S., Kapoor, G.S., Rohle, D., Turcan, S., Abdel-Wahab, O., Edwards, C.R., Khanin, R., Figueroa, M.G., Melnick, A., Wellen, K.E., O’Rourke, D.M., Berger, S.L., Chan, T.A., Levine, R.L., Mellinghoff, I.K. and Thompson, C.B. (2012) IDH1 mutation impairs demethylation and results in a block to cell differentiation. Nature, 483, 474-478. doi:10.1038/nature10860
- Wellen, K.E., Hatzivassiliou, G., Sachdeva, U.M., Bui, T.V., Cross, J.R. and Thompson, C.B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science, 324, 1076-1080. doi:10.1126/science.1164097
- Vander Heiden, M.G., Cantley, L.C. and Thompson, C.B. (2009) Understanding the Warburg effect: The metabolic requirements for cell proliferation. Science, 324, 1029- 1033. doi:10.1126/science.1160809
- Des Rosiers, C., Labarthe, F., Lloyd, S.G. and Chatham, J.C. (2011) Cardiac anaplerosis in health and disease. Cardiovascular Research, 90, 210-219. doi:10.1093/cvr/cvr055
- Owen, O.E., Kalhan, S.C. and Hanson, R.W. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. The Journal of Biological Chemistry, 277, 30409-30412. doi:10.1074/jbc.R200006200
- Marin-Valencia, I., Roe, C.R. and Pascual, J.M. (2010) Pyruvate carboxylase deficiency: Mechanism, mimics and anaplerosis. Molecular Genetics and Metabolism, 101, 9-17. doi:10.1016/j.ymgme.2010.05.004
- Jitrapakdee, S., St. Maurice, M., Rayment, I., Cleland, W.W., Wallace, J.C. and Attwood, P.V. (2008) Structure, mechanism and regulation of pyruvate carboxylase. Biochemistry Journal, 413, 369-387. doi:10.1042/BJ20080709
- Hohmeier, H.E., Mulder, H., Chen, G., Henkel-Rieger, R., Prentki, M. and Newgard, C.B. (2000) Isolation of INS- 1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes, 49, 424-430. doi:10.2337/diabetes.49.3.424
- Jitrapakdee, S., Vidal-Puig, A. and Wallace, J.C. (2006) Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cellular and Molecular Life Sciences, 63, 843- 854. doi:10.1007/s00018-005-5410-y
- Merritt, M.E., Harrison, C., Sherry, A.D., Malloy, C.R. and Burgess, S.C. (2011) Flux through hepatic pyruvate carboxylase and phosphoenolpyruvate carboxykinase detected by hyperpolarized 13 C magnetic resonance. Proceedings of the National Academy of Sciences USA, 108, 19084-19089. doi:10.1073/pnas.1111247108
- Hertz, L., Peng, L. and Dienel, G.A. (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycolgenolysis. Journal of Cerebral Bloodflow and Metabolism, 27, 219-249. doi:10.1038/sj.jcbfm.9600343
- Lapidot, A. and Gopher, A. (1994) Cerebal metabolic compartmentation. estimation of glucose flux via pyruvate carboxylase/pyruvate dehdrogenase by 13C NMR isotopomer analysis of D-[13C]glucose metabolites. Journal of Biological Chemistry, 269, 198-208.
- De la Rosa, V., Campos-Sandoval, J.A., Marín-Rufían, M., Cardona, C., Matés, J.M., Segura, J.A., Alonso, F.J. and Márquez, J. (2009) A novel glutaminase isoform in mammalian tissues. Neurochemistry international, 55, 76- 84. doi:10.1016/j.neuint.2009.02.021
- Prebil, M., Jensen, J., Zorec, R. and Kreft, M. (2011) Astrocytes and energy metabolism. Archives of Physiology and Biochemistry, 117, 64-69. doi:10.3109/13813455.2010.539616
- Guppy, M., Leedman, P., Zu, X. and Russel, V. (2002) Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. The Biochemical Journal, 364, 309-315.
- Whitaker-Menezes, D., Martinez-Outchoorn, U.E., Flomenberg, N., Birbe, R.C., Witkiweicz, A.K., Howell, A., Pavlides, S., Tsirigos, A., Ertel, A., Pestell, R.G., Broda, P., Minetti, C., Lisanti, M.P. and Sotgia, F. (2011) Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle, 10, 4047-4064. doi:10.4161/cc.10.23.18151
- Rodríguez-Enríquez, S., Vital-González, P.A., FloresRodríguez, F.L., Marín-Hernández, A., Ruiz-Azuara, L. and Moreno-Sánchez, R. (2006) Control of cellular proliferation by modulation of oxidative phosphorylation in human and rodent fast-growing tumor cells. Toxicology and Applied Pharmacology, 215, 208-217. doi:10.1016/j.taap.2006.02.005
- Martin, M., Beauvoit, M., Voisin, P.J., Canioni, P., Guérin, B. and Rigoulet, M. (1998) Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. Journal of Bioenergetics and Biomembranes, 30, 565-778. doi:10.1023/A:1020584517588
- Marin-Valencia, I., Yang, C., Mashimo, T., Cho, S., Baek, H., Yang, X.L., Rajagopalan, K.N., Maddie, M., Vemireddy, V., Zhao, Z., Cai, L., Good, L., Tu, B.P., Hatanpaa, K.J., Mickey, B.E., Matés. J.M., Pascual, J.M., Maher, E.A., Malloy, C.R., DeBerardinis, R.J. and Bachoo, R.M. (2012) Analysis of tumor metabolism reveals glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metabolism, 15, 827-837. doi:10.1016/j.cmet.2012.05.001
- Das, S.K., Eder, S., Schauer, S., Diwoky, C., Temmel, H., Guertl, B., Gorkiewicz, G., Tamilarasan, K.P., Kumari, P., Trauner, M., Zimmermann, R., Vesely, P., Haemmerle, G., Zechner, R. and Hoefler, G. (2011) Adipose triglyceride lipase contributes to cancer-associated cachexia. Science, 333, 233-238. doi:10.1126/science.1198973
- Nieman, K.M., Kenny, H.A., Penicka, C.V., Ladanyi, A., Buell-Gutbrod, R., Zillhardt, M.R., Romero, I.L., Carey, M.S., Mills, G.B., Hotamisligil, G.S., Yamada, S.D., Peter, M.E., Gwin, K. and Lengyel, E. (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine, 17, 1498-1503. doi:10.1038/nm.2492
- Rodríguez-Enríquez, S., Juárez, O., Rodríguez-Zavala, J.S. and Moreno-Sánchez, R. (2001) Multisite control of the Crabtree effect in ascites hepatoma cells. European Journal of Biochemistry, 268, 2512-2519. doi:10.1046/j.1432-1327.2001.02140.x
- Rossignol, R., Gillkerson, R., Aggeler, R., Yamagata, K., Remington, S.J. and Capaldi, R.A. (2004) Energy substrate modulates mitochondria structure and oxidative capacity in cancer cells. Cancer Research, 64, 985-993. doi:10.1158/0008-5472.CAN-03-1101
- Schroeder, T., Yuan, H., Viglianti, B.L., Peltz, C., Asopa, S., Vujaskovic, Z. and Dewhirst, M.W. (2005) Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Research, 65, 5163-5171. doi:10.1158/0008-5472.CAN-04-3900
- Sonveaux, P., Végran, F., Schroeder, T., Wergin, M.C., Verrax, J., Rabbani, Z.N., De Seadleer, C.J., Kennedy, K.M., Diepart, C., Jordan, B.F., Kelley, M.J., Gallez, B., Wahl, M.L., Feron, O. and Dewhirst, W. (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. The Journal of Clinical Investigation, 118, 3930-3942.
- Martinez-Outschoorn, U.E., Pestell, R.G., Howell, A., Tykocinski, M.L., Nagajyothi, F., Machado, F.S., Tanowitz, H.B., Sotgia, F. and Lisanti, M.P. (2011) Energy transfer in “parasitic” cancer metabolism: Mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle, 10, 4208-4216. doi:10.4161/cc.10.24.18487
- Pfeiffer, T., Schuster, S. and Bonhoeffer, S. (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science, 292, 504-507. doi:10.1126/science.1058079
- Vander Heiden, M.G., Locasale, J.W., Swanson, K.D., Sharfi, H., Heffron G.J., Amador-Noguez, D., Christofk, H.R., Wagner, G., Rabinowitz, J.D., Asara, J.M. and Cantley, L.C. (2010) Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science, 329, 1492-1499. doi:10.1126/science.1188015
- Locasale, J.W., Grassian, A.R., Melman, T., Lyssiotis, C.A., Mattaini, K.R., Bass, A.-J., Heffron, G., Metallo, C.M., Muranen, T., Sharfi, H., Sasaki, A.T., Anastasiou, D., Mullarky, E., Vokes, N.I., Sasaki, M., Beroukhim, R., Stephanopoulos, G., Ligon, A.H., Meyerson, M., Richardosn, A.L., Chin, L., Wagner, G., Asara, J.M., Brugge, J.S., Cantley, L.C. and Vander Heiden, M.G. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature Genetics, 43, 869-874. doi:10.1038/ng.890
- Hitosugi, T., Kang, S., Vander Heiden, M.G., Chung, T.W., Elf, S., Lythgoe, K., Dong, S., Lonial, S., Wang, X., Chen, G.Z., Xie, J., Gu, T.L., Polakiewicz, R.D., Roesel, J.L., Boggon, T.J., Khuri, F.R., Gilliland, D.G., Cantley, L.C., Kaufman, J. and Chen, J. (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Science Signaling, 2, ra73. doi:10.1126/scisignal.2000431
- Frezza, C., Zheng, L., Folger, O., Rajagopalan, K.N., McKenzie, E.D., Jerby, L., MIcaroni, M., Chaneton, B., Adam, J., Hedley, A., Kalna, G., Tomlinson, I.P., Pollard, P.J., Watson, D.G., DeBerardinis, R.J., Shlomi, T., Ruppin, E. and Gottlieb, E. (2011) Haem oxygenase is synthetically lethal with the tumor suppressor fumarate hydratase. Nature, 477, 225-228. doi:10.1038/nature10363
- Pelicano, H., Martin, D.S., Xu, R.H. and Huang, P. (2006) Glycolysis inhibition for anticancer treatment. Oncogene, 25, 4633-4646. doi:10.1038/sj.onc.1209597
- Sauer, L.A., Stayman, 3rd., J.W. and Dauchy, R.T. (1982) Amino acid, glucose and lactic acid accumulation in vivo by rat tumors. Cancer Research, 42, 4090-4097.
- DeBerardinis, R.J. and Cheng, T. (2010) Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene, 29, 313-324. doi:10.1038/onc.2009.358
- Pérez-Gómez, C., Campos-Sandoval, J.A., Alonso, F.J., Segura, J.A., Manzanares, E., Ruiz-Sánchez, P., González, M.E., Márquez, J. and Matés, J.M. (2005) Co-expression of glutaminase K and L isoenzymes in human tumour cells. The Biochemical Journal, 386, 535-542. doi:10.1042/BJ20040996
- Cassago, A., Ferreira, A.P., Ferreira, I.M., Fornezari, C., Gomes, E.R., Greene, K.S., Pereira, H.M., Garratt, R.C., Dias, S.M. and Ambrosio, C.L. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proceedings of the National Academy of Sciences of the United States of America, 109, 1092-1097. doi:10.1073/pnas.1112495109
- [61] Gómez-Fabre, P.M., Aledo, J.C., Del Castillo-Olivares, A., Alonso, F.J., Nuñez de Castro, I., Campos, J.A. and Márquez, J. (2000) Molecular cloning, sequencing and expression studies of the human breast cancer glutaminase. The Biochemical Journal, 345, 365-375. doi:10.1042/0264-6021:3450365
- [62] Szeliga, M., Sidoryk, M., Matyja, E., Kowalczyk, P. and Albrecht, J. (2005) Lack of expression of the liver-type glutaminase (LGA) mRNA in human malignant gliomas. Neuroscience Letters, 374, 171-173. doi:10.1016/j.neulet.2004.10.051
- [63] Matsuno, I. and Hirai, H. (1989) Glutamine synthetase and glutaminase activities in various hepatoma cells. Biochemistry International, 19, 219-225.
- [64] Colombo, S.L., Palacios-Callender, M., Frakich, N., De Leon, J., Schmitt, C.A., Boorn, L., Davis, N. and Moncada, S. (2010) Anaphase-promoting complex/cyclosomeCdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes, Proceedings of the National Academy of Sciences of the United States of America, 107, 18868-18873. doi:10.1073/pnas.1012362107
- [65] Colombo, S.L., Palacios-Callender, M., Frackich, N., Carcamo, S., Kovacs, I., Tudzarova, S. and Moncada, S. (2011) Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proceedings of the National Academy of Sciences of the United States of America, 108, 21069-21074. doi:10.1073/pnas.1117500108
- [66] Garedew, A., Andreassi, C. and Moncada, S. (2012) Mitochondrial dynamics, biogenesis, and function are coordinated with the cell cycle by APC/C CDH1. Cell Metabolism, 15, 466-479. doi:10.1016/j.cmet.2012.03.003
- [67] Wise, D.R., DeBerardinis, R.J., Mancuso, A., Sayed, N., Zhang, X.Y., Pfeiffer, H.K., Nissim, I., Daikhin, E., Yudkoff, M., McMahon, S.B. and Thompson, C.B. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America, 105, 18782-18787. doi:10.1073/pnas.0810199105
- [68] Li, F., Wang, Y., Zeller, K.I., Potter, J.J., Wonsey, D.R., O’Donell, K.A., Kim, J.W., Yustein, J.T., Lee, L.A. and Dang, C.V. (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and Cellular Biology, 25, 6225-6234. doi:10.1128/MCB.25.14.6225-6234.2005
- [69] DeBerardinis, R.J., Lum, J.J., Hatzivassiliou, G. and Thompson, C.B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metabolism, 7, 11-30. doi:10.1016/j.cmet.2007.10.002
- [70] Le, A., Lane, A.N., Hamaker, M., Bose, S., Gouw, A., Barbi, J., Tsukamoto, T., Rojas, C.J., Slusher, B.S., Zhang, H., Zimmerman, L.J., Liebler, D.C., Slebos, R.J., Lorkiewicz, P.K., Higashi, R.M., Fan, T.W. and Dang, C.V. (2012) Glucose-independent glucose metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15, 110-121. doi:10.1016/j.cmet.2011.12.009
- [71] Parlo, R.A. and Coleman, R.S. (1984) Enhanced rate of citrate export from cholesterol-rich hepatoma mitochondria. The truncated Krebs cycle and other metabolic ramifications of mitochondrial membrane cholesterol. The Journal of Biological Chemistry, 259, 9997-10003.
- [72] Fillipp, F.V., Scott, D.A., Ronai, Z.A., Osterman, A.L. and Smith, J.W. (2012) Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell & Melanoma Research, 25, 375-383. doi:10.1111/j.1755-148X.2012.00989.x
- [73] Seltzer, M.J., Bennett, B.D., Joshi, A.D., Gao, P., Thomas, A.G., Ferraris, D.V., Tsukamoto, T., Rojas, C.J., Slusher, B.S., Rabinowitz, J.D., Dang, C.V. and Riggins, G.J. (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Research, 70, 8981-8987. doi:10.1158/0008-5472.CAN-10-1666
- [74] Katt, W.P., Ramachandran, S., Erickson, J.W. and Cerione, R.A. (2012) Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Molecular Cancer Therapeutics, 11, 1269-1278. doi:10.1158/1535-7163.MCT-11-0942
- [75] Wang, J.B., Erickson, J.W., Fuji, R., Ramachandran, S., Gao, P., Dinavahi, R., Wilson, K.F., Ambrosio, A.L., Dias, S.M., Dang, C.V. and Cerione R.A. (2010) Targeting mitochondrial glutaminase activity oncogenic transformation. Cancer Cell, 18. 207-219. doi:10.1016/j.ccr.2010.08.009
- [76] Portais, J.C., Schuster, R., Merle, M. and Canioni, P. (1993) Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon [1-13C]glucose incubation. European Journal of Biochemistry, 217, 47-68. doi:10.1111/j.1432-1033.1993.tb18265.x
- [77] Hammond, K.D. and Balinsky, D. (1978) Activities of key gluconeogenic enzymes and glycogen synthase in rat and human livers, hepatomas, and hepatoma cell cultures. Cancer Research, 38, 1317-1322.
- [78] Sato, T., Kashima, K., Gamachi, A., Daa, T., Nakayama, I. and Yokoyama, S. (2002) Immunohistochemical localization of pyruvate carboxylase and carbamyl-phosphate synthetase I in normal and neoplastic human pancreatic tissues. Pancreas, 25, 130-135. doi:10.1097/00006676-200208000-00003
- [79] Bramwell, M. and Humm, S.M. (1992) Variations in the relative amount of biotin-containing enzymes present in both tumorigenic and non-tumorigenic hybrid cells and other cell lines. Biochimica et Biophysyca Acta, 1139, 115-121. doi:10.1016/0925-4439(92)90090-A
- [80] Liu, K.J., Kleps, R., Henderson, T. and Nyhus, L. (1991) 13C NMR study of hepatic pyruvate carboxylase activity in tumor rats. Biochemical and Biophysical Research Communications, 179, 366-371. doi:10.1016/0006-291X(91)91379-Q
- [81] Díaz-Ruiz, R., Rigoulet, M. and Devin, A. (2011) The Warburg and Crabtree effects: on the origin of cancer cell energy metabolism and of yeast glucose repression. Biochimica et Biophysica Acta, 1807, 568-576. doi:10.1016/j.bbabio.2010.08.010
- [82] Lee, S.Y., Jeon, H.M., Ju, M.K., Kim, C.H., Yoon, G., Han, S.I., Park, H.G. and Kang, H.S. (2012) Wnt/snail signaling regulates cytochrome c oxidase and glucose metabolism. Cancer Research, 72, 3607-3617. doi:10.1158/0008-5472.CAN-12-0006
- [83] Godlewski, J., Nowicki, N.O., Bronisz, A., Nouvo, G., Palatini, J., De Lay, M., Van Brocklyn, J., Ostrowski, M.C., Chiocca, E.A. and Lawler, S.E. (2010) MicroRNA- 451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Molecular Cell, 37, 620-632. doi:10.1016/j.molcel.2010.02.018
- [84] Kimura, Y., Kashima, K., Daa, T., Kondo, Y., Yada, K., Sasaki, A., Matsumoto, T., Kitano, S., Kubo, N. and Yokoyama, S. (2005) Biotin-rich intranuclear inclusions in morule-lacking adenocarcinoma of the gallbladder: A new category of “neoplastic/non-morular” lesions. Virchows Archives: An International Journal of Pathology, 446, 194-199.
- [85] Cheng, T., Sudderth, J., Yang, C., Mullen, A.R., Jin, E.S., Matés, J.M. and DeBerardinis, R.J. (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences of the United States of America, 108, 8674-8679. doi:10.1073/pnas.1016627108
- [86] Fan, T.W., Lane, A.N., Higashi, R.M., Faraq, M.A., Gao, H., Bousamra, M. and Miller, D.M. (2009) Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Molecular Cancer, 8, 41. doi:10.1186/1476-4598-8-41
- [87] Budczies, J., Denkert, C., Müller, B.M., Brockmöller, S.F., Klauschen, F., Györffy, B., Dietel, M., Richter-Ehrenstein, C., Marten, U., Salek, R.M., Griffin, J.L., Hilvo, M., Oresic, M., Wohlgemuth, G. and Fiehn, O. (2012) Remodeling of central metabolism in invasive breast cancer in invasive breast cancer compared to normal breast tissue: A GC-TOFMS based metabolomics study. BMC Genomics, 13, 334. doi:10.1186/1471-2164-13-334