International Journal of Organic Chemistry
Vol.3 No.1A(2013), Article ID:30588,16 pages DOI:10.4236/ijoc.2013.31A001
Application of Computer-Aided Drug Design to Traditional Chinese Medicine
State Key Laboratory of Pharmaceutical Biotechnology, College of Life Sciences, Nanjing University, Nanjing, China
Email: yangjie@nju.edu.cn
Copyright © 2013 Jie Yang. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 12, 2013; revised February 28, 2013; accepted April 5, 2013
Keywords: Traditional Chinese Medicine; Drug Target Discovery; Chinese Herbal Drugs; Drug Design; Virtual Screening
ABSTRACT
Computer-aided drug design (CADD) is an interdisciplinary subject, playing a pivotal role during new drug research and development, especially the discovery and optimization of lead compounds. Traditional Chinese Medicine (TCM) modernization is the only way of TCM development and also an effective approach to the development of new drugs and the discovery of potential drug targets (PDTs). Discovery and validation of PTDs has become the “bottle-neck” restricted new drug research and development and is urgently solved. Innovative drug research is of great significance and bright prospects. This paper mainly discusses the “druggability” and specificity of PTDs, the “druglikeness” of drug candidates, the methods and technologies of the discovery and validation of PTDs and their application. It is very important to achieve the invention and innovation strategy “from gene to drug”. In virtue of modern high-new technology, especially CADD, combined with TCM theory, research and develop TCM and initiate an innovating way fitting our country progress. This paper mainly discusses CADD and their application to drug research, especially TCM modernization.
1. Introduction
Traditional medicine plays an important role in health maintenance for the proples of Asia, and is becoming more frequently used in countries in the West [1]. Traditional Chinese medicine (TCM) is a system of ancient medical practice that differs in substance, methodology and philosophy to modern medicine. TCM-herbal mixtures developed though observation and experience accumulated over thousands of years, but with unknown mechanisms of action, while modern medicine consists of chemically purified compounds that have been discovered through scientific investigation and tested in controlled clinical trials [2]. For many years, people have been working to elucidate the material basis and molecular mechanism of TCM all the time. Chinese medicinal materials (CMMs) that originate from animal, mineral and plant have thousands years for diabetes therapy and accumulate a great deal of valuable clinical experience [3]. TCM includes unilateral and resipes, where the former involves single herb while the latter is composed of various ones [4]. Due to the complexity of TCM’s ingredients and its absorption and metabolism in the human body, as well as the complexity of compatibility, etc., it is so far difficult to provide a complete modern scientific foundation for TCM’s efficacy. To clarify the chemical component of TCM compounds as well as multi-target effect mechanism is the key to breaking the actuality of TCM research and development [5]. The use of TCM theory combined with modern science and technology is a development road to current TCM industry, but also an effective way to study new drugs.
Computer-aided drug design (CADD) is a specialized discipline that uses computational method to simulate drug-receptor interactions. On the basis of bioinformatics, chemical molecule database, protein structure databases, metabolic database, etc., CADD applies DOCK, molecular modeling, ligand-receptor free energy prediction, two-/three-dimensional quantitative structure-activity relationships (QSAR), pharmacophore models and other methods, to obtain structural information of drug interacted with protein, to analyse and provide PTDs in order to effectively predict and discover active (hit) compounds [6,7]. CADD represents a potentially useful tool for new drug research and development, and has made great inroads into improving the odds of finding bioactive leads [8]. Moreover, CADD is an important ligament connecting TCM with modern sciences. In particular, DOCK, similarity search, pharmacophore search, druglikeness screening, toxicity prediction, and other technology play stimulative roles in TCM compound research. With finishing of human genome project, CADD has been rapidly developed, especially combinatorial chemistry technology and life science research, becoming increasingly important role in discovery and optimization of lead compounds. Many well-known pharmaceutical companies have applied CADD technology to research and develop new drugs. For example, United Kingdom-based Glaxo Wellcome R & D Ltd. took methysergide (5-HT receptor antagonist) as a lead compound and further successfully developed a migraine headache treatment drug zolmitriptan (Zomig) through a series of active compounds found (such as sumatriptan) (Figure 1(a)) [9]. CADD can reduce the investment in drug development, economize scarce resources, decrease the waste of blind, but also significantly shorten the drug development cycle. This paper mainly discusses CADD in drug research, particularly the discovery and validation of drug effect targets and their application and prospects of TCM modernization.
2. Potential Drug Targets and Drug Candidates
During the 1990s, the pharmaceutical industry noticed that too many compounds were terminated in clinical development because of unsatisfactory pharmacokinetics (PK) [10]. Understanding the difference between biologically active small molecules and drugs became a priority in the drug discovery process, and the importance of addressing pharmacokinetic properties early during lead optimization is a clear result. The introduction of Lipinski’s “Rule of Five” (RO5) [11] associated with 90% of orally active drugs that achieved phase II status has initiated a profound shift in the thinking paradigm of medicinal chemists [10,12]. The Lipinski’s rules contains the following points: MW ≤ 500; ClogP ≤ 5; H-bond

Figure 1. Chemical structures of some active molecules.
donors ≤ 5; H-bond acceptors (sum of N and O atoms) ≤10; Polar surface area ≤ 140Å2 or sum of H-bond donors and acceptors ≤ 12; Rotatable bonds ≤ 10; and Remarks: no more than one violation, not applicable for substrates of transporters and natural products [13-15]. These concepts of “drug-likeness” and “druggability” have been extended to proteins and genes for target identification and selection [16]. For clarity, usually use the term “druggable” for proteins (targets) and apply the original, more appropriate term “drug-like” for compounds [10,11]. The phrase “drug-like” is becoming more widespread and it generally means “molecules which contain functional groups and/or have physical properties consist with the majority of known drugs” [16]. The drug-like moleculaes can be identified and obtained by analyzing the database of known drug library using a number of computational techniques (Figure 2). “Druglikeness” involves three aspects: physical and chemical properties (such as solubility, stability, etc.), topology characteristics, and pharmacokinetic properties (namely absorption, distribution, metabolism, excretion and toxicity, ADME/Tox) [13]. This statistical feature can be used in the early design and evaluation for compound libraries (combination compound libraries or virtual compound libraries), but we cannot distinguish the individual compounds while are “druglike” or “non-druglike” [14]. “Druglikeness” more used to assess the characteristics of compounds that may lead to failure in order to reduce the risk of drug research and development. Thus, “druglikeness” assessment usually follows Lipinski rules so that exclude those compounds with failure tendency, narrow the scope of screening and reduce the cost of drug research and development.
In 2002, Hopkins and Groom introduced the concept of the “druggable genome” [15]. Their purpose was to identify the limited set of molecular targets for which commercially viable, oral compounds can be developed. An estimated 60% of small molecule drug discovery projects fail in hit-to-lead because the biological target is found to be not “druggable” [17]. This is a combined attrition due to lack of lead matter from screening and difficulty in optimizing lead matter to yield reasonable drug leads. Druggability, the likelihood of being able to modulate a target with a small-molecule drug, is crucial in determining whether a drug discovery project progresses from “hit” to “lead” [18]. It is well-known that human genome consists of approximately 3000 - 10,000 of disease-related genes, and about 10% of genes related to disease have been verified by gene knockout technology [19]. By some estimates, only 10% of genes in the human genome represent druggable target, and only half of those are both druggable and relevant to disease [15]. It is important to be able to predict how druggable a novel target is in early drug discovery. Because such targets are expected to bind RO5-compliant compounds, they analyzed databases and used computational methods to identify all proteins belonging to families that have at least one member that has successfully been targeted by drug-like molecules [15,17,18]. The PTDs involved in the pharmaceutical industry should exist in the intersection of druggable genome and disease-related genome, only about 2% - 5% of the human genome. Moreover, the combination of proteins and small molecules with affinity make them druggable but not PTDs. The reason is that a potential drug target is only suitable for those disease-related proteins (specificity). Both druggability and specificity of the protein is critical for meeting requirements of a potential drug target. Assuming that druggability is shared among protein families and taking into account that a drug target needs to have the potential
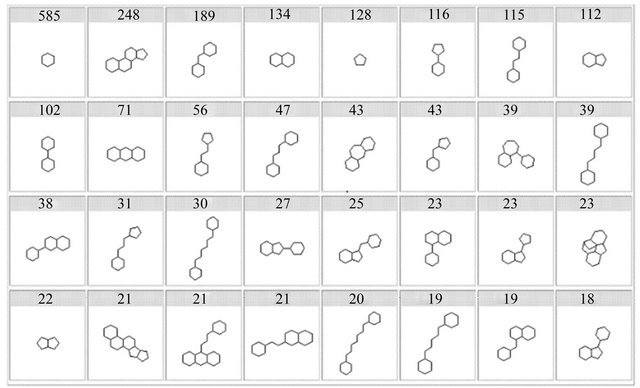
Figure 2. The most commonly frameworks in drugs [10].
to be disease-modifying, therefore, it is estimated that the human genome encodes 600 - 1500 targets for smallmolecule drugs.
3. Computer Virtual Screening
The ideal of target-based drug design is to discover and validate drug targetst. Drug targets currently contain enzymes, receptors, transport systems, DNA/RNA as well as the storage systems. Computer virtual screening includes receptor-based screening methods (namely molecular docking, DOCK) and ligand-based screening methods (viz. pharmacophore search). DOCK and reverse molecular docking (INVDOCK) methods usually used to carry out innovative drug design and discovery of PTDs [20]. The former is based on the critical target molecules in the disease development, the latter based on the structure of known active small molecules. The former is suitable for screening lead compounds from the groups of active ingredients from TCM prescription, the latter to carry out the effective screening of the target molecules for active ingredients from TCM prescription [21]. Virtual screening is the most successful CADD application technology, which must have compound databases. It has proved that it is a very important tool to detect small organic molecules in the process of biological systems [22]. In recent years, scientists began to try to search for TCM active compounds (chemical compositions) and develop major TCM compound databases, such as TCM chemical database (TCMD) and China natural products database (CNPD). Recently, scientists have found some active compounds interacted with a specific target from TCMD, CNPD and synthetic compound database by virtual screening. There are a lot of successful examples [23,24] as follows.
3.1. Potassium Channel Kv1.5 Blockers
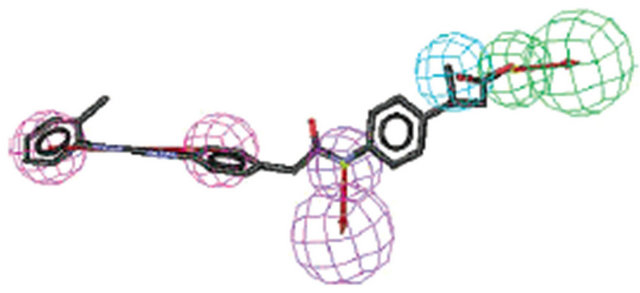
Atrial fibrillation and atrial flutter are the most frequent cardiac arrhythmias [23]. Clinical study found that atrial fibrillation can cause changes in atrial action potential (APD), effective refractory period (ERP) and its frequency adaptation, and atrial conduction velocity, namely electrical remodeling, which promots the onset and maintenance of atrial fibrillation. The ideal drug for treatment of atrial fibrillation should only affect the atrial ERP rather than cause serious ventricular arrhythmias. Voltage-gated potassium channels are transmembrane channels specific for potassium and sensitive to voltage changes in the cell’s membrane potential. Based on sequence homology of the hydrophobic transmembrane cores, the alpha subunits have been grouped into 12 classes labeled Kv1-12 [25]. Kv1.5 is regarded as a promising pharmacological target for the development of new atrial selective drugs with fewer side effects. Peukert and co-worker [23] designed, synthesized, screened, and successively found hit compound (IC50 9.5 μM), lead compound (IC50 4.8 μM), and candate compound (IC50 0.16 μM) (Figure 1(b)) through virtual screening and combinatorial chemistry techniques, taking Kv1.5 as a target, based on K1.5 channels expressed in Xenopus oocytes. The observed structure-activity relationship is described by a pharmacophore model that consists of three hydrophobic centers in a triangular arrangement (Figure 3(a)) [23]. The hydrophobic centers are matched by a phenyl or pyridyl ring of the bisaryl core and both ends of the side chains. The most potent compounds (e.g. active candidate compound) inhibited the Kv1.5 channel with sub-micromolar half-blocking concentrations and displayed 3-fold selectivity over Kv1.3 and no significant effect on the HERG (human ether a-go-go-related gene) channel and sodium currents. It is expected to become the new class III antiarrhythmic drugs doubly concerned about.
3.2. Cyclin-Dependent Kinase-2 Inhibitors
The fidelity of progression through the human cell cycle is interrogated at various checkpoints in order to ensure the integrity of daughter cells. If errors in DNA replication are detected, normal proliferating cells undergo programmed cell death (apoptosis) [24]. Transformed cells, however, gain a proliferative advantage by bypassing or overriding these checkpoints. Cyclin-dependent kinases (CDKs) are intimately associated with the regulation of cell cycle progression [26]. Cyclins, the activating subunits of CDKs, are expressed specifically during certain phases of the cycle. Thus D-type cyclins appear in G1 and associate with CDKs 4 and 6. Later cyclin E is expressed and forms an active complex with CDK2, an important drug target for treating cancer. The action of these complexes on various substrates, and the retinoblastoma tumor suppressor protein (pRb), in particular, permits passage into S phase by dissociating the complex between pRb and E2F transcription factors. Next in turn is cyclin A, which associates with CDK2 and maintains pRb hyperphosphorylation. Later in S phase, however, cyclin A/CDK2 fulfills another important function, i.e. phosphorylation and deactivation of E2F [24,27]. This activity is necessary for progression and inappropriate E2F transcriptional activity in late S phase constitutes an apoptotic trigger [28]. This suggests that inhibition of cyclin A/CDK2 during S phase would lead to S-phase arrest, inappropriately persistent E2F-1 activity, and apoptosis. It nevertheless remains somewhat unclear what selectivity profile a CDK inhibitor should have for optimal anticancer activity. CYC202 (R-roscovitine) (Figure 1(c)) is the most selective clinical CDK inhibitor whose main targets are CDK2 and CDK9 [29], as well as
 (a)
(a)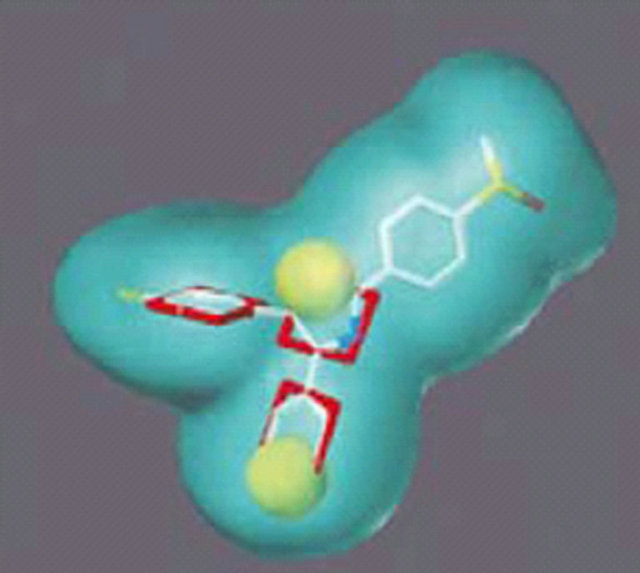 (b)
(b)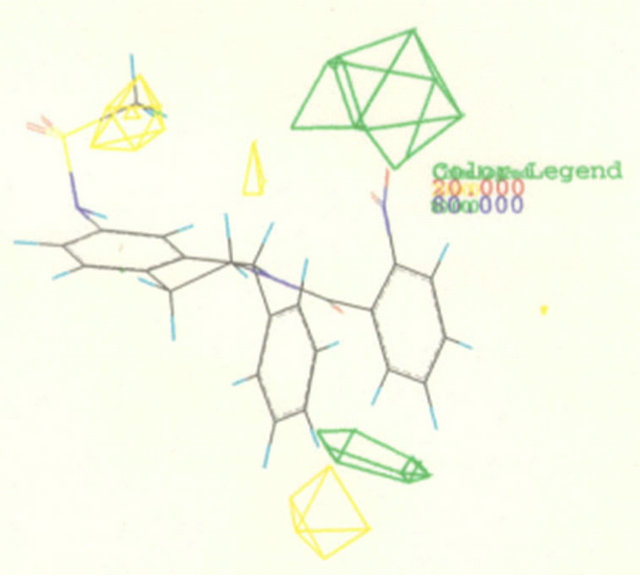 (c)
(c)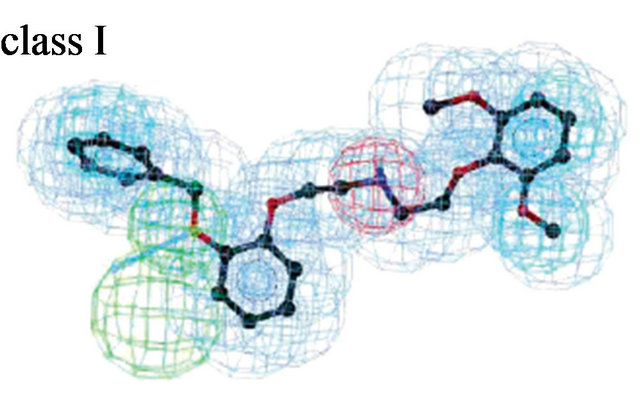 (d)
(d)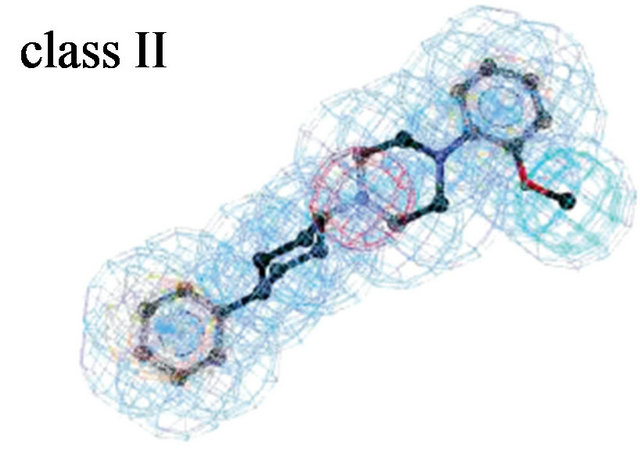 (e)
(e)
Figure 3. Pharmacophore models. (a) Pharmacophore model illustrated with active candidate compound [20]; (b) Structure of the most potent α4β1 antagonist (candidate compound) and its pharmacophore model: hydrogen-bond acceptors (green), hydrophobe (cyan), hydrogen-bond donors (purple) and superfeature (pink) [29]; (c) Structure of Type I TGFβ receptor kinase (TβRI) inhibitor (SB203580) and its pharmacophore model: shape (cyan sphere), aromaticity (red plane), and hydrogen-bond acceptors (yellow globe) [33]; (d) Catalyst pharmacophore models used for virtual screening of the Aventis compound repository. Shown are the two pharmacophore models (classes I and II) mapping two different classes of high-affinity ligands of the α1A receptor [35]; (e) CoMFA model taking compound 11 as a template [44].
by the hypothesis that CDK2 inhibition may provide a way of interfering selectively with cancer cells [30].
Wang and co-worker recently reported the identification of 2-anilino-4-(thiazol-5-yl) pyrimidines as CDK2 inhibitors using virtual screening methods [31]. The synthetic chemistry, the structure-guided design approach, and the SARs led to the discovery of the above ATPantagonistic CDK2 inhibitors. Furthermore, obtained 200 active compounds and found compound D with 2.0 nM Ki against CDK2 through virtual screening from more then 50,000 compound libraries [24]. Finally for the most potent CDK2 inhibitor (compound D) (Figure 1(c)) carried out a series of kinase-selective experiments and found compound D with 4.0 nM Ki against CDK9, with lower Ki values against GSK3β (Ki 20.0 nM), CDK1 (Ki 80.0 nM), CDK4 (Ki 53.0 nM), CDK7 (Ki 70.0 nM) and Abl (Ki 160.0 nM), respectively, but with higher Ki values (Ki > 1000 nM) against PKA, PKCα, ERK-2, Akt/PKB, p38 and p70S6, respectively. A further study found that the compound can block G1/S phase cell cycle progreeion, prevent the induction of CDK2 and G2/M phase, can reduce the mitotic index, and induce cell death.
3.3. Nonpeptidic Antigen α4β1 Antagonists
The antigen α4β1 (very late antigen-4, VLA-4) plays an important role in the migration of white blood cells to sites of inflammation. It has been implicated in the pathology of a variety of diseases including asthma, multiple sclerosis, and rheumatoid arthritis [32,33]. α4β1-mediated leukocyte recruitment and activation, media release, and apoptosis inhibition plays a central role in immune pathological processes. α4β1 interacts with its ligands VCAM-1 (vascular cell adhesion molecular-1) and fibronectin by Leu-Asp-Val motif, which has become the PTDs of the treatment of many diseases. Singh and co-worker [32] described a series of potent inhibitors of α4β1 that were discovered using computational screening for replacements of the peptide region of an existing tetrapeptide-based α4β1 inhibitor (4-[N’-(2- methylphenyl)-ureido]phenylacetyl-Leu-Asp-Val, compound 1, IC50 value 0.6 nM) derived from fibronectin (Figure 1(d)). The search query was constructed using a model of this inhibitor that was based upon the X-ray conformation of the related integrin-binding region of VCAM-1. The computational screen identified 12 reagents from a virtual library of 8624 molecules as satisfying the model and their synthetic filters and screened 416 hit compounds, including 170 nonpeptidic α4β1 inhibitors. All of the synthesized compounds tested inhibit α4β1 association with VCAM-1, with the most potent compound (compound 2) having an IC50 of 1.3 nM, comparable to the starting compound (Figure 1(d)). A 3D QSAR of these α4β1 antagonists was generated using CATALYST module of Insight II software (Figure 3(b)). The most potent compound was evaluated in a sheep model of asthma, and a 30 mg nebulized dose was able to inhibit early and late airway responses in allergic sheep following antigen challenge and prevented the development of nonspecific airway hyperresponsiveness to carbachol. This approach should be useful in identification of nonpeptidic α4β1 inhibitors with improved pharmacokinetic properties relative to their peptidic counterparts.
3.4. Type I TGFβ Receptor Kinase Inhibitors
Transforming growth factor (TGF) β1, TGFβ2 and TGFβ3 are multifunctional cytokines that play a critical role in the regulation of production, degradation and accumulation of extracellular matrix proteins [34]. The TGFβ pathway plays a pivotal role in progressive fibrotic diseases of the lung, liver and kidney that are major causes of morbidity and mortality, and represents an exciting target for the development of novel anti-fibrotic agents. TGFβ1 is closely related to cell proliferation and tumor cells angiogenesis. The complex of TGFβ with the TGFβ Type II receptor (TβRII) binds and activates TβRI (Type I TGFβ receptor kinase) by phosphorylating the GS region. The TβRI kinase is an important and novel drug target for fibrosis and cancer and further for a virtual screening strategy to discover novel inhibitors.
Singh J. and co-worker took a known TβRI inhibitor SB203580 (IC50 30.0 μM) [35] as a lead compound and carried out shape-based virtual screening (Figure 1(e)). The pharmacophoric query was built using the ViewHypothesis workbench within Catalyst using the conformation of SB203580 reported in the X-ray complex with p38 (pdbcode 1a9u) (Figure 3(c)) and the shape component of the query was built using the CatShape module within Catalyst [36]. Finally screen 87 active compounds from 20,000 compounds by DOCK and a potent TβRI inhibitor HTS466284 was identified which inhibited autophosphorylation with an IC50 of 27 nM. Moreover, the pyrazole N2 and quinoline nitrogen formhydrogen of HTS466284 bonds with Lys232 side chain nitrogen and His283 backbone amide NH of TβRI, respectively, commonly existing in known kinase inhibitor complexes [36]. There are two additional hydrogenbonds seen in the complex that were not part of the query. The first involves the N1 of the pyrazole ring, which acts as a hydrogen bond donor with one of the carboxylate oxygens from Asp351. The second additional interaction involves the 2-pyridyl nitrogen that forms a water-mediated network of hydrogen bonds with the enzyme. The inhibitor and receptor form a tetrahedral complex with the water molecule. The water molecule donates hydrogen bonds to the pyridyl nitrogen of HTS466284 and the carboxyl oxygen of Glu245 and accepts hydrogen bonds fromth e phenol of Tyr249 and the backbone NH of Asp351.
3.5. Adrenergic (α1A) Receptor Antagonists
G-protein coupled receptors (GPCRs) form a large protein family that plays an important role in many physiological and pathophysiological processes. The alpha1 adrenergic receptors are involved in blood pressure maintenance by modulating the vascular muscle tone. They are subdivided into the alpha1A, alpha1B and alpha1D adrenoreceptor subtypes [37]. Antagonists of the alpha1 adrenergic receptors such as indoramin and prazosin are employed as antihypertensitive agents. In addition, alpha1A (α1A) antagonists such as alfuzosin and prazosin are thought to be effective in the management of benign prostatic hypertrophy.
Molecular structure of the alpha1A receptor was generated by homology module based on the X-ray structure of bovine rhodopsin (PDB code: 1f88). Docked known α1A antagonists into their receptor and produced threedimensional pharmacophore models (Figure 3(d)) [38]. A virtual screening of the company’s compound collection was performed to test how well this model is suited to identify α1A antagonists. Applied a hierarchical virtual screening procedure guided by 2D filters and threedimensional pharmacophore models. The 23,000 filtered compounds were docked into the α1A homology model with GOLD and scored with PMF. 990 druglike molecules were found by Catalyst. From the top-ranked compounds, 80 diverse compounds were tested in a radioligand displacement assay. 37 compounds revealed Ki values better than 10 μM; three potential compounds (compound 3 - 5) with very low nM Ki, of which compound 4 binds with 1.4 nM to the alpha1A receptor (Figure 1(f)). The findings suggest that rhodopsin-based homology models may be used as the structural basis for GPCR lead finding and compound optimization.
4. Discovery and Validation of PTDs Based on TCM Active Ingredients
The validation of pharmacological activities of new compounds extracted from TCM is a complex matter. For example, Tibetan medicine ceratostigma has antiherpes simplex I virus (HSV-1) activity besides a strong antibacterial effect. The studies of its molecular mechanism have shown that it interfered with adsorption of HSV-1 to host cells, selectively inhibit synthesis of DNA and protein of HSV-1, and further inhibit gene transcription of HSV-1 glycoprotein D (gD) and expression of mRNA. Tibet Institute of Plateau Biology have isolated 23 kinds of compounds from tibetan medicine ceratostigma, including major components plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) and plumbagic acid (β-(2,3-dihydroxy benzoyl) butyric acid) (Figure 1(g)). Plumbagin has effect on acute promyelocytic leukemia (APL), can inhibit the proliferation of APL cell line NB4 and induce cell apoptosis and cell cycle arrest at G2/M phase [39]. It can inhibit the activity of lymphocytic leukemia cells of mouse P388, and even reverse the experimental tumors in rats. Plumbagic acid has antitussive, expectorant, and antibacterial effects, etc. Shanghai Institute of Mmateria Medica, Chinese Academy of Sciences, have applied INVDOCK to search the target protein matched with the compounds from built protein structure database. Moreover, succeeded in finding the potential target molecules of plumbagin, namely alkane hydrogen peroxide reductase (AhpC), and initially speculated that plumbagin can play anti-Hp (helicobacter pylori) and anti-tumor activities as AhpC inhibitor [40].
Triterpenes isolated from Ganoderma lucidum could inhibit the growth of numerous cancer cell lines and were thought to be the basis of the anticancer effects of G. lucidum, including ganodenic acid A, B, C, D and F [21]. Ganodenic acid D (GAD) is one of the major components in Ganoderma triterpenes (Figure 1(g)). Flow cytometric analysis and DNA fragmentation analysis indicated that GAD induced G2/M cell cycle arrest and apoptosis [21,41]. Guo DA group have identified and confirmed proteins altered in expressional level after GAD exposure of cells using two-dimensional gel electrophoresis, MALDI-TOF MS/MS (mass spectrometry) and Western blotting [21]. The cytotoxic effect of GAD was associated with regulated expression of 21 proteins. Furthermore these possible GAD target-related proteins were evaluated by an in silico drug target searching program, INVDOCK. The INVDOCK analysis results suggested that GAD could bind six isoforms of 14-3-3 protein family, annexin A5, and aminopeptidase B. The direct binding affinity of GAD toward 14-3-3 zeta (PDB code: 1qja) was confirmed in vitro using surface plasmon resonance biosensor analysis. In addition, the intensive study of functional association among these 21 proteins revealed that 14 of them were closely related in the protein-protein interaction network. They had been found to either interact with each other directly or associate with each other via only one intermediate protein from previous protein-protein interaction experimental results.
5. Discovery of New Compounds Possessing Anti-Platelet Aggregation Activity
Cardiovascular and cerebrovascular diseases are still the main cause of morbidity and mortality in the world. The formation of platelet aggregates is an important pathogenetic factor in widespread cardiovascular disease. The sudden occlusion of an arterial vessel by formation of a thrombotic plug is the crucial event leading to deficient oxygen supply of important target organs like the heart or the brain. Human blood platelets play a significant role not only in normal hemostasis but also in arterial thrombosis, particularly under conditions of high shear stress [42]. Currently used antiplatelet drugs, including aspirin, ticlopidine, and others, are effective against certain but not all of the many endogenous platelet activators. Because of their limited efficacy, a significant number of serious thromboembolic complications still occur, highlighting the need for a more effective therapy. Here, we have designed, synthesized and appraised a series of 1,2,3,4-tetrahydroisoquinoline derivates possessing antiplatelet aggregation activity based on the structural modification of isoquinoline alkaloids as well as CADD, but also have designed and discovered new guided fibrinogen receptor atagonists (novel recombinant fusion proteins) from decorsin, a 39-residue peptide isolated from the North American leech Macrobdella decora.
5.1. (±)-1-Phenyl-2-Substituted-7-Sulfonylamide/ Amide-1,2,3,4-Tetrahydroisoquinolines
Alkaloids are a rich source of bioactive products, and tetrahydroisoquinoline compounds exhibit a wide range of biological activities, such as antimalarial, antiviral, antipoliovirus and anti-HIV activities as well as antiplatelet and vasorelaxing effects [43]. Among these isoquinoline derivatives, aporphinoids [43] that are mainly encountered in some archaic botanical families appear to be increasingly investigated for biological activity. For instance, recent experiments on tetrahydroisoquinoline alkaloids (such as tetrandrine, protoberberine, dauricine, reticuline) (Figure 4) have focused on promising antiplatelet aggregation effect [44].
Trimetoquinol (TMQ) is a prototype of the tetrahydroisoquinoline class of compounds that is structurally distinct from catecholamines. The β-adrenoceptor activeties of TMQ isomers and selected derivatives were evaluated on human β-adrenoceptor subtypes expressed in Chinese hamster ovary cells [45]. TMQ has activity as a beta-adrenergic agonist and as a platelet antiaggregatory agent [46]. Some reports have focused on the mechanism of inhibition of platelet function by TMQ and its analogs. Based on its competitive and stereoselective inhibition of thromboxane mimetic agents, TMQ was proposed as an endoperoxide/thromboxane A2 receptor antagonist; however, this mechanism has been questioned. A radiolabeled TMQ analog with high specific activity would aid in the elucidation of the actual mechanism of action. In the current research, modifications of the trimethoxy ring system of TMQ have been investigated. Replacement of one or two of the methoxy groups with iodine atoms leads to retention of platelet antiaggregatory activity and agonist blocking activity. Thus, these analogs have promise as potential radioligands since iodide exchange labeling can provide 125I-labeled compounds. Further, replacement of a methoxy group with either a nitro or amino functionality leads to decreased activity in
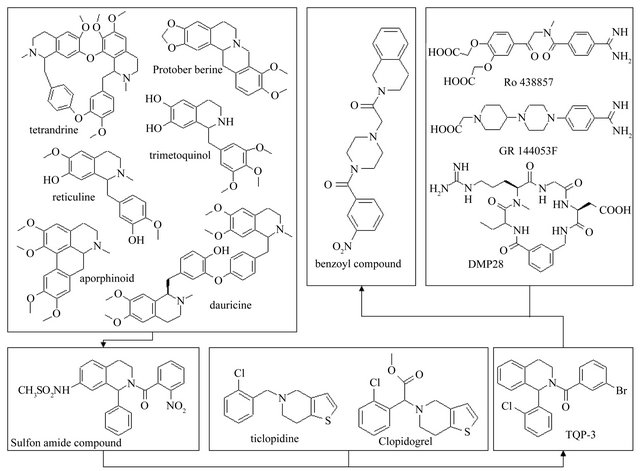
Figure 4. Design of 1,2,7-substituted-1,2,3,4-tetrahydroisoquinoline derivates possessing antiplatelet aggregation activities.
platelet systems. These results suggest that the putative sites of interaction for the trimethoxy ring system of TMQ in platelet systems will tolerate large, lipid soluble groups but will not tolerate large changes in the electronic characteristics of the ring system.
Thus, a series of (±)-1-phenyl-2-substituted-7-sulfonylamide/amide-1,2,3,4-tetrahydroisoquinoline derivatives were designed and synthesized. Fifteen analogs proved to be potential antiplatelet aggregation agents, and compound 11 (Figure 4) [47] which inhibits ADP-induced human platelet aggregation with IC50 values of approximately 22.90 nM was the most active.
5.2. 1-o-Chlorophenyl-2-Substituted- 1,2,3,4-Tetrahydroisoquinoline Derivatives
Ticlopidine and clopidogrel are orally active antiplatelet agents that block ADP-mediated platelet activation [48]. The therapeutic efficacy of ticlopidine might be associated not only with its delayed antiplatelet effects but also with its immediate thrombolytic action, which is likely to be mediated by endothelial prostacyclin and tissue plasminogen activator rather than by platelet mechanisms [49]. Like ticlopidine, clopidogrel is inactive in vitro and must be administered i.v. or orally to exhibit antiaggregatory and antithrombotic activities [50]. The irreversible modification of the ADP receptor site could be explained by the formation of a disulfide bridge between the reactive thiol group of the active metabolite and a cysteine residue of the platelet ADP receptor.
A series of 1-o-chlorophenyl-2-substituted-1,2,3,4-tetrahydroisoquinoline derivatives were designed and synthesized in order to search for a new type of platelet antiaggregants on the basis of the principles of isosterism and rational drug design, and in view of the structural features of ticlopidine, clopidogrel, as well as CoMFA results of 15 synthesized antiplatelet aggregating agents (±)-1-phenyl-2-substituted-7-sulfonylamide/amide-1,2,34-tetrahydroisoquinoline. The CoMFA study reveals that the steric interactive energy is a major contribution to antiplatelet aggregatory activity, and the sulfonylamide group is necessary for activity (Figure 3(e)) [47]. An area accommodating a small group exists near the 7 position of the tetrahydroisoquinoline nucleus. Smaller substituent at the 7 position of the tetrahydroisoquinoline nucleus, favors platelet aggregation inhibitory activity. It seems to show that removing a substituent from the 7 position of the tetrahydroisoquinoline nucleus would improve antiplatelet aggregation activity. So, we designed a type of 7-unsubstituted-1,2,3,4-tetrahydroisoquinoline derivatives, which is similar to ticlopidine. Four analogs proved to be potential antiplatelet aggregation agents, and TQP-3 (No. patents: ZL03132021.X) (Figure 4) that inhibits ADP-induced human platelet aggregation with IC50 values of approximately 0.206 nM was the most active.
Moreover, we have predicted the most probable active sites of ADP receptor (such as P2Y12) using the scoring matrix, which include three distant areas: “head area” (LGTGPLRTFV, 87 - 96), “middle area” (VGLITNGLAM, 38 - 47, and LGAKILSVVI, 139 - 148), and “bottom area” (RTRGVGKVPR, 222 - 231). Subsequently built the structural model of P2Y12 (PDB code 1VZ1) [51] using Homology module of InsightII software, which was docked with ATP/ADP in comparison with P2Y1 (PDB code 1ddd). As a comparison, we docked its antagonists, such as ticlopidine and clopidogrel, to the most probable sites and calculated their intermolecular energy. Our results imply that P2Y12 has the potential to be inhibited by ADP/ATP analogs, and it suggests that P2Y12 acts as a target of new drugs that inhibit platelet aggregation.
5.3. 2-(4-Substituted-Piperazin-1-Ylacetyl)- 1,2,3,4-Tetrahydroisoquinoline Derivatives
Ligand binding to integrins initiates intracellular signals that are crucial for cellular growth and differentiation. Binding of integrinαIIbβ3 (the glycoprotein IIb-IIIa complex, GP IIb/IIIa, fibrinogen receptor) to the symmetrical molecules fibrinogen and/or vWF in solution was shown to be essential for platelet aggregation and thrombus growth [52]. In addition, a variety of other platelet activators, including adenosine diphosphate (ADP), collagen, epinephrine, thromboxane A2, plateletactivating factor (PAF), serotonin or thrombin, might be involved in the physiological process of platelet stimulation leading finally to the functionalization ofαIIbβ3 and to platelet aggregation. Therefore, the blockade ofαIIbβ3 might constitute a superior approach in effectively preventing arterial thrombus formation. There are someα IIbβ3 antagonists, such as a systematically active peptide analog (DMP 728) of RGD motif [53], decorsin of 39- residue RGD-protein [54], and nonpeptide antagonists (Ro 438857 and GR144053F) [52,55]. A series of 2-(4- substituted-piperazin-1-ylacetyl)-1,2,3,4-tetrahydroisoquinoline derivatives [56] were designed and synthesized in order to search for new type of platelet antiaggregants based on a phenylpiperazine scaffold as well as TQP-3.
To identify potential candidates for antiplatelet drugs, human αIIbβ3 (GPIIb/IIIa) was expressed in Chinese hamster ovary (CHO) cells [57], which was validated by tetrapeptide RGDS (Arg-Gly-Asp-Ser) with IC50 of 0.057 mM, supported by Basanis results [58]. The ability of 2-(4-substituted-piperazin-1-ylacetyl)-1,2,3,4-tetrahydroisoquinoline derivatives to inhibit fibrinogen binding to αIIbβ3 based on the CHO cell model was measured by flow cytometry using GPIIb/IIIa assay, and the IC50 values of compounds were 0.166, 0.037, 0.311, 0.025, 0.034, and 0.184 mM, respectively [59]. Our research results indicated that the compounds with phenylsulfonyl and benzoyl groups at position 4 of piperazine showed higher IC50 values of inhibiting ADP-induced human platelet aggregation. Particularly benzoyl compound (Figure 4) possessed IC50 value of approximately 6.84 nM.
Similarly, we also built the structural model of human GPIIb/IIIa (PDB code 1UV9) [60,61] using Homology module. Additionally, a complex model of GPIIb/IIIa with the benzoyl compound revealed that the pharmacophore included m-nitro group of 4-benzene-piperazine, the nitrogen atom in the piperazine group, and 2-nitrogen of 1,2,3,4-tetrahydroisoquinoline nucleus, interacted with the hydroxyl groups of Thr125 of β3 and Tyr166 of α2b by hydrogen bonds and the carboxyl group at side chain of Asp179 of α2b in the fashion of electrostatic interaction [57]. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide) assays showed that benzoyl compounds possess potential anti-cancer activities, suggesting a potential role of integrin-guided signal pathway in cancer therapy. Further evaluation is under investigation.
5.4. Design, Expression, Purification and Activity Assay of New Recombinant Antagonists of Fibrinogen Receptor
Decorsin is isolated from the North American leech Macrobdella decora and acts as an antagonist of fibrinogen receptor on platelets, consisting of 39 amino acids including six cysteine and six praline residues, as well as the sequence Arg-Gly-Asp (RGD), a proposed recognition site of many adhesion proteins [62]. Annexin V is able to recognize stimulated platelets. It can assemble into highly ordered arrays on phospholipid membranes and has proven to preferentially bind to negatively charged phospholipids like PS (phosphatidylserine), which is greatly exposed on their membranes when platelets are activated, in the presence of Ca2+ [63]. During the aggregation of platelets, the restriction of PS to the inner surface of the plasma membrane bilayer is lost [64]. Hence it makes Annexin V a highly efficient arrow to recognize the stimulated platelets.
Two recombinant proteins, Annexin V plus Decorsin (AnnV-D39) and Annexin V plus the C terminal 27 amino acids variant of Decorsin (AnnV-D27), were constructed, expressed, and purified [65,66]. Platelet Aggregation assay results appear that AnnV-D39 shows good anti-platelet aggregation activity, but AnnV-D27 no such activities in any platelet aggregation assay test. And computational simulations reveal that AnnV-D39 showed good anti-platelet aggregation activity as a new antagonist of fibrinogen receptor, while Annv-D27 needs remodification [61]. Despite the AnnV-D39 fusion is more than decorsin, the former maintains the binding sites of decorsin interacton with its receptor, which contains Asp10, Arg28-Asp33, and Tyr37-Glu39. The addition of Annexin V could not influence the interaction between its decorsin fraction with its receptor GPIIb/IIIa due to the linkage peptide (GGGGSGGGGS). Although the AnnV-D27 fusion is similar to the AnnV-D39 fusion, there are differences between them, where the former is in shortage of the linkage peptide and the N-terminal segment of decorsin whose one residue (Asp10) contribution to its interaction with GPIIb-IIIa. Meanwhile, these complex models suggest that decorsin plays a role in antiplatelet aggregation not only by its RGD motif interaction with its GPIIb/IIIa receptor, but also by other residues, especially Asp10 of its N-terminal segment and Tyr37-Glu39 of its C-terminal segment. On the other hand, the linkage peptide acts as avoidance of influence and blockage between domains of fusion. This is the cause that AnnV-D39 shows good anti-platelet aggregation activity.
6. Application to TCM Modernization
TCM modernization refers to use modern science and technology to research and develop modern TCM, including the elucidation of the efficacy and mechanism (modernization), large-scale production (industrializetion), and internationally competitive TCM preparations accepted by the international markets (internationalizetion). TCM compound prescription is a complex system, characterized by multi-enzyme, multi-target and multicomponent, whose mechanism is very complicated [67]. It is necessary to carry out the following research: systematic research of TCM efficacy components at multidimensional and multi-mode levels using new technologies and new methods; the relationship among main components, structure, conformation and pharmacological activity after the preparation of TCM efficacy components and after taking the drug; and the relationship among TCM chemical composition, a variety of chemical information (including composition, structure, conformation, morphology and the process of change), and their efficacy and activity [68]. CADD is an effective way to research on modernization of Chinese medicine. By understanding the information of compound-related components, use computer simulation and screening methods to explore its possible mechanism, and validate in vitro and in vivo activities. This will greatly reduce the manpower and material inputs, facilitate the implementation of the experimental process, and quickly achieve the purpose of the experiment.
6.1. Application to the Study of Active Ingredients of TCM Compound Si-Ni-San
Si-Ni-San, consisting of bupleurum (Chinese Thorowax Root, Chai Hu), peony (Radix Paeoniae, Mu Dan), zhishi (Immature Bitter Orange, citrus aurantium), and glycyrrhiza (Radix Glycyrrhizae Preparata, Gan Cao), is a traditional Chinese compound prescription. It commonly used in the treatment of chronic hepatitis, cholecystitis, cholelithiasis, gastric ulcer, gastritis, gastrointestinal neurosis, accessories inflammation, tubal obstruction, acute mastitis, etc. besides type 2 diabetes [69]. Xu Q et al. [70] have demonstrated the inhibitory effects of SiNi-San on picryl chloride (2,4,6-trinitrochlorobenzene)- induced ear contact sensitivity (PCl-CS) mice, T cellmediated delayed-type hypersensitivity (DTH) model. This study aimed to evaluate the role of the four major constituents contained in the prescription (saikosaponins, paeoniflorin, naringin and glycyrrhizin) in the inhibitory effect (Figure 1(h)). When administered during the induction phase, saikosaponin A and glycyrrhizin showed significant inhibitory effects, while paeoniflorin and naringin did not. These components in Si-Ni-San also inhibited the activation and proliferation of T lymphocytes as well as the production of cytokines such as tumour necrosis factor-alpha and interferon-gamma to different extents. Saikosaponin A and paeoniflorin dose-dependently reduced the splenocyte adhesion to type I collagen, while glycyrrhizin only showed a slight tendency. Furthermore, treatment with glycyrrhizin or saikosaponin A, rather than paeoniflorin or naringin, moderately inhibited the matrix metalloproteinase (MMP)-2 activity of the splenocytes from PCl-CS mice, and the combination of all four components showed a strong inhibition against MMP-2. Moreover, the components markedly decreased the serum level of nitric oxide in PCl-sensitized mice. The results indicated that saikosaponin A and glycyrrhizin may be the major contributors in the alleviation effect of Si-Ni-San on contact sensitivity, and paeoniflorin and naringin may exhibit a co-operative effect.
The effects of Si-Ni-San and herbal drug combinations (bupleurum-Peony, bupleurum-zhishi and peony-glycyrrhiza) on metalloproteinase activity and adhesion of mouse spleen cells activated by concanavalin A (Con A) were studied in vitro [71]. The activities of MMP-2 and MMP-9, and spleen cell adhesion were measured by gelatin zymography assay and violet crystal staining, respectively. The ethanol extracts of Si-Ni-San and drug combinations significantly inhibited the activities of MMP-2 and MMP-9, and the adhesion to type I collagen of mouse spleen cells. However, the extracts did not inhibit the activities of metalloproteinases produced by spleen cells isolated from normal mice. Among the drug combinations, the effects of bupleurum-peony were similar to those of Si-Ni-San. The results suggest that SiNi-San may inhibit the over-activation of spleen cells through the downregulation of metalloproteinases and inhibition of cell adhesion to the extracellular matrix. This effect was mainly exhibited by the combination between bupleurum and peony.
Moreover, the study above initially explored the synergistic action of the active ingredients of Si-Ni-San and their associated proteins, and provides a basis for seeking potential drug target. Thus, according to the related proteins of important parts of DTH process, screen and predict the PTDs of saikosaponin A, paeoniflorin, naringin and glycyrrhizin by INVDOCK, and make experimental verification. Then, simulate the relationship of competition and collaboration between the various active ingredients of Si-Ni-San using computer simulation technology, and further validate and explain the interaction among saikosaponin A, paeoniflorin, naringin and glycyrrhizin through molecular biology experiments.
6.2. Application to the Study of Drugs for Treating AIDS
Some TCM extracts, such as trichosanthin (TCS), the plant protein MAP30 from balsam pear (Momordica charantia), and some polysaccharides from glycyrrhiza (Radix Glycyrrhizae Preparata), ganoderma lucidum (Leyss. Fr.), and lentinus edodes (Shiitake Mushroom), respectively, is an important means of AIDS treatment. Trichosanthin, an abortifacient, immunosuppressive and anti-tumor protein purified from the traditional Chinese herb medicine Tian Hua Fen, is a potent inhibitor against HIV-1 replication and kill infected cells [72]. Under normal enzymatic digestion conditions, trichosanthin as a ribosome-inactivating protein (RIP) cleaves the supercoiled double-stranded DNA to produce nicked circular and linear DNA. Trichosanthin has no effect on linear double-stranded DNA. Neither does it convert relaxed circular duplex DNA into a supercoiled form in the presence of ATP. Thus trichosanthin is not a DNA gyrase. However, trichosanthin can cleave the relaxed circular DNA into a linear form, indicating that both the circular as well as the supercoiled forms are essential for trichosanthin recognition. In addition, trichosanthin contains one calcium metal ion per protein molecule, which presumably is related to its endonucleolytic activity. Similarly, MAP30 (Momordica Anti-HIV Protein, MW 30 kD) as a ribosome-inactivating protein exhibits potent activity against HIV-1, yet little toxicity to uninfected cells and intact animals. The antiviral action of MAP30 has been demonstrated in acutely and chronically infected cells. In addition, MAP30 inhibits the function of eukaryotic ribosomal RNA and DNA topoisomerase in messenger RNA translation, and elevated T4 cell ratio [73]. MAP30 is capable of relaxing and decatenating supercoiled DNA, as well as catalyzing double-stranded breakage of DNA, resulting in topologically inactive products. The dual capability of MAP30 to act on both DNA and RNA substrates may be related to its potent anti-HIV action at multiple stages of the viral life cycle.
Polysaccharides are highly branched polymeric carbohydrate structures, formed of repeating units (either monoor di-saccharides) joined together by glycosidic bonds. Glycyrrhiza polysaccharide is mainly composed of glucose, arabinose and galactose while lentinan mostly includes pyran-glucan. Ganoderma lucidum polysaccharides contain a few of arabinose, xylose, fucose, rhamnose, galactose and other monosaccharides besides glucose. Atomic force microscope (AFM) observations revealed that the sugar chains of glycyrrhiza polysaccharides showed close multi-strand helical structures [74], while ganoderma lucidum polysaccharides did helical three-dimensional structures composed of three single sugar chains, similar to those of triple-stranded DNA [75-77]. The former can enhance immune function, stimulate the reticuloendothelial system, and enhance the natural killer cell activity, the latter has important roles in improvement of microcirculation and regulating T and B cells, can stimulate macrophages and natural killer cells, and promote mononuclear cell secretion of interleukins and tumor necrosis factor to suppress excessive immune response.
There are a number of TCM also can regulate immune function. Coix Seed (Semen Coicis, Yi Yi Ren) can increase in T-cell while ginseng (Panax ginseng, Ren Shen) and Codonopsis pilosulae (Radix Codonopsis pilosulae, Dang Shen) increase in white blood cell. Mushroom (Lentinus edodes, Lentinula edodes, Xiang Gu) can enhanced neutrophil phagocytosis, astragali radix (English: milk-vetch root; Chinese: Huang Qi) and semen lablab bean (White Hyacinth Bean, Bai Bian Dou) induce production of interferon (IFN). Cassia Twig (Ramulus Cinnamomi, Gui Zhi), Rhizoma Polygonati (Polygonatum sibiricum, Huang Jing), and mushrooms strengthen antibody and complement activities, Radix Astragali (Membranous Milkvetch Root, Huang Qi) and Epimedium Herb (Shorthorned Epimedium Herb, Yin Yang Huo) boost the immune system. Many HIV (human immunodeficiency virus) patients will soon relapse although they can be controlled below the detection limit by taking the above drugs. This may be related to the third variable (V3) loop of HIV glycoprotein gp120 [78,79]. The entry of primate immunodeficiency viruses into target cells depends on a sequential interaction of the gp120 envelope glycoprotein with the cellular receptors, CD4 and co-receptor, members of the chemokine receptor family (CCR5 or CXCR4) [80,81]. The gp120 V3 region variation has been implicated in chemokine receptor binding, but the use of CCR5 by diverse primate immunodeficiency viruses suggests the involvement of an additional, conserved gp120 element [77]. Through the use of gp120 mutants, a highly conserved gp120 structure was shown to be critical for CCR5 binding. This structure is located adjacent to the V3 loop and contains neutralization epitopes induced by CD4 binding. This conserved element may be a useful target for pharmacologic or prophylactic intervention in HIV infections.
In traditional Chinese prescription, Minor Bupleurum Combination (Xiao Chai Hu Tang), consisting of Bupleurum Radix (Chai Hu) 28.26%, Pinellia Tuber (Ban Xia) 19.56%, Panax Ginseng (Ren Shen) 10.87%, Glycyrrhiza Radix (Gan Cao) 10.87%, Scutellaria Radix (Huang Qin) 10.87%, Ziziphus Fructus (Da Zao) 10.87%, Zingiber Officinale Radix (Sheng Jiang) 8.70%, have a stronger inhibitory effect on HIV. It will significantly heighten lymphocytes, auxiliary T cells and natural killer cell activities.
It is repoted that HIV-1 exploits multiple host proteins during infection. Harvard Medical School researchers have found HIV-related PTDs by genome-wide RNA interference (RNAi) screening system, small interfering RNA (siRNA) library (Thermo Fisher Scientific, Dharmacon Products, Lafayette, Co.) [82]. They performed a large-scale siRNA screen to identify host factors required by HIV-1 through silencing more than 21,000 human genes and blocking gene expression. Finally identified more than 250 HIV-dependency factors (HDFs), namely 273 proteins associated with HIV replication, including 36 known HIV-related genes. These proteins participate in a broad array of cellular functions and implicate new pathways in the viral life cycle. Further analysis revealed previously unknown roles for retrograde Golgi transport proteins (Rab6 and Vps53) in viral entry, a karyopherin (TNPO3) in viral integration, and the Mediator complex (Med28) in viral transcription. Transcriptional analysis revealed that HDF genes were enriched for high expression in immune cells, suggesting that viruses evolve in host cells that optimally perform the functions required for their life cycle. This effort illustrates the power with which RNAi and forward genetics can be used to expose the dependencies of human pathogens such as HIV, and in so doing identify potential targets for therapy. This greatly expands the number of HIV potential targets, provides new programs for the treatment of AIDS, and changes people’s general understanding of HIV therapeutic targets. That lead HIV therapeutic target proteins from the virus itself toward the host proteins will reduce the drug resistance caused by virus mutation, give us another way to look for the new drug targets capable of disrupting the interaction of viruses and proteins and killing the HIV virus, and bring the dawn of hope to create a new AIDS “magic recipes”.
7. Conclusion
As the therapeutic effect of TCM is a multi-component, multi-path, multi-functional, multi-target approach of integrated regulation, it may be difficult to determine their true targets of many TCM ingredients. Therefore, the study of TCM and its chemical constituents conduct component-wide qualitative and quantitative data analysis by chemomics technology, including integration of modern separation and analysis, comparative chemical analysis methods, and in vitro and in vivo metabolic analysis. The biological effects of various parts of TCM should be evaluated at the whole, organizational, cellular, and molecular levels, and comprehensively analyzed by computer to obtain valuable information. To explore the mechanism of Chinese medicine using computer simulation not only can compensate for the deficiencies of pharmacology experimental methods, but more importantly establish new concepts and advanced research tools in TCM research areas. This is helpful to provide new ideas and new ways for TCM research, is expected to make modernization of TCM into a track of healthy development, and provide strong support of modern science and technology for TCM into the world.
8. Acknowledgements
This work was supported by a grant from National Key Technology R & D Program (2008-BAI51B01).
REFERENCES
- F. Cheung, “TCM: Made in China,” Nature, Vol. 480, No. 7378, 2011, pp. S82-S83. doi:10.1038/480S82a
- P. Tian, “Convergence: Where West meets East,” Nature, Vol. 480, No. 7378, 2011, pp. S84-S86. doi:10.1038/480S84a
- J. Yang, “Systematical Analysis of the Application of Chinese Traditional Medicine Informatics to Diabetes Proved Recipes,” Journal of Diabetes Research & Clinical Metabolism, Vol. 1, No. 1, 2012, p. 10. doi:10.7243/2050-0866-1-10
- J. Yang, “Application of Chinese Traditional Medicine Informatics to Diabetes Proved Recipes,” World Clinical Drugs, Vol. 29, No. 2, 2008, pp. 94-100.
- J. Yang, “Idea and Strategy for New Drug Research,” World Science-Technology R & D, Vol. 21, No. 5, 1999, pp. 64-66.
- J. Yang, W. Y. Hua and S. X. Peng, “New trend of CADD—Receptor Structure-Based Drug Design,” Journal of China Pharmaceutical University, Vol. 24, No. 3, 1993, pp. 187-192.
- J. Yang and W. Y. Hua, “Basic Pharmacophore for Some Antithrombotic Agents with Combined Thromboxane Receptor Antagonists (TXRA)/Thromboxine Synthase Inhibitor (TXSI) Activities,” Drug Development Research, Vol. 39, No. 2, 1996, pp. 197-200. doi:10.1002/(SICI)1098-2299(199610)39:2<197::AID-DDR14>3.0.CO;2-9
- P. J. Meek, Z. W. Liu, L. F. Tian, C. Y. Wang, W. J. Welsh and R. J. Zauhar, “Shape Signatures: Speeding up Computer Aided Drug Discovery,” Drug Discovery Today, Vol. 11, No. 19-20, 2006, pp. 895-904. doi:10.1016/j.drudis.2006.08.014
- The International 311C90 Long-term Study Group, “The Long-Term Tolerability and Efficacy of Oral Zolmitriptan in the Acute Treatment of Migraine,” Headache, Vol. 38, No. 3, 1998, pp. 173-183. doi:10.1046/j.1526-4610.1998.3803173.x
- T. H. Keller, A. Pichota and Z. Yin, “A Practical View of ‘Druggability’,” Current Opinion in Chemical Biology, Vol. 10, No. 4, 2006, pp. 357-361. doi:10.1016/j.cbpa.2006.06.014
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, “Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings,” Advanced Drug Delivery Reviews, Vol. 46, No. 1-3, 2001, pp. 3-26. doi:10.1016/S0169-409X(00)00129-0
- C. A. Lipinski, “Leadand Drug-Like Compounds: The Rule-of-Five Revolution,” Drug Discovery Today: Technologies, Vol. 1, No. 4, 2004, pp. 337-341. doi:10.1016/j.ddtec.2004.11.007
- W. P. Waters and M. A. Mureko, “Prediction of DrugLikeness,” Advanced Drug Delivery Reviews, Vol. 54, No. 3, 2002, pp. 255-271. doi:10.1016/S0169-409X(02)00003-0
- Y. Sugiyama, “Druggability: Selecting Optimized Drug Candidates,” Drug Discovery Today, Vol. 10, No. 23-24, 2005, pp. 1577-1579. doi:10.1016/S1359-6446(05)03675-5
- I. Muegge, “Selection Criteria for Drug-Like Compounds,” Medicinal Research Reviews, Vol. 23, No. 3, 2003, pp. 302- 32l. doi:10.1002/med.10041
- A. L. Hopkins and C. R. Groom, “The Druggable Genome,” Medicinal Research Reviews, Vol. 1, No. 9, 2002, pp. 727-730. doi:10.1038/nrd892
- D. Brown and G. Superti-Furga, “Rediscovering the Sweet Spot in Drug Discovery,” Drug Discovery Today, Vol. 8, No. 23, 2003, pp. 1067-1077. doi:10.1016/S1359-6446(03)02902-7
- J. Owens, “Target Validation: Determining Druggability,” Nature Reviews Drug Discovery, Vol. 6, No. 3, 2007, pp. 187. doi:10.1038/nrd2275
- D. W. Walke, C. Han, J. Shaw, E. Wann, B. Zambrowicz and A. Sands, “In Vivo Drug Target Discovery: Identifying the Best Targets from the Genome,” Current Opinion in Biotechnology, Vol. 12, No. 6, 2001, pp. 626-631. doi:10.1016/S0958-1669(01)00271-3
- Y. Z. Chen and D. G. Zhi, “Ligand-Protein Inverse Docking and Its Potential Use in the Computer Search of Protein Targets of a Small Molecule,” Proteins, Vol. 43, No. 2, 2001, pp. 217-226. doi:10.1002/1097-0134(20010501)43:2<217::AID-PROT1032>3.0.CO;2-G
- Q. X. Yue, Z. W. Cao, S. H. Guan, X. H. Liu, L. Tao, W. Y. Wu, Y. X. Li, P. Y. Yang, X. Liu and D. A. Guo, “Proteomic Characterization of the Cytotoxic Mechanism of Ganoderic Acid D and Computer Automated Estimation of the Possible Drug-Target Network,” Molecular & Cellular Proteomics, Vol. 7, No. 5, 2008, pp. 949-961. doi:10.1074/mcp.M700259-MCP200
- B. R. Stockwell, “Exploring Biology with Small Organic Molecules,” Nature, Vol. 432, No. 7019, 2004, pp. 846- 854. doi:10.1038/nature03196
- S. Peukert, J. Brendel, B. Pirard, A. Brüggemann, P. Below, H. W. Kleemann, H. Hemmerle and W. Schmidt, “Identification, Synthesis, and Activity of Novel Blockers of the Voltage-Gated Potassium Channel Kv1.5,” Journal of Medicinal Chemistry, Vol. 46, No. 4, 2003, pp. 486- 498. doi:10.1021/jm0210461
- S. Wang, C. Meades, G. Wood, A. Osnowski, S. Anderson, R. Yuill, M. Thomas, M. Mezna, W. Jackson, C. Midgley, G. Griffiths, I. Fleming, S. Green, I. McNae, S. Y. Wu, C. McInnes, D. Zheleva, M. D. Walkinshaw and P. M. Fischer, “2-Anilino-4-(Thiazol-5-yl)Pyrimidine CDK Inhibitors: Synthesis, SAR Analysis, X-Ray Crystallography, and Biological Activity,” Journal of Medicinal Chemistry, Vol. 47, No. 7, 2004, pp. 1662-1675. doi:10.1021/jm0309957
- G. A. Gutman, K. G. Chandy, S. Grissmer, M. Lazdunski, D. McKinnon, L. A. Pardo, G. A. Robertson, B. Rudy, M. C. Sanguinetti, W. Stuhmer and X. Wang, “International Union of Pharmacology. LIII. Nomenclature and Molecular Relationships of Voltage-Gated Potassium Channels,” Pharmacological Reviews, Vol. 57, No. 4, 2005, pp. 473-508. doi:10.1124/pr.57.4.10
- M. Malumbres and M. Barbacid, “To Cycle or Not to Cycle: A Critical Decision in Cancer,” Nature Reviews Cancer, Vol. 1, No. 3, 2001, pp. 222-231. doi:10.1038/35106065
- M. Kitagawa, H. Higashi, I. Suzuki-Takahashi, K. Segawa, S. K. Hanks, Y. Taya, S. Nishimura and A. Okuyama, “Phosphorylation of E2F-1 by Cyclin A-Cdk2,” Oncogene, Vol. 10, No. 2, 1995, pp. 229-236.
- W. Krek, G. Xu and D. M. Livingston, “Cyclin A-Kinase Regulation of E2F-1 DNA Binding Function Underlies Suppression of an S Phase Checkpoint,” Cell, Vol. 83, No. 7, 1995, pp. 1149-1158. doi:10.1016/0092-8674(95)90141-8
- S. J. McClue, D. Blake, R. Clarke, A. Cowan, L. Cummings, P. M. Fischer, M. MacKenzie, J. Melville, K. Stewart, S. Wang, N. Zhelev, D. Zheleva and D. P. Lane, “In Vitro and in Vivo Antitumor Properties of the Cyclin Dependent Kinase Inhibitor CYC202 (R-Roscovitine),” International Journal of Cancer, Vol. 102, No. 5, 2002, pp. 463-468. doi:10.1002/ijc.10738
- J. A. Lees and R. A. Weinberg, “Tossing Monkey Wrenches into the Clock: New Ways of Treating Cancer,” Proceedings of the National Academy of Sciences, Vol. 96, No. 8, 1999, pp. 4421-4223. doi:10.1073/pnas.96.8.4221
- S. Y. Wu, I. McNae, G. Kontopidis, S. J. McClue, C. McInnes, K. J. Stewart, S. Wang, D. I. Zheleva, H. Marriage, D. P. Lane, P. Taylor, P. M. Fischer and M. D. Walkinshaw, “Discovery of a Novel Family of CDK Inhibitors with the Program LIDAEUS: Structural Basis for Ligand-Induced Disordering of the Activation Loop,” Structure, Vol. 11, No. 4, 2003, pp. 399-410. doi:10.1016/S0969-2126(03)00060-1
- J. Singh, H. Van Vlijmen, Y. Liao, W. C. Lee, M. Cornebise, M. Harris, I. H. Shu, A. Gill, J. H. Cuervo, W. M. Abraham and S. P. Adams, “Identification of Potent and Novel a4b1 Antagonists Using in Silico Screening,” Journal of Medicinal Chemistry, Vol. 45, No. 14, 2002, pp. 2988-2993. doi:10.1021/jm020054e
- D. Seiffge, “Protective Effects of Monoclonal Antibody to VLA-4 on Leukocyte Adhesion and Course of Disease in adjuvant Arthritis in Rats,” The Journal of Rheumatology, Vol. 23, No. 12, 1996, pp. 2086-2091.
- J. B. Massague, S. W. Blain and R. S. Lo, “TGF-ß Signaling in Growth Control, Cancer, and Heritable Disorders,” Cell, Vol. 103, No. 2, 2000, pp. 295-309. doi:10.1016/S0092-8674(00)00121-5
- P. A. Eyers, M. Craxton, N. Morrice, P. Cohen and M. Goedert, “Conversion of SB 203580-Insensitive MAP Kinase Family Members to Drug-Sensitive Forms by a Single Amino-Acid Substitution,” Chemistry & Biology, Vol. 5, No. 6, 1998, pp. 321-328. doi:10.1016/S1074-5521(98)90170-3
- J. Singh, C. E. Chuaqui, P. A. Boriack-Sjodin, W.-C. Lee, T. Pontz, M. J. Corbley, H.-K. Cheung, R. M. Arduini, J. N. Mead, M. N. Newman, J. L. Papadatos, S. Bowes, S. Josiah and L. E. Ling, “Successful Shape-Based Virtual Screening: The Discovery of a Potent Inhibitor of the Type I TGFbeta Receptor Kinase (TbetaRI),” Bioorganic & Medicinal Chemistry Letters, Vol. 13, No. 24, 2003, pp. 4355-4359. doi:10.1016/j.bmcl.2003.09.028
- H. Zhong and K. P. Minneman, “Alpha1-Adrenoceptor Subtypes,” European Journal of Pharmacology, Vol. 375, No. 1-3, 1999, pp. 261-276. doi:10.1016/S0014-2999(99)00222-8
- A. Evers and T. Klabunde, “Structure-Based Drug Discovery Using GPCR Homology Modeling: Successful Virtual Screening for Antagonists of the Alpha1a Adrenergic Receptor,” Journal of Medicinal Chemistry, Vol. 48, No. 4, 2005, pp. 1088-1097. doi:10.1021/jm0491804
- Y. L. Zhao and D. P. Lu, “In Vitro Effect of Plumbagin in Human Acute Promyelocytic Leukemia Cells,” Journal of Experimental Hematology, Vol. 14, No. 2, 2006, pp. 208- 210.
- J. W. Chen, C. M. Sun, W. L. Sheng, Y. C. Wang and W. J. Syu, “Expression Analysis of Up-Regulated Genes Responding to Plumbagin in Escherichia coli,” Journal of Bacteriology, Vol. 188, No. 2, 2006, pp. 456-463. doi:10.1128/JB.188.2.456-463.2006
- L. Ma, F. Wu and R. Y. Chen, “Analysis of Triterpenoids in Ganoderma Lucidum,” Acta Pharmaceutica Sinica, Vol. 38, No. 1, 2003, pp. 50-52.
- T. Weller, L. Alig, M. H. Muller, W. C. Kouns and B. Steiner, “Fibrinogen Receptor Antagonists—A Novel Class of Promising Antithrombotics,” Drugs Future, Vol. 19, No. 5, 1994, pp. 461-476.
- J. Boustie, J. L. Stigliani, J. Montanha, M. Amoros, M. Payard and L. Girre, “Antipoliovirus Structure-Activity Relationships of Some Aporphine Alkaloids,” Journal of Natural Products, Vol. 61, No. 4, 1998, pp. 480-484. doi:10.1021/np970382v
- J. F. Wu and T. P. Liu, “Effects of Berberine on Platelet Aggregation and Plasma Levels of TXB2 and 6-KetoPGFla in Rats with Reveksible Middle Cerebral Arteky Occlusion,” Acta Pharmaceutica Sinica, Vol. 30, No. 2, 1995, pp. 98-102.
- A. A Konkar, S. S. Vansal, G. Shams, P. F. Fraundorfer, W. P. Zheng, V. I. Nikulin, J. De Los Angeles, R. H. Fertel, D. D. Miller and D. R. Feller, “β-Adrenoceptor Subtype Activities of Trimetoquinol Derivatives: Biochemical Studies on Human β-Adrenoceptors Expressed in Chinese Hamster Ovary Cells,” Journal of Pharmacology and Experimental Therapeuties, Vol. 291, No. 2, 1999, pp. 875-883.
- M. W. Harrold, M. A. Gerhardt, K. Romstedt, D. R. Feller and D. D. Miller, “Synthesis and Platelet Antiaggregatory Activity of Trimetoquinol Analogs as Endoperoxide/Thromboxane A2 Antagonists,” Drug Design and Delivery, Vol. 1, No. 3, 1987, pp. 193-207.
- J. Yang, W. Y. Hua, F. X. Wang, Z. Y. Wang and X. Wang, “Design, Synthesis, and Inhibition of Platelet Aggregation for Some 1-o-Chlorophenyl-1,2,3,4-Tetra-Hydroisoquinoline Derivatives,” Bioorganic & Medicinal Chemistry, Vol. 12, No. 24, 2004, pp. 6547-6557. doi:10.1016/j.bmc.2004.09.028
- S. T. Gardell, “Ticlopidine and Clopidogrel: Antithrombotic Agents that Block ADP-Mediated Platelet Activation”, Drug Discovery and Design, Vol. 1, No. 3, 1993, pp. 521-526. doi:10.1007/BF02171863
- R. J. Gryglewski, R. Korbut, J. Swies, E. Kostka-Trabka, K. Bieron and J. Robak, “Thrombolytic Action of Ticlopidine: Possible Mechanisms,” European Journal of Pharmacology, Vol. 308, No. 1, 1996, pp. 61-67. doi:10.1016/0014-2999(96)00256-7
- A. A. Weber, S. Reimann and K. Schror, “Specific Inhibition of ADP-Induced Platelet Aggregation by Clopidogrel in Vitro,” British Journal of Pharmacology, Vol. 126, No. 2, 1999, pp. 415-420. doi:10.1038/sj.bjp.0702276
- C. Y. Zhan, J. Yang, X. C. Dong and Y. L. Wang, “Molecular Modeling of Purinergic Receptor P2Y12 and Interaction with Its Antagonists,” Journal of molecular graphics & modelling Volume, Vol. 26, No. 1, 2007, pp. 20-31.
- D. A. Handley, “Developmental Preclinical Therapies and Clinical Strategies in the Treatment of Vascular Restenosis,” Drugs Future, Vol. 20, No. 3, 1995, pp. 265-290.
- S. A. Mousa, J. M. Bozarth, M. S. Forsythe, S. M. Jackson, A. Leamy, M. M. Diemer, R. P. Kapil, R. M. Knabb, M. C. Mayo and S. K. Pierce, “Antiplatelet and Antithrombotic Efficacy of DMP 728, a Novel Platelet GPIIb/ IIIa Receptor Antagonist,” Circulation, Vol. 89, No. 1, 1994, pp. 3-12. doi:10.1161/01.CIR.89.1.3
- A. M. Krezel, J. S. Ulmer, G. Wagner and R. A. Lazarus, “Recombinant Decorsin: Dynamics of the RGD Recognition Site,” Protein Science, Vol. 9, No. 8, 2000, pp. 1428- 1438. doi:10.1110/ps.9.8.1428
- J. H. van Maarseveen, J. A. J. den Hartog, K. Tipker, J. H. Reinders, J. Brakkee, U. Schon, W. Kehrbach and C. G. Kruse, “Design and Synthesis of an Orally Active GPIIb/ IIIa Antagonist Based on a Phenylpiperazine Scaffold,” Bioorganic & Medicinal Chemistry, Vol. 8, No. 12, 1998, pp. 1531-1536. doi:10.1016/S0960-894X(98)00257-1
- Z. Y. Wang, C. Z. Wang, F. X. Wang and J. Yang, “Synthesis and Platelet Aggregation Inohibitory Activity of 1,2,3,4-Tetrahydroisoquinoline Derivatives,” Chinese Journal of Pharmaceuticals, Vol. 36, No. 5, 2005, pp. 261- 263.
- J. Yang, J. Yao, J. Chen, X. N. Wang, T. Y. Zhu, L. L. Chen and P. Chu, “Construction of Drug Screening Cell Model and Application to New Compounds Inhibiting FITC-Fibrinogen Binding to CHO Cells Expressing Human αIIbβ3,” European Journal of Pharmacology, Vol. 618, No. 1-3, 2009, pp. 1-8. doi:10.1016/j.ejphar.2009.07.011
- R. B. Basani, D. L. French, G. Vilaire, D. L. Brown, F. Chen, B. S. Coller, J. M. Derrick, T. K. Gartner, J. S. Bennett and M. Poncz, “A Naturally Occurring Mutation Near the Amino Terminus of Alpha IIb Defines a New Region Involved in Ligand Binding to Alpha IIbbeta 3,” Blood, Vol. 95, No. 1, 2000, pp. 180-188.
- J. B. Yang, J. Yao, L. L. Chen and J. Yang, “The AminoTerminal Domain of Integrin β3 Functions as a Transcriptional Activator in Yeast,” Molecular and Cellular Biochemistry, Vol. 288, No. 1-2, 2006, pp. 1-5. doi:10.1007/s11010-005-9078-2
- [61] J. Yang, C. Y. Zhan, X. C. Dong, K. Yang and F. X. Wang, “Interaction of Human Fibrinogen Receptor (GPIIbIIIa) with Decorsin,” Acta Pharmacologica Sinica, Vol. 25, No. 8, 2004, pp. 1096-1104.
- [62] J. Yang, C. Y. Zhan, X. C. Dong, J. Yao, K. Yang and Z. C. Hua, “Structure and Function of AnnexinV-Decorsin Fusion,” Journal of Molecular Structure: Theochem, Vol. 730, No. 1-3, 2005, pp. 227-233. doi:10.1007/s11010-005-9078-2
- [63] P. A. Lapchak and D. M. Araujo, “Therapeutic Potential of Platelet Glycoprotein IIb/IIIa Receptor Antagonists in the Management of Ischemic Stroke,” Journal of Biological Chemistry, Vol. 271, No. 30, 1996, pp. 17785- 17790.
- [64] J. F. Tait and D. Gibson, “Phospholipid Binding of Annexin V: Effects of Calcium and Membrane Phosphotidylserine Content,” Archives of Biochemistry and Biophysics, Vol. 298, No. 1, 1992, pp. 187-191. doi:10.1016/0003-9861(92)90111-9
- [65] E. M. Bevers, P. Comfurius and R. F. Zwaal, “Changes in Membrane Phospholipid Distribution during Platelet Activation,” Biochimica et Biophysica Acta (BBA)-Biomembranes, Vol. 736, No .1, 1983, pp. 57-66. doi:10.1016/0005-2736(83)90169-4
- [66] J. B. Yang, J. Yao, K. Yang, Z. C. Hua and J. Yang, “Expression, Purification and Activity Assay of New Recombinant Antagonists of Fibrinogen Receptor,” American Journal of Biochemistry and Biotechnology, Vol. 1, No. 2, 2005, pp. 69-73.
- [67] J. Yao, K. Yang, J. B. Yang, Z. C. Hua and J. Yang, “Expression, Purification and Activity Assay of Two New Recombinant Antagonists of Fibrinogen Receptor,” Journal of China Pharmaceutical University, Vol. 36, No. 4, 2005, pp. 173-178.
- [68] X. J. Xu, “Study on Computer Simulation for Chinese Traditional Compound Medicine,” Progress in Chemistry, Vol. 11, No. 2, 1999, pp. 202-204.
- [69] X. R. Wang, D. H. Sun, C. X. Zhuang and X. Chen, “Considerations of Basic Research on Modernization of Chinese Traditional Medicine,” Progress in Chemistry, Vol. 19, No. 2, 1999, pp. 197-199.
- [70] J. Morello, S. Rodriguez-Novoa, I. Jimenez-Nacher and V. Soriano, “Drug Interactions of Tipranavir, a New HIV Protease Inhibitor,” Drug Metabolism Letters, Vol. 1, No. 1, 2007, pp. 81-84. doi:10.2174/187231207779814256
- [71] L. Zhang, Y. Dong, Y. Sun, T. Chen and Q. Xu, “Role of Four Major Components in the Effect of Si-Ni-San, a Traditional Chinese Prescription, against Contact Sensitivity in Mice,” Journal of Pharmacy and Pharmacology, Vol. 58, No. 9, 2006, pp. 1257-1264. doi:10.1211/jpp.58.9.0013
- [72] Y. Sun and Q. Xu, “Effects of Si-Ni-San, a Traditional Chinese Formula, and Its Drug-Pairs on Activities of Metalloproteinases and Adhesion of Mouse Spleen Cells Activated by Concanavalin A,” Chinese Journal of Natural Medicines, Vol. 1, No. 2, 2003, pp. 103-106.
- [73] M. X. Li, H. W. Yeung, L. P. Pan and S. I. Chan, “Trichosanthin, a Potent HIV-1 Inhibitor, Can Cleave Supercoiled DNA in Vitro,” Nucleic Acids Research, Vol. 19, No. 22, 1991, pp. 6309-6312. doi:10.1093/nar/19.22.6309
- [74] S. Lee-Huang, H. C. Chen, H. F. Kung and P. L. Huang, “MAP30, an Anti-HIV Protein, Inhibits Both Ribosomal RNA Function and DNA Topological Inter-Conversions,” International AIDS Conference, Berlin, 6-11 June 1993, p. 467.
- [75] R. G. Sun and J. Zhang, “A Study of Helical Structure of Glycyrrhiza Polysaccharides by Atomic Force Microscope,” Acta Chimica Sinica, Vol. 64, No. 24, 2006, pp. 2467-2472.
- [76] J. Yang, X. C. Dong and Y. Leng, “Conformation Biases of Amino Acids Based on Tripeptide Microenvironment from PDB database,” Journal of Theoretical Biology, Vol. 240, No. 3, 2006, pp. 374-384. doi:10.1016/j.jtbi.2005.09.025
- [77] J. Yang, X. C. Dong and Y. Leng, “Application of FTTP to Alpha-Helix or Beta-Strand Motifs,” Journal of Theoretical Biology, Vol. 242, No. 1, 2006, pp. 199-219. doi:10.1016/j.jtbi.2006.02.014
- [78] J. Yang and C. Q. Liu, “Stability and Molecular Modeling of Triplex DNA Inhibiting DNA Binding Protein Binding to the Core Promoter of Hepatitis B Virus,” Progress in Biochemistry and Biophysics, Vol. 27, No. 3, 2000, pp. 283-286.
- [79] C. D. Rizzuto, R. Wyatt, N. Hernandez-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson and J. Sodroski, “A Conserved HIV gp120 Glycoprotein Structure Involved in Chemokine Receptor Binding,” Science, Vol. 280, No. 5371, 1998, pp. 1949-1953. doi:10.1126/science.280.5371.1949
- [80] J. Yang and C. Q. Liu, “Molecular Modeling on Human CCR5 Receptors and Complex with CD4 Antigens and HIV-1envelope Glycoprotein gp120,” Acta Pharmacologica Sinica, Vol. 20, No. 1, 2000, pp. 29-34.
- [81] J. Yang, Y. W. Zhang, J. F. Huang, Y. P. Zhang and C. Q. Liu, “Structure Analysis of CCR5 from Human and Primates,” Journal of Molecular Structure: Theochem, Vol. 505, No. 1-3, 2000, pp. 199-210. doi:10.1016/S0166-1280(99)00393-0
- [82] J. Yang and C. Q. Liu, “Interaction between Humaninterleukin-16 and CD4 Receptor of HIV-1,” Acta Pharmacologica Sinica, Vol. 21, No. 6, 2000, pp. 547- 553.
- [83] A. L. Brass, D. M. Dykxhoorn, Y. Benita, N. Yan, A. Engelman, R. J. Xavier, J. Lieberman and S. J. Elledge, “Identification of Host Proteins Required for HIV Infection through a Functional Genomic Screen,” Science, Vol. 319, No. 5865, 2008, pp. 921-926. doi:10.1126/science.1152725