International Journal of Organic Chemistry
Vol. 2 No. 2 (2012) , Article ID: 20371 , 10 pages DOI:10.4236/ijoc.2012.22016
Electron Transport Materials: Synthesis, Properties and Device Performance
1Energy and Environment Directorate, Pacific Northwest National Laboratory, Richland, USA
2Department of Chemistry, Fort Lewis College, Durango, USA
Email: Asanga.Padmaperuma@PNNL.gov
Received January 25, 2012; revised February 25, 2012; accepted March 7, 2012
Keywords: Electron Transport; OLED; Triplet Energy
ABSTRACT
We report the design, synthesis and characterization, thermal and photophysical properties of two silane based electron transport materials, dibenzo[b,d]thiophen-2-yltriphenylsilane (Sif87) and (dibenzo[b,d]thiophen-2-yl)diphenylsilane (Sif88) and their performance in blue organic light emitting devices (OLEDs). The utility of these materials in blue OLEDs with iridium (III) bis[(4,6-difluorophenyl)-pyridinato-N,C2’]picolinate (Firpic) as the phosphorescent emitter was demonstrated. Using the silane Sif87 as the electron transport material (ETm), an EQE of 18.2% was obtained, with a power efficiency of 24.3 lm/W (5.8 V at 1 mA/cm2), in a heterostructure. When Sif88 is used, the EQE is 18.5% with a power efficiency of 26.0 lm/W (5.5 V at 1 mA/cm2).
1. Introduction
A crucial roadblock in the application of organic light emitting devices (OLEDs) in both displays and lighting is the lack of a highly efficient and stable blue electrophosphorescent system. To efficiently transfer charge to the phosphorescent dopant, both the singlet and triplet excited states of the host material must be higher in energy than the triplet excited state of the dopant [1,2]. Development of efficient blue OLEDs based on this technology has been particularly challenging because the host material must possess triplet exciton energy >2.75 eV to achieve efficient (exothermic) energy transfer to a blue dopant. Furthermore, to prevent exciton loss into surrounding charge-transport layers, excitons must be confined within the emissive layer which requires higher triplet energy in the surrounding transport materials than that of the phosphorescent emitter to prevent quenching of the dopant emission [3]. As a result, for a blue phosphorescent device, all charge transport materials must have high triplet energy.
In general, the mobility of holes appears higher than that of electrons in most organic semiconducting materials due to electron trapping [4,5]. In order to further improve charge balance, efficiency and stability in an OLED, the development of efficient electron transporting materials (ETm) is still a major objective. In pursuit of this goal, several candidate materials have been developed such as, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP) [6,7], 2,20,20-(1,3,5-phenylene)-tris(1- phenyl-1H-benzimidazole) (TBPI) [8], 3-(4-biphenyl-yl)- 4-phenyl-5-(4-tert-butylphenyl)-1,2,4-triazole (TAZ) [9], and aluminum(III) bis(2-methyl-8-quinolinato)-4-phenylphenolate (BAlq) [10]. However further improvement of light emitting efficiency while maintaining the operational stability of the electron transporting materials remains a challenge. In this regard, several guidelines have been established based on the general requirements for the properties of component materials in blue OLEDs: high electron mobility, wide energy bandgap (Eg) and high triplet energy (ET) to confine excitons and a deep highest occupied molecular orbital (EHOMO) energy, to block holes.
We have already demonstrated organic phosphine oxide (PO) materials with excellent electron injection and hole and exciton blocking capabilities as well as good morphological stability [11-14]. In addition, PO materials can be designed to have high triplet energies and adjustable HOMO and LUMO energies for optimizing electron injection into, and hole and exciton blocking for, a variety of wide band gap host materials. However their stability in a device is still a concern, so we are exploring different classes of compounds in order to maximize both efficiency and stability. One of the challenges in designing suitable electron transporting materials for blue electrophosphorescence, is to achieve a high ET and high electron mobility in the same molecule. Silanes appear to provide a means of preventing conjugation in a molecule and keeping the triplet energy high, while bulking up the molecule via substitution.
There have been many reports of incorporating hole transport moieties, such as triphenylamine and carbazole into silane containing compounds, however studies of electron deficient moieties containing silanes are rather sparse [15]. In this work, we explore the influence of a slightly electron deficient moiety such as dibenzothiophene, on the chemical, physical and electronic properties of the materials containing them. Herein we report the synthesis, characterization and properties of two silane based electron transporting materials, dibenzo[b,d]- thiophen-2-yltriphenylsilane (Sif87) and (dibenzo[b,d]- thiophen-2-yl)diphenylsilane (Sif88) with deep HOMO energy levels and triplet energies greater than 2.7 eV. The chemical structures of these two materials are illustrated in Figure 1.
2. Experimental Section
2.1. Synthesis and Characterization
All chemicals used in synthetic procedures were obtained from Aldrich Chemical Co. and used as received unless noted otherwise. All reactions were performed in ovendry glassware, under argon. Thermal analysis by differential scanning calorimetry (DSC) was obtained using a Netzsch simultaneous analyzer (STA400). From these studies, both melting point and glass transition temperatures for Sif87 and Sif88 were determined. Sublimed samples (5 mg) were placed in aluminum pans and heated at 20˚C/min under N2 gas at a flow rate of 50 mL/min. Indium metal was used as the temperature standard. NMR spectra were obtained using a Varian Oxford 500 MHz spectrometer at the following frequencies: 499.8 MHz (1H) and 125.7 MHz (13C). Tetramethylsilane was used as an internal reference for 1H and 13C spectra. Mass spectra were obtained using dopant assisted, atmospheric pressure photo ionization using an ABI QTRAP
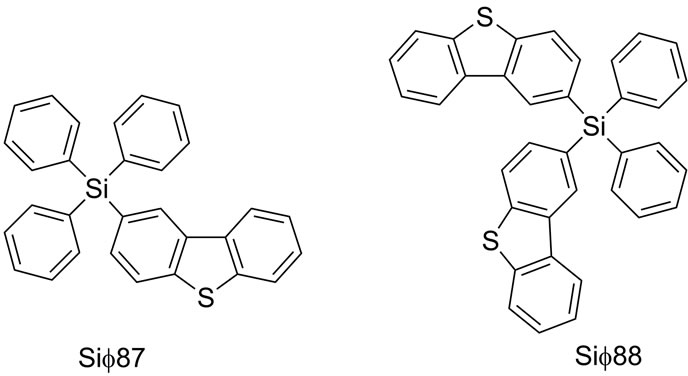
Figure 1. Chemical structures of the electron transport materials.
mass spectrometer. Elemental analysis was performed by Columbia Analytical Services, Tuscon, AZ, USA. X-Ray crystal data were collected on a Bruker-AXS Kappa APEX II CCD diffractometer with 0.71073 Å Mo Kα radiation. The data sets were treated with SADABS absorption corrections based on redundant multi scan data [16]. For each of the crystal structure studies, a single crystal was mounted using NVH immersion oil onto a nylon fiber and cooled to the data collection temperature of 100(2) K. The starting material 2-bromobenzothiophene was prepared from commercially available dibenzothiophene via published procedures [17].
Synthesis of dibenzo[b,d]thiophen-2-yltriphenylsilane, Sif87. The intermediate 2-bromodibenzo[b,d]thiophene (2.0 g, 7.60 mmol, 1 eq) was dissolved in 40 mL of anhydrous THF and cooled to –78˚C. To the well stirred mixture 1.6 M BuLi was added (5.0 mL, 7.98 mmol, 1.05 eq) slowly via syringe. The resulting mixture became yellow but stayed homogeneous. After stirring at –78˚C for one hour, triphenylcholorosilane (2.24 g, 7.60 mmol, 1.0 eq) was added in one portion. The reaction mixture was stirred for 3 h at –78˚C, then allowed to warm up to room temperature where it was stirred overnight. Next day, the clear yellow mixture was quenched with MeOH and concentrated to a residue. TLC indicated the presence of one major species with Rf = 0.7 (5% ethyl acetate in hexanes) in a relatively clean mixture. The residue was slurried in CH2Cl2 and MeOH (1:3) and stirred at around 40˚C for 15 minutes. The flask was chilled in ice, and the white precipitate was isolated by filtration and washed with cold MeOH. The solid (2.74 g, 81%) was further purified by repetitive vacuum sublimations, to yield 1.4 g of a white solid (3× sublimed, 41% overall). Mp: 150˚C (DSC, 10 K/min, N2). 1H NMR (CDCl3, 295 K, 500 MHz): δ 8.35 (s, 1H), 8.00 (d, J = 7.7 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.68 - 7.60 (m, 6H), 7.42 - 7.35 (m, 12H). 13C NMR (CDCl3, 295 K, 125 MHz): 142.31, 140.23, 137.55, 136.38, 136.22, 135.27, 135.22, 130.86, 130.77, 130.61, 129.10, 127.93, 125.51, 123.91, 123.54, 122.78. Anal. Calcd for C30H22SSi: C, 81.40; H, 5.01; S, 7.24; Si, 6.34. Found: C, 81.06; H, 5.01; S, 7.41; Si, 6.53. M/z calculated: 442.12 (100.0%), 443.12 (38.3%), 444.12 (9.8%), 444.13 (5.2%), 445.12 (2.8%); Found: 442.3 (100%), 443.3 (47.17%), 444.3 (20.20%), 445.3 (5.31%).
Synthesis of bis(dibenzo[b,d]thiophen-2-yl)diphenylsilane, Sif88. The intermediate 2-bromodibenzo[b,d]- thiophene (3.0 g, 11.4 mmol, 1 eq) was dissolved in 50 mL of anhydrous THF and cooled to –78˚C. To the well stirred mixture 1.6 M BuLi was added (7.5 mL, 12.0 mmol, 1.05 eq) slowly via syringe. The resulting mixture became yellow but remained homogeneous. After stirring at –78˚C for one hour, diphenyldicholorosilane (1.0 mL, 5.13 mmol, 0.45 eq) was added via syringe in one portion. The reaction mixture was stirred for 3 h at –78˚C, then allowed to warm up to room temperature where it was stirred overnight. Next day, the clear yellow mixture was quenched with MeOH and concentrated to a residue. TLC indicated the presence of one major species with Rf = 0.6 (5% ethyl acetate in hexanes) along with other byproducts. The major fraction was isolated by column chromatography on silica gel (5% ethyl acetate and hexanes) to result in a white foamy material. The chemically pure material was further purified by sublimation (3×) to yield 1.2 g of a white glassy solid in 42% overall yield. Tg = 70˚C 1H NMR (CDCl3, 295 K, 500 MHz): 8.41 (s, 2H), 8.00 (d, J = 7.8 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.85 (d, J = 7.89Hz, 2H), 7.69 (d, J = 7.6 Hz, 6H ), 7.49 (t, J = 7.3 Hz, 2H), 7.43 (t, J = 7.2 Hz, 6H), 7.36 (t, J = 7.0Hz, 2H). 13C NMR (CDCl3, 295 K, 125 MHz): 142.44, 140.24, 137.60, 136.37, 136.31, 135.29, 135.23, 130.99, 130.81, 130.59, 129.21, 127.97, 125.55, 123.91, 123.65, 122.83. Anal. Calcd. for C36H24S2Si: C, 78.79; H, 4.41; S, 11.69; Si, 5.12. Found: C, 78.77; H, 5.24; S, 11.68; Si, 3.24. M/z calculated: 548.11 (100.0%), 549.11 (45.6%), 550.10 (9.0%), 550.12 (7.5%), 550.11 (6.1%), 551.11 (5.1%), 551.12 (1.3%), 552.11 (1.2%); Found: 548.3 (100%), 549.3 (52.8%), 550.3 (28.5%), 551.3 (9.4%), 552.3 (2.7%).
2.2. Computational Methods
All calculations were performed with the NWChem computational package [18]. Molecular orbitals and bond lengths were visualized using Extensible Computational Chemistry Environment (ECCE), [19] a component of the Molecular Science Software Suite (MS3) developed at Pacific Northwest National Laboratory. Geometry optimization and electronic properties were computed via DFT using the B3LYP hybrid functionals [20-24]. For all calculations the Pople’s standard split-valence plus polarization 6-31G* [25,26] basis set was used.
2.3. Photophysical Characterization
UV-visible absorption spectra were collected on a Varian Cary 5 UV-Vis-NIR spectrophotometer. Emission spectra were collected on a Horiba Jobin-Yvon Fluorolog Tau spectrofluorometer equipped with a Xenon lamp. Solution spectra were collected in 1 cm path length quartz cells for the concentration range 10–5 - 10–3 M. Phosphorescence spectra were obtained in CH2Cl2 at 77 K at an excitation wavelength of 280 nm and time delay of 300 µs using a nanosecond optical parametric oscillator/amplifier operating at a 10 Hz repetition rate. The output was directed onto the sample and emission was collected at right angles to the excitation and focused into a 1/8 m monochromator with a gated intensified CCD camera to record the spectra. The gate of the CCD camera could be set to reject scattered laser light and short-lived luminescence, allowing the observation of long-lived luminescence
2.4. Electrochemical Characterization
Cyclic voltammetry was performed using a Princeton Applied Research EG&G potentiostat/galvanostat model 263 Å, with a silver wire as the pseudo-reference electrode, a Pt wire as the counter electrode, and glassy carbon as the working electrode. Reduction potentials were determined in anhydrous and degassed DMF with 80 mM tetra(n-butyl)-ammonium hexafluorophosphate, 3 mM ferrocene, and 7 mM sample (scan speeds of 20 - 300 mV/s from –2.9 to +1.3 V). Reduction potentials were recorded relative to a ferrocenium/ferrocene (Fc+/Fc) redox couple used as the internal reference.
2.5. OLED Characterization
Compounds HM-A1 (4-(diphenylphosphoryl)-N,N-diphenylaniline), Sif87 and Sif88 were prepared in house and purified by vacuum sublimation. TAPC (1,1-bis[(di-4- tolylamino)phenyl]cyclohexane) was purchased from TCI America and vacuum-sublimed twice before use in the deposition system. OLEDs were fabricated on glass substrates pre-coated with a 100 nm thick layer of indium tin oxide having a sheet resistance of 15 Ohms/sq. The substrates were cleaned with detergent and water followed by organic solvents, and then treated using UV ozone immediately prior to device fabrication. As the first step to fabricate the organic stack, a thin layer of hole injection material was formed on the ITO surface by spin-coating Plexcore OC ink (purchased from Aldrich and diluted with methanol at 1:1 ratio) at 5000 rpm. All organic and cathode layer depositions were performed by thermal evaporation in a high vacuum system (base pressure < 3 × 10–7 Torr). Film thicknesses were measured using a quartz crystal oscillator and calibrated using ellipsometry. Current-voltage characteristics were measured with an Agilent Technologies 4155B semi-conductor parameter analyzer. Light output was measured using a Si photodetector placed behind the OLED. No corrections were made for light waveguided in the organic thin films or the substrate. Electroluminescence spectra were recorded with an Acton Research Spectrum M CCD mounted to an Acton Research monochromator.
3. Results and Discussion
Synthesis of the electron transport materials was accomplished via established procedures [27]. To prepare the common intermediate 2-bromodibenzothiophene, dibenzothiophene was treated with bromine as shown in Scheme 1.
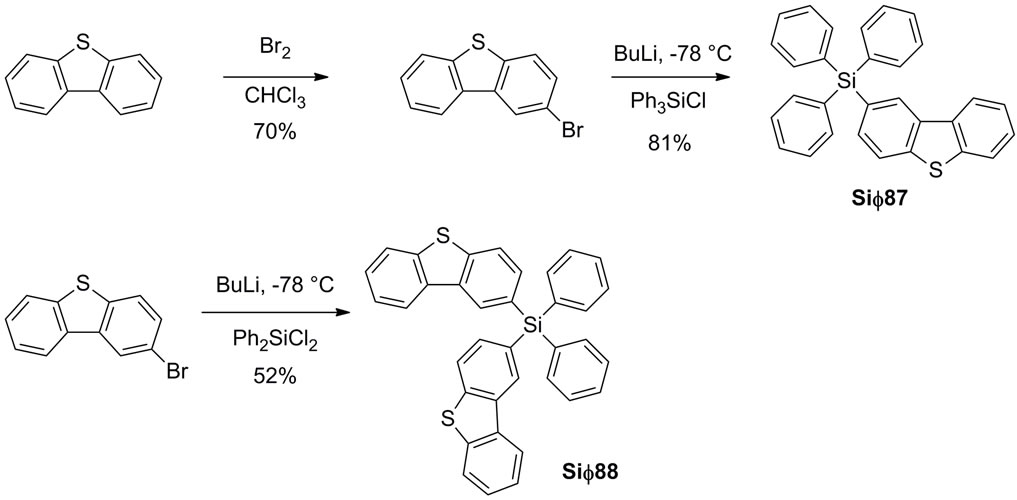
Scheme 1. Synthetic route for the silane based electron transport materials.
The product was contaminated with 4-bromoand 2,8- dibromodibenzothiophene analogs, and the majority of by-products could be removed by crystallization from chloroform. However, due to our stringent purity requirements for our final product, the intermediate had to be sublimed at least two times, until no impurities were detected by 1H NMR.
A lithium-halogen exchange reaction between the 2- bromodibenzothiophene intermediate and n-butyllithium, followed by reaction with either triphenylchlorosilane or diphenyldichlorosilane, gave the desired products Sif87 and Sif88 respectively in reasonable yields. Interestingly, even when chemically pure, the two analogs appeared to have different morphology which was counterintuitive. The mono dibenzothiophene (DBT) analog was a white solid, while the bis DBT analog was a foam. After further purification, by sublimation, Sif88 became a glass, while Sif87 became a crystalline solid. The reason this is significant, is because their different morphologies might impact their transport properties. In order to better understand differences in morphology, we performed XRay studies. X-ray quality crystals of both Sif87 and Sif88 were grown by slow evaporation from ethyl acetate, over a few days. A drawing of each molecule showing the assigned numbering scheme is shown in Figure 2.
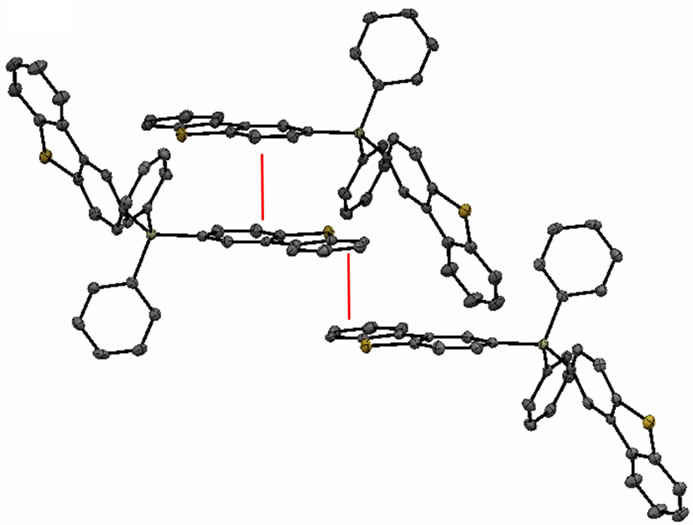
The structure confirms that each molecule contains a silicon atom bound to either three phenyl rings and one dibenzothiophene (DBT) group in the case of Sif87, or two phenyl rings and two DBT groups in the case of Sif88. The crystal packing in each structure reveals a number of close intermolecular contacts. The packing structure in Sif87 consists of two DBT units in a slipped pi-stack, 3.267 Å apart. Additionally, these slipped pistacks interact in a face-edge manner with a phenyl group of a second two-molecule unit, with a distance of 2.87 Å (Figure 3).
In the case of Sif88 which contains two DBT units per molecule, one DBT is involved in an infinite slipped
 (a)
(a)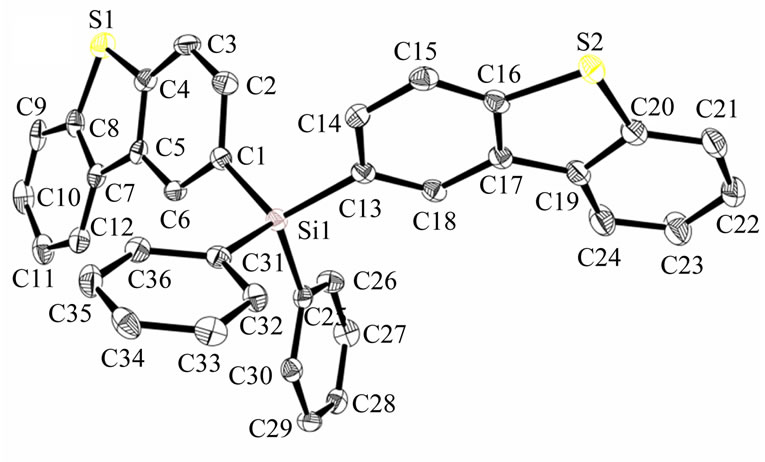 (b)
(b)
Figure 2. Assigned numbering scheme for (a) Sif87 and (b) Sif88.

Figure 3. Packing structure of Sif87.
pi-stack that alternates between 3.283 and 3.297 Å apart (Figure 4(a)). The second DBT unit on each molecule is involved only in face-to-edge interactions with two separate phenyl groups from another molecule. These interactions range from 2.698 to 2.880 Å apart (Figure 4(b)). Full crystallographic data is contained in the supplemental material.
The X-Ray data coupled with the different morphologies of the two analogs, suggests that the two analogs have different packing patterns, and as a result might have different charge transport properties in an OLED. Due to the pi-stacking of the Sif88 analog, this compound is expected to have better charge transporting properties than the Sif87.
The molecular and electronic structures of the silane based electron transport materials were investigated by computational methods. The gas phase structure was computed by geometry optimization at B3LYP/6-31G* level. The orbital amplitude plots for the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of Sif87, and Sif88 are depicted in Figure 5 and are shown to be localized predominately on the dibenzothiophene moiety.
 (a)
(a)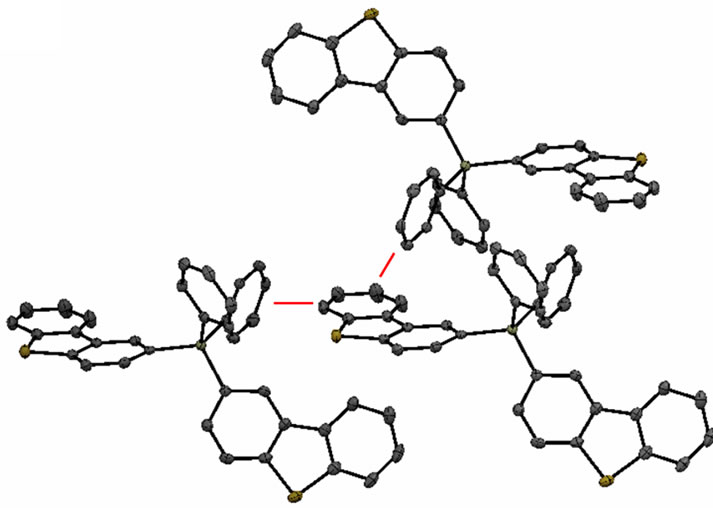 (b)
(b)
Figure 4. Packing structure of Sif88.
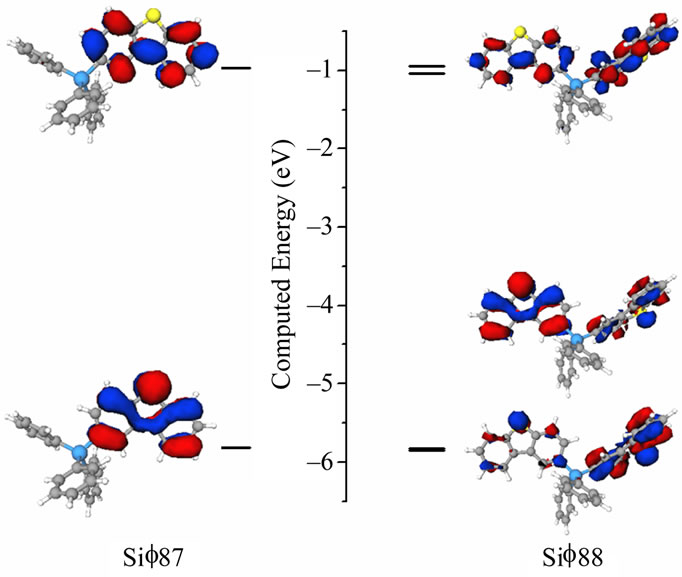
Figure 5. HOMO and LUMO plots for Sif87 (left) and Sif88 (right).
As a result, for the molecule Sif88, which incorporates two DBT moieties, there are two occupied MOs with similar energies. Sif88 has a slightly shallower ELUMO than Sif87 and the same trend applies to the EHOMO. The predicted ELUMO/EHOMO values for the host material HM-A1 are –0.77/–5.19 eV [28], while the new compounds have an EHOMO slightly deeper than –5.8 eV (Table 1).
The EHOMO predicted values suggest that the new materials are suitable for hole blocking and confinement of holes in the emissive layer. The predicted barrier for electron injection is low as well, indicating that the silanes have favorable ELUMO for electron injection in to the emissive layer.
To experimentally confirm the trends in predicted orbital energies, it is necessary to carry out cyclic voltammetry (CV). The experimental electrochemical reduction potential is converted to the LUMO energy relative to vacuum, using a reference compound as described in [29]. For our purposes, 4,4-(N,N-dicarbazoyl)biphenyl (CBP) was used for the reference with E1/2red (CBP) = –2.77 V and ELUMO = –2.7 eV according to [29]. Due to the deep HOMO level of these molecules electrochemical oxidation is difficult to obtain in most common solvents. As a result, only the reduction potentials are listed in Table 1. The electrochemically estimated ELUMO and the onset of optical absorption (Eg) measured in solution were used to estimate the EHOMO state. Both blocking materials have EHOMOs deep enough to be useful as hole blocking materials and the data is consistent with the theoretical prediction.
Time dependent density functional theory (TD-DFT, B3LYP/6-31G*) studies were carried out on Sif87 and Sif88 to predict electronic properties (Table 1). The lowest energy (non-zero) singlet transitions for both molecules are between the HOMO and the LUMO level.

Table 1. Theoretical and experimental electronic properties of Sif87 and Sif88.
The said transition (S1¬S0) for Sif87 and Sif88 are centered at 4.17 eV (oscillator strength, f – 0.02145) and 4.16 eV respectively (oscillator strength, f – 0.01899). This data predicts that both molecules will have absorbance and emission profiles dominated by the DBT moiety. Moreover, the energy of these transitions for the two molecules will be similar to each other. Lowest energy triplet transitions (T1¬S0) for Sif87 and Sif88 are centered at 3.14 eV predicting that the two molecules will also have emission phosphorescent profiles.
The electronic absorption spectra for the two molecules are shown in Figure 6(a), and are compared to the corresponding spectra for the DBT chromophore (inset).
The electronic transitions in DBT have previously been assigned according to the symmetry elements of the molecule [30-32]. Substitution at the DBT 2-position by aryl silanes does not strongly polarize the long axis of the molecule, nor increase the electron delocalization length of the active chromophore. As a consequence, both Sif87 and Sif88 exhibit a well-defined absorption spectrum similar to that of DBT with a small red shift of all bands (by ~280 to 500 cm–1) (see Figure 6). Comparing the molar extinction co-efficient (ε) values for Sif87, and Sif88 we can state that the as expected the ε value increases as the number of DBT rings increase in the molecule.
The room temperature fluorescence and low temperature phosphorescence spectra of Sif87 and Sif88 are shown in Figures 6(b) and (c). The two compounds have identical profiles, both at room temperature singlet emission and low temperature triplet emission. The silanes exhibit strong and structured UV emission peaking at 349 nm with a small Stoke’s shift (cm–1) and a narrow full width at half max (FWHM). The energy of the singlet state (ES) can be estimated from the absorbance and emission spectra as described by Turro [33], and the ES for the Sif87 and Sif88 are estimated to be 3.75 eV. Phosphorescence spectra of the silanes were obtained in CH2Cl2 at 77 K and are also shown in Figure 6(c). The triplet energies (ET) of organic molecules can be determined from the ω0-0 transitions identified as the highestenergy band of their phosphorescence spectra. Here, Sif87 and Sif88 have similar ω0,0 peaks at 418 nm corresponding to an ET of 2.97 eV. This is particularly significant, since these compounds were designed for blue phosphorescent OLEDs, where a high triplet energy emitter is required.
The silane based molecules described here were studied in simple heterostructure blue OLEDs as electron transport/hole blocking materials. Phosphine oxide based ambipolar host molecule HM-A18 was used as the host for the phosphorescent emitter FIrpic and either Sif87 or Sif88 was used as the hole blocking/electron transporting layer in the OLED stack. PO15, a high performance phosphine oxide based ETm was also used in the devices for comparison purposes (Device structure—ITO/35 nm TAPC/15 nm 5% wt. FIrpic: HM-A1/50 nm ETm/1 nm LiF/100 nm Al, where ETm is 50 nm Sif87, 50 nm Sif88, or 50 nm PO15, 45 nm Sif87/5 nm PO15, or Sif88/5 nm PO15). The thickness of the transport layers was optimized for FIrpic: HM-A1 system and was reported previously [34]. Figure 7 shows current density as a function of voltage (Figure 7(a)), external quantum efficiency as a function of current density (Figure 7(b)) for these OLEDs and the electroluminescent spectra at 10 mA/cm2 (Figure 7(c)).
A summary of OLED operation properties can also be found in Table 2.
The emission for these devices came exclusively from FIrpic, suggesting the ability of the silane materials to transport electrons as well as to block holes (Figure 7(c)). Unfortunately, the drive voltages obtained using either 50 nm Sif87 or 50 nm Sif88 as the ETm are high and there is virtually no emission.
The high drive voltage obtained using the 50 nm Sif ETms is a combination of low charge mobility and/or a large barrier for electron charge injection. Unlike the phosphine oxide based ETm we have reported previously
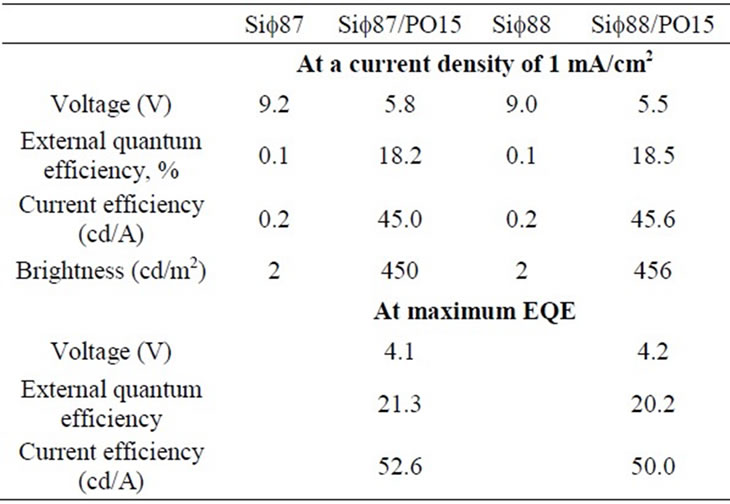
Table 2. Device performance of OLEDs containing the materials under study with and without an injection layer.
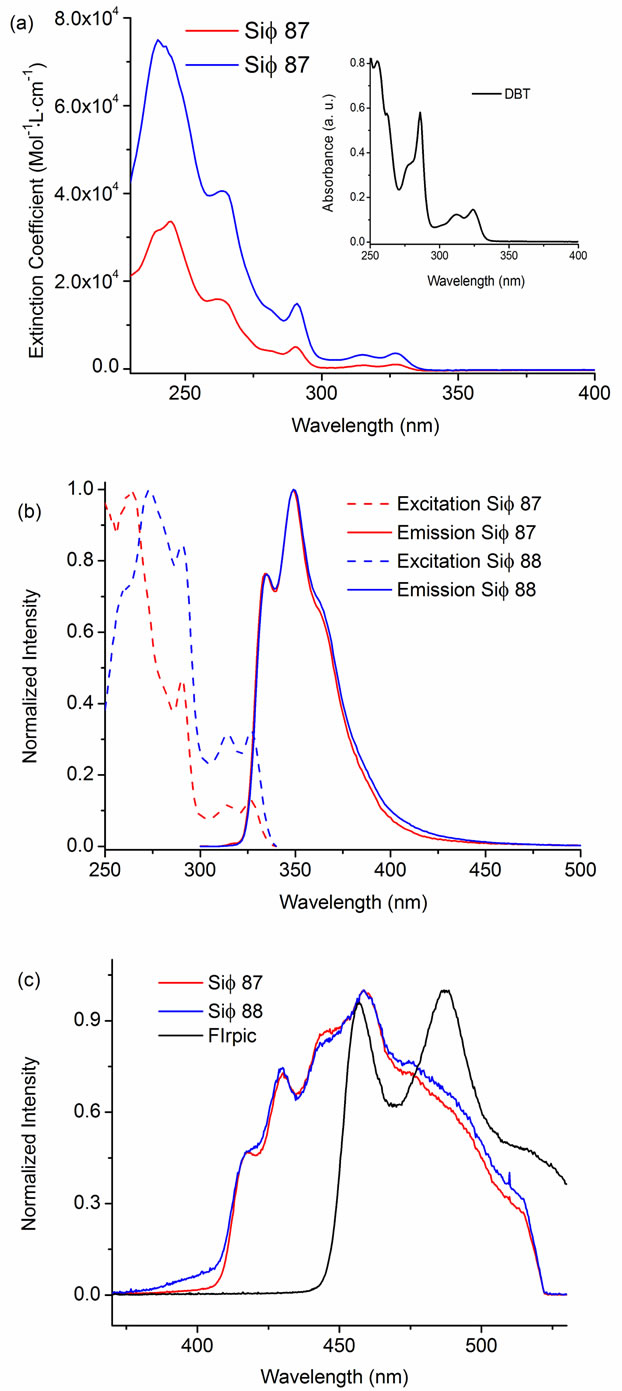
Figure 6. (a) Absorbance spectra; (b) Room temperature excitation and emission of Sif87 and Sif88; (c) Low temperature phosphorescence spectra for Sif87 and Sif88 and FIrpic.
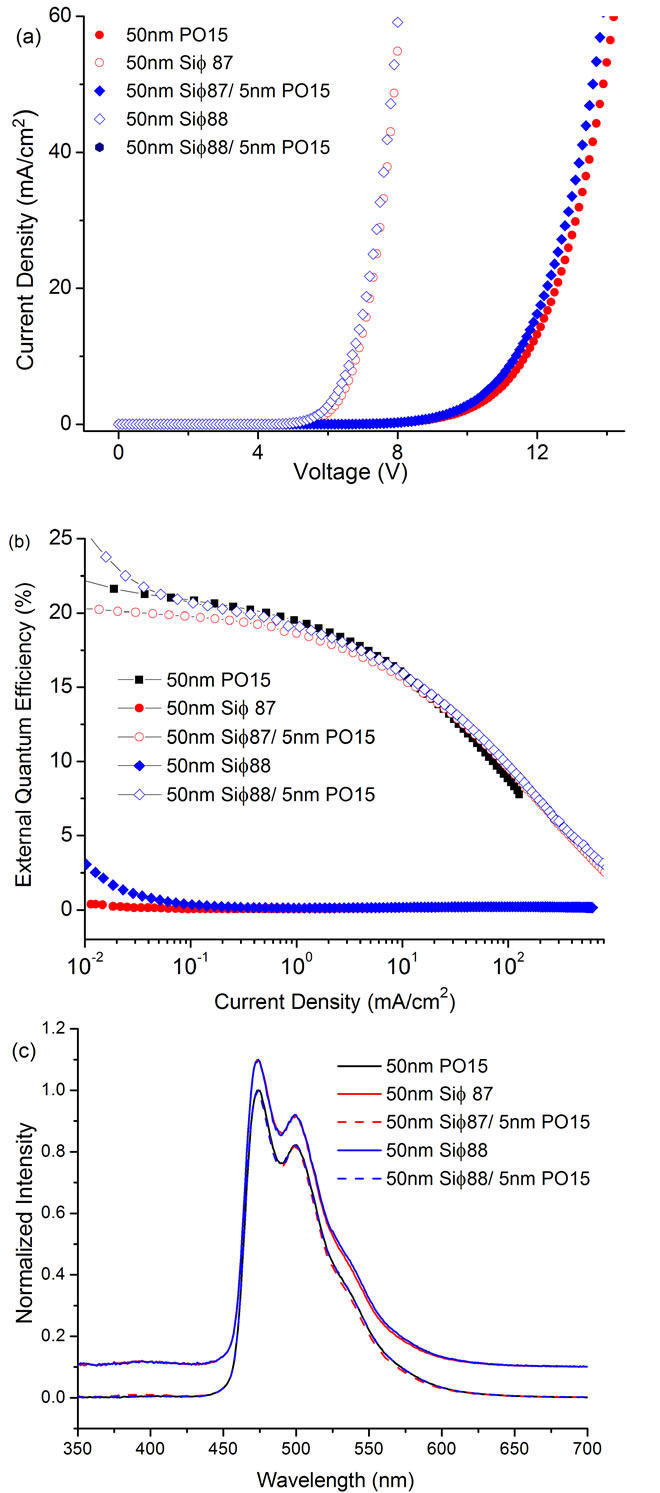
Figure 7. (a) Current density vs voltage; and (b) EQE vs current density for blue phosphorescent OLEDs fabricated using Sif87 and Sif88 as the transporting material; (c) Electroluminescence spectra. Device structure: ITO/35 nm TAPC/15 nm 5% wt. FIrpic: HM-A1/45 nm ETL/5 nm PO15/1 nm LiF/100 nm Al.
[35,36], the silane-based ETms lack the ability to coordinate metal ions which could lead to poor electron injection at the cathode/ETL interface. As reported in [37], this interaction between the ETL material and the cathode could enhance electron injection. To enhance electron injection into the silane-based ETms, a thin layer (5 nm) of PO15 was inserted between the ETm layer and the cathode. As seen in Figures 7(a) and (b), the introduction of the PO15 interlayer reduces the operating voltage by 3.5 V.
The emission efficiency also increases significantly demonstrating that there is a large barrier for electron injection into the Sif ETms. In fact, when the 5 nm PO15 injection layer is used, the OLEDs with both Sif87 and Sif88 demonstrate similar EQE to PO15. But the operation voltage for the devices with 45 nm Sif ETms/5 nm PO15 is still ~1.5V higher than for 50 nm PO15 devices. This drive voltage difference could be caused by either the lower bulk transport mobility of electrons in the ETm (Sif87, Sif88) or the extra injection barrier for electrons to transfer from the ETm to the host (HM-A1). Experiments are ongoing to differentiate the effects of these two possible factors. There is however a slight difference in drive voltage between the two analogs: Sif88 has a slightly lower voltage whether used alone or in conjunction with PO15, than Sif87. X-Ray data does indicate that due to packing, Sif88 ought to be the better electron transport among the two compounds. In the described architecture, the silanes appear to have similar transport properties, with a very slight advantage of Sif88 over Sif87.
4. Conclusion
The synthesis and characterization of two silane containing electron transport/hole blocking materials are reported. The ETms have sufficiently deep HOMO energies to block holes and high enough triplet energies to prevent exciton quenching. These materials have the chemical functionality to transport electrons and were used as the electron transport/hole blocking layer in blue electrophosphorescent OLED devices. The two materials contain one or two dibenzothiophene units, and their electronic properties are determined by these moieties. As a result, they have similar electronic properties as well as similar device performance in a heteroarchitecture. Using the silane Sif87 as ETm, an EQE of 18.2% was obtained, with a power efficiency of 24.3 lm/W (5.8 V at 1 mA/ cm2), in a heterostructure. When Sif88 is used, the EQE is 18.5% with a power efficiency of 26.0 lm/W (5.5 V at 1 mA/cm2). The only significant difference between the analogs is their packing pattern as determined by the X-Ray data. The crystal packing of Sif88 (infinite pi stacking of one of the BDT units) suggest that this compound would be a better electron transport than Sif87, and that would be evident from the voltage values. OLED data illustrates in the particular device structure presented herein, that there is potentially a slight difference between the two, with a slight electron transport preference of Sif88 over Sif87 (voltage 5.5 V versus 5.8 V).
5. Acknowledgements
This work was funded by the Solid Sate Lighting Program of the US Department of Energy, within the Building Technologies Program (BT), (Award No. M68004043) and managed by the National Energy Technology Laboratory (NETL). A portion of this research was performed using Environmental Molecular Sciences Laboratory (EMSL), which is a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and is located at Pacific Northwest National Laboratory (PNNL). The authors would like to thank Dr. Alan Joly (low temperature triplet emission measurements) and Dr. Sarah Burton (NMR instrument) for their kind assistance. Pacific Northwest National Laboratory (PNNL) is operated by Battelle Memorial Institute for the US Department of Energy (DOE) under Contract No. DE-AC06-76RLO1830.
Supporting Information Available: 1H and 13C, NMR spectra for all novel compounds, X-ray crystallography tables, mass spectra and solution cyclic voltammetry data.
REFERENCES
- M. P. Blochwitz, M. Hofmann and K. Leo, “Non-Polymeric OLEDs with a Doped Amorphous Hole Transport Layer and Operating Voltages down to 3.2 V to Achieve 100 cd/m2,” Synthetic Metals, Vol. 127, No. 1-3, 2002, pp. 169-173. doi:10.1016/S0379-6779(01)00616-6
- D. Gebeyehu, K. Walzer, G. He, M. Pfeiffer, K. Leo, J. Brandt, A. Gerhard, P. Stöβel and H. Vestweber, “Highly Efficient Deep-Blue Organic Light-Emitting Diodes with Doped Transport Layers,” Synthetic Metals, Vol. 148, No. 2, 2005, pp. 205-2011. doi:10.1016/j.synthmet.2004.09.024
- C. Adachi, R. Kwong, P. I. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, “Endothermic Energy Transfer: A Mechanism For Generating Very Efficient High-Energy Phosphorescent Emission in Organic Materials,” Applied Physics Letters, Vol. 79, No. 13, 2001, pp. 2082-2085. doi:10.1063/1.1400076
- H. Antoniadis, M. A. Abkowitz and B. R. Hsieh, “Carrier Deep—Trapping Mobility—Lifetime Products in Poly(pphenylene vinylene),” Applied Physics Letters, Vol. 65, No. 16, 1994, pp. 2030-2032. doi:10.1063/1.112784
- R. J. Holmes, B. W. D’Andrade, S. R. Forrest, X. Ren, J. Li and M. E. Thompson, “Efficient, Deep-Blue Organic Electrophosphorescence by Guest Charge Trapping,” Applied Physics Letters, Vol. 83, No. 18, 2003, pp. 3818- 3820. doi:10.1063/1.1624639
- Y. Zheng, S.-H. Eom, N. Chopra, J. Lee, F. So and J. Xue, “Efficient Deep-Blue Phosphorescent Organic Light-Emitting Device with Improved Electron and Exciton Confinement,” Applied Physics Letters, Vol. 92, No. 22, 2008, Article ID 223301. doi:10.1063/1.2937403
- S.-J. Yeh, M.-F. Wu, C.-T. Chen, Y.-H. Song, Y. Chi, M.-H. Ho, S.-F. Hsu and C. H. Chen, “New Dopant and Host Materials for Blue-Light-Emitting Phosphorescent Organic Electroluminescent Devices,” Advanced Materials, Vol. 17, No. 3, 2005, pp. 285-289. doi:10.1002/adma.200401373
- M.-H. Tsai, H.-W. Lin, H.-C. Su, T.-H. Ke, C.-C. Wu, F.-C. Fang, Y.-L. Liao, K.-T. Wong and C.-I. Wu, “Highly Efficient Organic Blue Electrophosphorescent Devices Based on 3,6-Bis(triphenylsilyl)carbazole as the Host Material,” Advanced Materials, Vol. 18, No. 9, 2006, 1216-1220. doi:10.1002/adma.200502283
- S. Tokito, T. Iijima, Y. Suzuki, H. Kita, T. Tsuzuki and F. Sato, “Confinement of Triplet Energy on Phosphorescent Molecules for Highly-Efficient Organic Blue-Light-Emitting Devices,” Applied Physics Letters, Vol. 83, No. 3, 2003, pp. 569-571. doi:10.1063/1.1594834
- P. A. Vecchi, A. B. Padmaperuma, H. Qiao, L. S. Sapochak and P. E. Burrows, “A Dibenzofuran-Based Host Material for Blue Electrophosphorescence,” Organic Letters, Vol. 8, No. 19, 2006, pp. 4211-4214. doi:10.1021/ol0614121
- A. B. Padmaperuma, L. S. Sapochak and P. E. Burrows, “New Charge Transporting Host Material for Short Wavelength Organic Electrophosphorescence: 2,7-Bis(diphenylphosphine oxide)-9,9-dimethylfluorene,” Chemistry of Materials, Vol. 18, No. 9, 2006, pp. 2389-2396. doi:10.1021/cm0600677
- L. S. Sapochak, A. B. Padmaperuma, P. A. Vecchi, H. Qiao and P. E. Burrows, “Design Strategies for Achieving High Triplet Energy Electron Transporting Host Materials for Blue Electrophosphorescence,” Proceedings of SPIE, May 2006, pp. 1-13.
- T. Matsushima and C. Adachi, “Extremely Low Voltage Organic Light-Emitting Diodes with P-Doped Alpha-Sexithiophene Hole Transport and N-Doped Phenyldipyrenylphosphine Oxide Electron Transport Layers,” Applied Physical Letters, Vol. 89, No. 25, 2006, Article ID 253506. doi:10.1063/1.2410236
- T. Oyamada, H. Sasabe, C. Adachi, S. Murase, T. Tominaga and C. Maeda, “Extremely Low-Voltage Driving of Organic Light-Emitting Diodes with a Cs-Doped Phenyldipyrenylphosphine Oxide Layer as an Electron-Injection Layer,” Applied Physics Letters, Vol. 86, No. 3, 2005, Article ID 033503. doi:10.1063/1.1852707
- L. Xiao, S.-J. Su, A. Agata, H. Lan and J. Kido, “Nearly 100% Internal Quantum Efficiency in an Organic BlueLight Electrophosphorescent Device Using a Weak Electron Transporting Material with a Wide Energy Gap,” Advanced Materials, Vol. 21, No. 12, 2009, pp. 1271-1274. doi:10.1002/adma.200802034
- “SADABS Version 2006: An Empirical Absorption Correction Program,” Bruker AXS Inc., Madison, 2006.
- O. Shimomura, T. Sato, I. Tomita, M. Suzuki and T. Endo, “Synthesis and Radical Polymerization of 2-Vinyldibenzothiophene,” Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 35, No. 14, 1997, pp. 2813-2819. doi:10.1002/(SICI)1099-0518(199710)35:14<2813::AID-POLA1>3.0.CO;2-R
- E. J. Bylaska, et al., “NWChem, A Computational Chemistry Package for Parallel Computer, Version 5.1,” A Modified Version, Pacific Northwest National Laboratory, Richland, 2007.
- G. Black, et al., “ECCE, A Problem Solving Environment for Computational Chemistry, Software Version 6.0,” Pacific Northwest National Laboratory, Richland, 2009.
- A. D. Becke, “Density-Fnnctional Exchange-Energy Approximation with Correct Asymptotic Behavior,” Physical Reviews A, Vol. 38, No. 6, 1988, pp. 3098-3100. doi:10.1103/PhysRevA.38.3098
- J. P. Perdew, “Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas,” Physical Reviews B, Vol. 33, No. 12, 1986, pp. 8822-8824. doi:10.1103/PhysRevB.33.8822
- C. Lee, W. Yang and R. G. Parr, “Development of the Colic-Salvetti Correlation-Energy Formula into a Functional of the Electron Density,” Physical Reviews B, Vol. 37, No. 2, 1988, pp. 785-789. doi:10.1103/PhysRevB.37.785
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, “Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr,” Chemical Physics Letters, Vol. 157, No. 3, 1989, pp. 200-206. doi:10.1016/0009-2614(89)87234-3
- A. D. Becke, “Density-Functional Thermochemistry. III. The Role of Exact Exchange,” Journal of Chemical Physics, Vol. 98, No. 7, 1993, pp. 5648-5652. doi:10.1063/1.464913
- W. J. Hehre, R. Ditchfield and J. A. Pople, “Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules,” Journal of Chemical Physics, Vol. 56, No. 5, 1972, pp. 2257-2261. doi:10.1063/1.1677527
- M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, “Self-Consistent Molecular Orbital Methods. XXIII. A PolarizationType Basis Set for Second-Row Elements,” Journal of Chemical Physics, Vol. 77, No. 7, 1982, pp. 3654-3665. doi:10.1063/1.444267
- D. Tanaka, Y. Agata, T. Takeda, S. Watanabe and J. Kido, “Ultra High Efficiency Green Organic Light-Emitting Devices,” Japanese Journal of Applied Physics, Vol. 46, 2007, pp. L117-L119. doi:10.1143/JJAP.46.L117
- E. Polikarpov, P. K. Koech, L. Wang, J. S. Swensen, L. Cosimbescu, J. E. Rainbolt, A. L. Von Ruden, D. J. Gaspar and A. B. Padmaperuma, “Controlling Charge Transport in Blue OLEDs by Chemical Functionalization of Host Materials,” Journal of Photonics for Energy, Vol. 1, 2011, Article ID 011007-1. doi:10.1117/1.3528499
- D. Kolosov, V. Adamovich, P. I. Djurovich, M. E. Thompson and C. Adachi, “1,8-Naphthalimides in Phosphorescent Organic Leds: The Interplay Between Dopant, Exciplex, and Host Emission,” Journal of the American Chemical Society, Vol. 124, No. 33, 2002, pp. 9945-9954. doi:10.1021/ja0263588
- A. Bree and R. Zwarich, “Electronic Spectra of Dibenzothiophene,” Spectrochimca Acta, Vol. 27A, No. 4, 1971, pp. 621-630. doi:10.1016/0584-8539(71)80264-7
- E. Castellucci, P. Foffi and P. R. Salvi, “The Two-Photon Fluorescence Excitation Spectrum of Dibenzothiophene,” Chemical Physics, Vol. 63, No. 3, 1981, pp. 437-443. doi:10.1016/0301-0104(81)87018-8
- J. Spanget-Larsen and E. W. Thulstrup, “The Electronic Transitions of Dibenzothiophene: Linear Dichroism Spectroscopy and Quantum Chemical Calculations,” Journal of Molecular Structure, Vol. 661-662, 2003, pp. 603-610. doi:10.1016/S0022-2860(03)00509-X
- N. J. Turro, “Modern Molecular Photochemistry,” University Science Books, Sausalito, 1991, pp. 146-148.
- E. Polikarpov, J. S. Swensen, N. Chopra, F. So and A. B. Padmaperuma, “An Ambipolar Phosphine Oxide-Based Host for High Power Efficiency Blue Phosphorescent Organic Light Emitting Devices,” Applied Physics Letters, Vol. 94, No. 22, 2009, Article ID 223304. doi:10.1063/1.3148642
- A. L. Von Ruden, L. Cosimbescu, E. Polikarpov, P. K. Koech, J. S. Swensen, J. T. Darsell and A. B. Padmaperuma, “Phosphine Oxide Based Electron Transporting and Hole Blocking Materials for Blue Electrophosphorescent Organic Light Emitting Devices,” Chemistry of Materials, Vol. 22, No. 20, 2010, pp. 5678-5686. doi:10.1021/cm1013653
- E. Polikarpov, J. S. Swensen, L. Cosimbescu, P. K. Koech, J. E. Rainbolt and A. B. Padmaperuma, “Emission Zone Control in Blue Organic Electrophosphorescent Devices through Chemical Modification of Host Materials,” Applied Physics Letters, Vol. 96, No. 5, 2010, Article ID 053306. doi:10.1063/1.3298556
- S. Madhusoodhanan, E. Polikarpov, S. Garon and M. E. Thompson, “Crown Ether Dopants to Improve n-Type Doping of Electron Transport Layers,” Proceedings of the International Symposium on Super-Functionality Organic Devices, IPAP Conferences Series 6, 30 March 2005, pp. 110-113.