International Journal of Organic Chemistry
Vol.2 No.1(2012), Article ID:17841,6 pages DOI:10.4236/ijoc.2012.21001
Synthesis and Plasma Stability of Disulfide-Bridged Cyclic Endomorphin-1 Derivatives
School of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, Australia
Email: i.toth@uq.edu.au
Received December 12, 2011; revised January 14, 2012; accepted January 26, 2012
Keywords: Endomorphin-1; Opioid Peptide; Cyclisation; Disulfide
ABSTRACT
Endomorphin-1 is an endogenous opioid peptide that mediates pain relief through interaction with the µ-opioid receptor in the central nervous system. To enhance the metabolic stability of this tetrapeptide, cyclisation through the formation of a disulfide bridge between the side chains of cysteine residues added to the sequence was explored. A further increase in stability was achieved through N-terminal modification with lipoamino acid and lactose succinamic acid, and the inclusion of D-amino acids. The latter also provided an alternative spatial arrangement of the aromatic side chains. The lipidated cyclic derivatives were insoluble in aqueous buffer, however, the cyclic peptides and glycopeptides showed greatly improved stability towards enzymatic degradation in human plasma.
1. Introduction
The use of opium, the dried sap isolated from the opium poppy (Papaver somniferum), for pain relief dates back thousands of years. Although it contains more than 50 different alkaloids, morphine, which was first isolated by Sertürner in 1804 [1], is the most abundant and potent of these. It is still in use for the relief of severe pain despite a plethora of side effects such as respiratory depression, constipation, tolerance and physical dependence.
Morphine acts on the µ-opioid receptor in the central nervous system, and research efforts towards finding an adequate replacement for treatment of pain without the undesired side effects now include the endogenous ligands of this receptor type. The opioid peptides endomorphin-1 (YPWF-NH2, 1) and endomorphin-2 (YPFFNH2) exhibit great affinity and selectivity for the µ- opioid receptor [2]. However, their usefulness is limited due to the drawbacks associated with all small peptides, mainly poor metabolic stability and low membrane permeability.
To address these shortcomings and explore structureactivity relationships, a variety of chemical modifications has been investigated. These include the substitution of one or more amino acids in the sequence by D-amino acids [3-5], β-amino acids [6-8] and other non-natural amino acids, for example 2,6-dimethyl tyrosine [9,10], or the addition of sugar moieties or lipoamino acids [11,12].
Although cyclisation of peptides is a widely used strategy for enhancing stability as well as lipophilicity
[13,14], not many cyclic analogues of the endomorphins have been synthesised so far. Cardillo, Gentilucci et al. [15-17] synthesised a series of diastereoand enantiomeric cyclic endomorphin-1 derivatives in which Tyr1 and Phe4 are connected through glycine by amide bonds. All of these peptides showed decreased µ-opioid receptor affinity in comparison with endomorphin-1 which was attributed to the lack of a terminal amine. However, the best peptide, c[YpwFG], exhibited affinity in the nanomolar range, possibly indicating a suitable spatial arrangement of the amino acid side chains in cyclic endomorphin derivatives.
The strong analgesic effect of a cyclic analogue of endomorphin-2, Y-c[DFFK]-NH2, is in disagreement with its poor affinity for the opioid receptors. Data indicated that it elicits its effect by stimulating the release of dynorphin A which acts on the κ-opioid receptor [18]. However, the substitution of the amino acid in position 2 by its enantiomer resulted in a potent analgesic with high µ-opioid receptor affinity [19].
Here, we describe the synthesis of a series of new cyclic analogues of endomorphin-1 with two cysteine amino acids incorporated into the sequence to allow for cyclisation via a disulfide-bridge. Some of the peptide sequences include D-amino acids for increased stability and to test alternative spatial arrangements. We also present lipidated and glycosylated derivatives of these peptides, as these modifications were shown to enhance stability [11,20-22].
2. Results and Discussion
2.1. Synthesis
Fmoc solid-phase peptide synthesis was used to synthesise the cyclic endomorphin-1 derivatives 2-13 (Table 1). Initial attempts at using lysine and glutamic acid to close the ring via an amide bond resulted in very poor yields under several different reaction conditions. As the formation of cyclic disulfides from thiol-containing amino acids was previously used successfully to increase the stability and selectivity of opioid peptides [23,24], we decided to use two cysteine residues for ring closure instead. Cyclisation was carried out on the resin using an excess of iodine in DMF. All peptides were purified to a single peak in two different RP-HPLC conditions and provided the expected [M + H]+ peak in mass spectrometry (Table 1). The C10-lipoamino acid was incorporated as a racemic mixture, giving rise to two diastereomeric peptides which were not separated prior to carrying out stability studies. While compounds 2-7 were obtained in reasonable yields (8% - 31%), peptides 8-13 were more challenging to synthesise and purify. Many side products were formed and although not all of them were identified, it was notable that dimeric structures containing two disulfide bonds were formed. Surprisingly, these persisted even when a low loading resin (0.34 mmol/g) was used.
Two different series of cyclic derivatives were synthesised. In peptides 2-7 the four-amino acid-sequence of endomorphin-1 is conserved and the two cysteine residues that enable cyclisation are flanking this sequence, resulting in a 20-membered ring. Two diastereomeric derivatives were synthesised, with the first one containing all amino acids in the L-configuration (2). The other diastereomer included the D-enantiomers of Trp and Pro (3), as this particular substitution pattern was found to result in the most favourable spatial arrangement of the aromatic side chains by Cardillo and Gentilucci et al. [15-17]. Contrary to their cyclic endomorphin-1 derivatives, however, the free terminal amine, which was shown to be of vital importance for receptor binding and agonist acitivity [25], was conserved in these peptides. Lipidated (4, 5) and glycosylated derivatives (6, 7) of these cyclic peptides were also synthesised.
In the other series of cyclic peptides Pro3 of the native endomorphin sequence was replaced with D-cysteine. This allowed for cyclisation with the side chain of Lcysteine added at the C-terminus giving a 14-membered ring. Compound 8 contains Trp in the L-configuration, and in analogue 9 this is replaced by D-Trp. To further
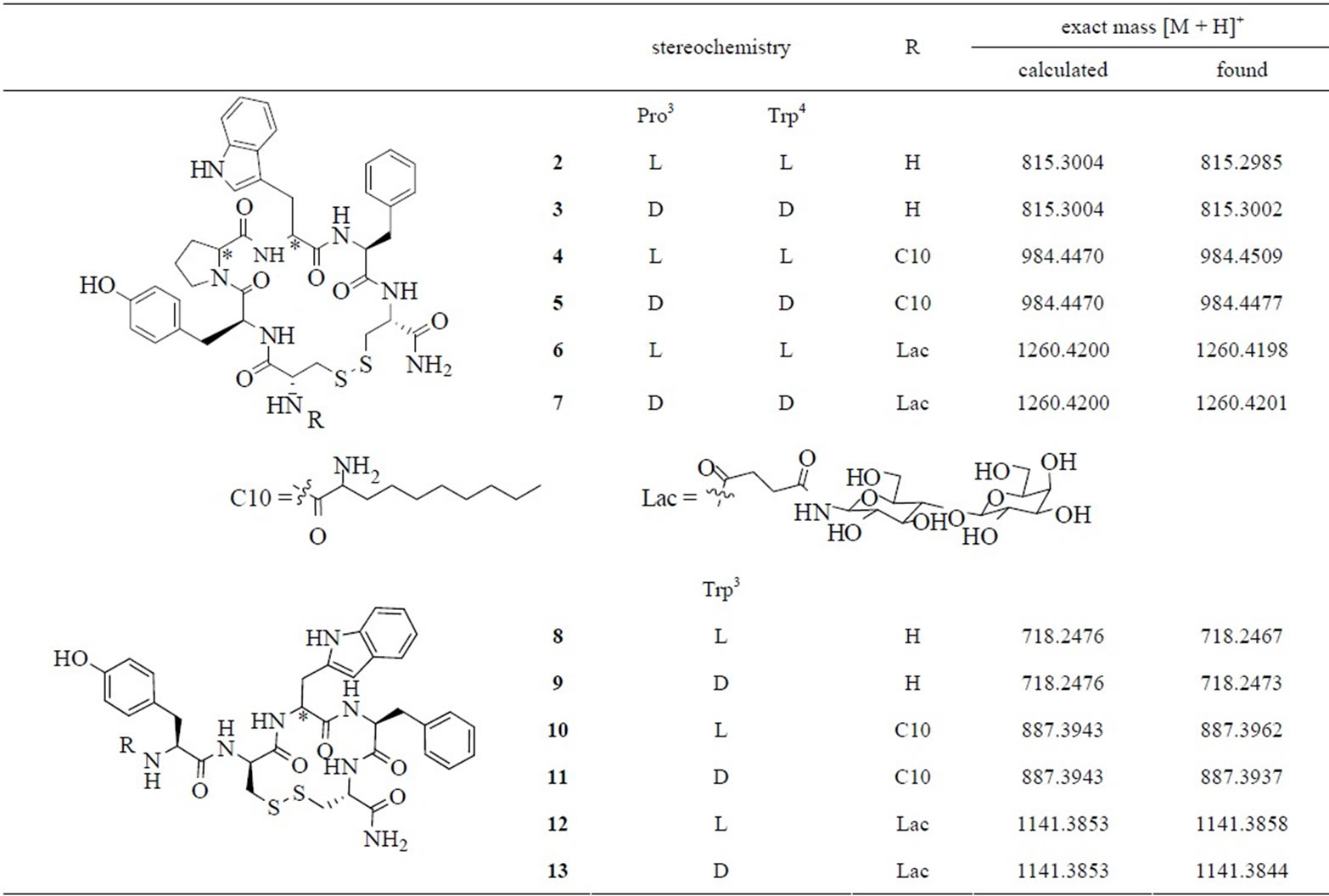
Table 1. The cyclic disulfide-bridged derivatives of endomorphin-1 synthesised for this study.
increase stability, derivatives of peptides 8 and 9 containing lipoamino acid (10, 11) or lactose (12, 13) were also synthesised.
2.2. Stability in Human Plasma
The stability of the cyclic endomorphin derivatives was investigated using fresh human plasma from healthy volunteers. This was mixed with solutions containing the cyclic disulfide-bridged peptides and incubated at 37˚C. Samples were taken at predetermined time points and the amount of intact peptide remaining was analysed using HPLC to determine the half-life (Figure 1). For purposes of comparison and to ensure enzyme activity, the stability of endomporphin-1 (1) was also determined and, at a half-life of 5.6 min, found to be consistent with literature reports [11,26].
The stability of all lipidated derivatives (4, 5, 10 and 11) could not be evaluated due to their low solubility. Even after addition of up to 5% DMSO these peptides were not dissolved in sufficient concentration for the experiment, and addition of a higher percentage of DMSO would interfere with enzyme activity. The linear C10-lipoamino acid-functionalised endomorphin-1 derivative is soluble after minimal addition of DMSO (1%, unpublished data), indicating that the cyclisation of endomorphin-1 resulted in a significant increase in lipophilicity.
All tested cyclic endomorphin derivatives showed an increased half-life in comparison with the native peptide (Figure 1), with values ranging from 37.1 min for analogue 2 to 152.3 min for analogue 13. Peptides 2 and 6, containing only L-amino acids, showed the smallest improvement in enzymatic stability, while the other peptides, which all included at least one D-amino acid, showed considerably greater stability. This is consistent with the paucity of enzymes capable of hydrolysing peptide bonds with amino acids in the D-configuration [27]. Past attempts at exploiting this fact by substituting Lfor D-amino acids in linear endomorphin derivatives were unsuccessful [3,4]. The substitutions resulted in binding affinities that were lower by up to three orders of magnitude, and structural comparisons with the native peptides suggested that the spatial arrangement of the side chains was unfavourable for receptor binding. Cyclisation, however, is an excellent way of overcoming this limitation [15-17].
Analogues 6 and 7 as well as 12 and 13 were glycosylated with the disaccharide lactose at the N-terminus. Previously, it was shown that in comparison with the native peptide only a slight reduction in binding affinity was caused by a similar modification with glucose [11]. It is also known that opioid peptides benefit from glycosylation [28,29] as it facilitates active transport across
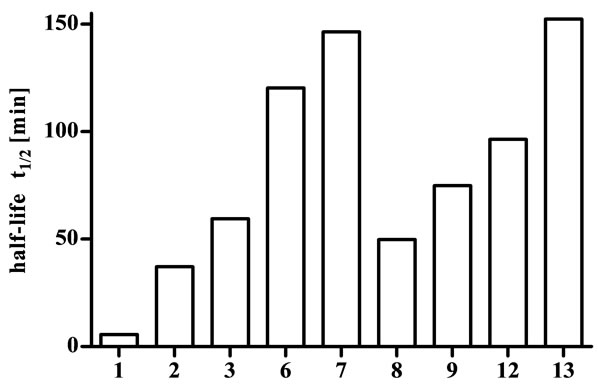
Figure 1. Half-lives of endomorphin-1 (1) and cyclic derivatives in human plasma.
the blood-brain barrier. Additionally, in a study of enkephalin-based opioid glycopeptides, Elmagbari et al. concluded that modifications with disaccharides resulted in the most potent compounds [30]. Therefore, it is promising that the half-lives of the cyclic lactose derivatives are in the range between 96.4 min and 152.3 min. This suggests that these peptides may be stable enough to be active after peripheral administration and we are currently investigating the activity of all cyclic endomorphin derivatives.
3. Conclusions
In this study, we demonstrated a facile route to new cyclic endomorphin-1 derivatives, by forming a disulfide bridge between the side chains of two cysteine amino acids that were incorporated into the peptide. Different ring sizes, 14- or 20-membered rings, were achieved by careful positioning of the cysteines, and the spatial arrangement of the side chains was varied by using amino acids in the Lor the D-configuration. Furthermore, cyclic peptides functionalised with lipoamino acids or carbohydrates were shown to be easily accessible.
Cyclisation was exploited extensively for increasing the bioavailability of other opioid peptides such as enkephalin but is underutilised in endomorphin research. Here, we provided further support for the application of this technique by showing that the increase in lipophilicity conferred by cyclisation was accompanied by an increase in stability in comparison with the native peptide.
4. Experimental
4.1. General
Dimethylformamide (DMF), trifluoroacetic acid (TFA) and piperidine of peptide synthesis grade were purchased from Merck Biosciences (Kilsyth, VIC, Australia). HPLCgrade acetonitrile was purchased from RCI Labscan Ltd. (Bangkok, Thailand). Fmoc-protected amino acids and Rink amide MBHA resin (100 - 200 mesh, 0.3 - 0.8 mmol/g loading) were obtained from Novabiochem (Melbourne, VIC, Australia). All other chemicals were purchased from Sigma Aldrich. The Boc-protected C10- lipoamino acid was synthesised from its bromide alkyl precursor following a published procedure [31].
Preparative HPLC was carried out on a Waters 600 controller and pump with a 490E programmable multiwavelength detector, and analytical HPLC was performed on an Agilent 1100 system fitted with a binary pump, autosampler and a detector set to a wavelength of 214 nm. Electrospray ionisation mass spectrometry was carried out on a PE Sciex API3000 triple quadrupole mass spectrometer, using a mixture of solvent A (0.1% acetic acid in water) and B (0.1% acetic acid in 9:1 acetonitrile/water) at 0.05 ml/min.
4.2. Synthesis
Peptides were assembled on Rink amide MBHA resin using HBTU (4 eq.)/DIPEA(6 eq.) activation and the in situ neutralisation protocol for Fmoc chemistry [32]. The following protected amino acids (4.2 eq.) were coupled twice for at least one hour: Fmoc-Cys(Trt)-OH, Fmoc-DCys(Trt)-OH, Fmoc-Phe-OH, Fmoc-Trp(Boc) -OH, FmocD-Trp(Boc)-OH, Fmoc-Pro-OH, Fmoc-D-Pro-OH, FmocTyr(tBu)-OH and Boc-C10-Laa. Per-O-acetylated lactose succinamic acid (1.5 eq.) was activated with 1.4 eq. HBTU and 3 eq. DIPEA and coupled over night, followed by deacetylation with 75% hydrazine hydrate in MeOH (2 × 15 min, 1 × 30 min). After both cysteine molecules had been added to the sequence, cyclisation was carried out on the resin with iodine (12 eq.) in DMF for two hours.
When construction of the peptide was complete, the resin was washed with DMF, DCM and MeOH and dried under vacuum for at least two hours. Cleavage of the peptide was achieved by treatment of the resin with TFA/triisopropylsilane/water (95:2.5:2.5) for two hours. The solvent was removed under a stream of air and the crude peptide precipitated by addition of cold diethyl ether, dissolved in acetonitrile/water/TFA (50:50:0.1) and lyophilised.
The crude peptides were purified by preparative RPHPLC using a Vydac C18 column (22 × 250 mm) with HPLC gradient A (Table 2, 4, 5, 10, 11) or HPLC gradient B (Table 3, all other peptides) at a flow rate of 10 ml/min. The purity of peptides in the collected fractions was confirmed by ESI-MS and analytical RP-HPLC using a Vydac C18 column (5 µm, 4.6 × 250 mm) and a gradient of 100% A to 100% B over 30 minutes at a flow rate of 1 ml/min. Fractions containing pure peptides were combined and lyophilised.
4.3. Plasma Stability Assay
Human plasma was collected from healthy adults with their informed written consent under approval of The
Table 2. HPLC gradient A.
Table 3. HPLC gradient B.
University of Queensland Medical Research Ethics Committee (project number: 2006000950). A solution of 1 mg/ml of each peptide was prepared in PBS containing 5% DMSO, and 300 μl aliquots were mixed with 300 μl of pre-warmed (37˚C) plasma. At selected time points (5, 10, 20, 30, 40, 60, 90 and 120 min), samples (50 μl) were collected and mixed with acetonitrile (75 μl) to precipitate plasma proteins which were removed by centrifugation at 10,000 rpm for 10 minutes. The supernatant was analysed for the amount of peptide remaining by RPHPLC using a Vydac C18 column (5 µm, 4.6 × 250 mm) and an elution gradient of 0% - 100% solvent B over 20 min (Solvent A: 0.1% TFA in water; solvent B: 0.1% TFA in 90% acetonitrile/water) at a flow rate of 1.5 ml/min. Data was analysed with GraphPad Prism 5 using the “One phase exponential decay” function.
5. Acknowledgements
We are grateful to Mr. Graham Macfarlane for assistance with high resolution mass spectrometry. This research was supported by an NHMRC Development Grant 519827.
REFERENCES
- S. Morimoto, K. Suemori, J. Moriwaki, F. Taura, H. Tanaka, M. Aso, M. Tanaka, H. Suemune, Y. Shimohigashi and Y. Shoyama, “Morphine Metabolism in the Opium Poppy and Its Possible Physiological Function—Biochemical Characterization of the Morphine Metabolite, Bismorphine,” Journal of Biological Chemistry, Vol. 276, No. 41, 2001, pp. 38179-38184. doi:10.1074/jbc.M107105200
- J. E. Zadina, L. Hackler, L. J. Ge and A. J. Kastin, “A Potent and Selective Endogenous Agonist for the MuOpiate Receptor,” Nature, Vol. 386, No. 6624, 1997, pp. 499-502. doi:10.1038/386499a0
- Y. Okada, A. Fukumizu, M. Takahashi, Y. Shimizu, Y. Tsuda, T. Yokoi, S. D. Bryant and L. H. Lazarus, “Synthesis of Stereoisomeric Analogues of Endomorphin-2, H-Tyr-Pro-Phe-Phe-NH2, and Examination of Their Opioid Receptor Binding Activities and Solution Conformation,” Biochemical and Biophysical Research Communications, Vol. 276, No. 1, 2000, pp. 7-11. doi:10.1006/bbrc.2000.3416
- M. G. Paterlini, F. Avitabile, B. G. Ostrowski, D. M. Ferguson and P. S. Portoghese, “Stereochemical Requirements for Receptor Recognition of the Mu-Opioid Peptide Endomorphin-1,” Biophysical Journal, Vol. 78, No. 2, 2000, pp. 590-599. doi:10.1016/S0006-3495(00)76619-7
- J. Fichna, J. C. Do-Rego, P. Kosson, J. Costentin and A. Janecka, “Characterization of Antinociceptive Activity of Novel Endomorphin-2 and Morphiceptin Analogs Modified in the Third Position,” Biochemical Pharmacology, Vol. 69, No. 1, 2005, pp. 179-185. doi:10.1016/j.bcp.2004.09.011
- G. Cardillo, L. Gentilucci, P. Melchiorre and S. Spampinato, “Synthesis and Binding Activity of Endomorphin-1 Analogues Containing Beta-Amino Acids,” Bioorganic & Medicinal Chemistry Letters, Vol. 10, No. 24, 2000, pp. 2755-2758. doi:10.1016/S0960-894X(00)00562-X
- G. Cardillo, L. Gentilucci, A. R. Qasem, F. Sgarzi and S. Spampinato, “Endomorphin-1 Analogues Containing BetaProline Are Mu-Opioid Receptor Agonists and Display Enhanced Enzymatic Hydrolysis Resistance,” Journal of Medicinal Chemistry, Vol. 45, No. 12, 2002, pp. 2571- 2578. doi:10.1021/jm011059z
- S. Spampinato, A. R. Qasem, M. Calienni, G. Murari, L. Gentilucci, A. Tolomelli and G. Cardillo, “Antinociception by a Peripherally Administered Novel Endomorphin-1 Analogue Containing Beta-Proline,” European Journal of Pharmacology, Vol. 469, No. 1-3, 2003, pp. 89-95. doi:10.1016/S0014-2999(03)01736-9
- Y. Okada, Y. Fujita, T. Motoyama, Y. Tsuda, T. Yokoi, T. Y. Li, Y. Sasaki, A. Ambo, Y. Jinsmaa, S. D. Bryant and L. H. Lazarus, “Structural Studies of [2',6'-DimethylL-tyrosine(1)]endomorphin-2 Analogues: Enhanced Activity and cis Orientation of the Dmt-Pro Amide Bond,” Bioorganic & Medicinal Chemistry, Vol. 11, No. 9, 2003, pp. 1983-1994. doi:10.1016/S0968-1896(03)00068-3
- Y. Jinsmaa, E. Marczak, Y. Fujita, K. Shiotani, A. Miyazaki, T. Y. Li, Y. Tsuda, A. Arnbo, Y. Sasaki, S. D. Bryant, Y. Okada and L. H. Lazarus, “Potent in Vivo Antinociception and Opioid Receptor Preference of the Novel Analogue [Dmt(1)]endomorphin-1,” Pharmacology, Biochemistry and Behavior, Vol. 84, No. 2, 2006, pp. 252- 258. doi:10.1016/j.pbb.2006.05.005
- Y. Koda, M. Del Borgo, S. T. Wessling, L. H. Lazarus, Y. Okada, I. Toth and J. T. Blanchfield, “Synthesis and in Vitro Evaluation of a Library of Modified Endomorphin-1 Peptides,” Bioorganic & Medicinal Chemistry, Vol. 16, No. 11, 2008, pp. 6286-6296. doi:10.1016/j.bmc.2008.04.020
- Y. Koda, M. T. Liang, J. T. Blanchfield and I. Toth, “In Vitro Stability and Permeability Studies of Liposomal Delivery Systems for a Novel Lipophilic Endomorphin-1 Analogue,” International Journal of Pharmaceutics, Vol. 356, No. 1-2, 2008, pp. 37-43. doi:10.1016/j.ijpharm.2007.12.036
- C. Adessi and C. Soto, “Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability,” Current Medicinal Chemistry, Vol. 9, No. 9, 2002, pp. 963- 978. doi:10.2174/0929867024606731
- L. Gentilucci, R. De Marco and L. Cerisoli, “Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization,” Current Pharmaceutical Design, Vol. 16, No. 28, 2010, pp. 3185-3203. doi:10.2174/138161210793292555
- G. Cardillo, L. Gentilucci, A. Tolomelli, R. Spinosa, M. Calienni, A. R. Qasem and S. Spampinato, “Synthesis and Evaluation of the Affinity toward Mu-Opioid Receptors of Atypical, Lipophilic Ligands Based on the Sequence c[-Tyr-Pro-Trp-Phe-Gly-],” Journal of Medicinal Chemistry, Vol. 47, No. 21, 2004, pp. 5198-5203. doi:10.1021/jm0498811
- L. Gentilucci, A. Tolomelli and F. Squassabia, “Topological Exploration of Cyclic Endomorphin-1 Analogues, Structurally Defined Models for Investigating the Bioactive Conformation of MOR Agonists,” Protein and Peptide Letters, Vol. 14, No. 1, 2007, pp. 51-56. doi:10.2174/092986607779117218
- L. Gentilucci, F. Squassabia, R. Demarco, R. Artali, G. Cardillo, A. Tolomelli, S. Spampinato and A. Bedini, “Investigation of the Interaction between the Atypical Agonist c[YpwFG] and MOR,” FEBS Journal, Vol. 275, No. 9, 2008, pp. 2315-2337. doi:10.1111/j.1742-4658.2008.06386.x
- A. Janecka, J. Fichna, R. Kruszynski, Y. Sasaki, A. Ambo, J. Costentin and J. C. Do-Rego, “Synthesis and Antinociceptive Activity of Cyclic Endomorphin-2 and Morphiceptin Analogs,” Biochemical Pharmacology, Vol. 71, No. 1-2, 2005, pp. 188-195. doi:10.1016/j.bcp.2005.10.018
- R. Perlikowska, J. C. Do-Rego, A. Cravezic, J. Fichna, A. Wyrebska, G. Toth and A. Janecka, “Synthesis and Biological Evaluation of Cyclic Endomorphin-2 Analogs,” Peptides, Vol. 31, No. 2, 2010, pp. 339-345. doi:10.1016/j.peptides.2009.12.002
- I. Toth, J. P. Malkinson, N. S. Flinn, B. Drouillat, A. Horvath, J. Erchegyi, M. Idei, A. Venetianer, P. Artursson, L. Lazorova, B. Szende and G. Keri, “Novel Lipoamino Acidand Liposaccharide-Based System for Peptide Delivery: Application for Oral Administration of Tumor-Selective Somatostatin Analogues,” Journal of Medicinal Chemistry, Vol. 42, No. 19, 1999, pp. 4010- 4013. doi:10.1021/jm9910167
- P. Simerska, P. M. Moyle and I. Toth, “Modern Lipid-, Carbohydrate-, and Peptide-Based Delivery Systems for Peptide, Vaccine, and Gene Products,” Medicinal Research Reviews, Vol. 31, No. 4, 2009, pp. 520-547. doi:10.1002/med.20191
- R. Polt and M. M. Palian, “Glycopeptide Analgesics,” Drugs of the Future, Vol. 26, No. 6, 2001, pp. 561-576. doi:10.1358/dof.2001.026.06.668342
- J.-P. Meyer, N. Collins, F.-D. Lung, P. Davis, T. Zalewska, F. Porreca, H. I. Yamamura and V. J. Hruby, “Design, Synthesis, and Biological Properties of Highly Potent Cyclic Dynorphin a Analogs. Analogs Cyclized between Positions 5 and 11,” Journal of Medicinal Chemistry, Vol. 37, No. 23, 1994, pp. 3910-3917. doi:10.1021/jm00049a010
- H. I. Mosberg, R. Hurst, V. J. Hruby, K. Gee, H. I. Yamamura, J. J. Galligan and T. F. Burks, “Bis-Penicillamine Enkephalins Possess Highly Improved Specificity toward Delta Opioid Receptors,” Proceedings of the National Academy of Sciences USA, Vol. 80, No. 19, 1983, pp. 5871-5874. doi:10.1073/pnas.80.19.5871
- P. W. Schiller, G. Weltrowska, T. M. D. Nguyen, C. Lemieux, N. N. Chung and Y. X. Lu, “Conversion of Delta-, Kappaand Mu-Receptor Selective Opioid Peptide Agonists into Delta-, Kappaand Mu-Selective Antagonists,” Life Sciences, Vol. 73, No. 6, 2003, pp. 691-698. doi:10.1016/S0024-3205(03)00389-8
- W. Pan and A. J. Kastin, “From MIF-1 to Endomorphin: The Tyr-MIF-1 Family of Peptides,” Peptides, Vol. 28, No. 12, 2007, pp. 2411-2434. doi:10.1016/j.peptides.2007.10.006
- R. Yamada and Y. Kera, “D-Amino Acid Hydrolysing Enzymes,” EXS, Vol. 85, 1998, pp. 145-155. doi:10.1007/978-3-0348-8837-0_9
- R. Polt, F. Porreca, L. Z. Szabo, E. J. Bilsky, P. Davis, T. J. Abbruscato, T. P. Davis, R. Horvath, H. I. Yamamura and V. J. Hruby, “Glycopeptide Enkephalin Analogs Produce Analgesia in Mice—Evidence for Penetration of the Blood-Brain-Barrier,” Proceedings of the National Academy of Sciences of the USA, Vol. 91, No. 15, 1994, pp. 7114-7118. doi:10.1073/pnas.91.15.7114
- S. Horvat, “Opioid Peptides and Their Glycoconjugates: Structure-Activity Relationships,” Current Medicinal Chemistry: Central Nervous System Agents, Vol. 1, No. 2, 2001, pp. 133-154. doi:10.2174/1568015013358581
- N. O. Elmagbari, R. D. Egleton, M. M. Palian, J. J. Lowery, W. R. Schmid, P. Davis, E. Navratilova, M. Dhanasekaran, C. M. Keyari, H. I. Yamamura, F. Porreca, V. J. Hruby, R. Polt and E. J. Bilsky, “Antinociceptive Structure-Activity Studies with Enkephalin-Based Opioid Glycopeptides,” Journal of Pharmacology and Experimental Therapeutics, Vol. 311, No. 1, 2004, pp. 290-297. doi:10.1124/jpet.104.069393
- W. A. Gibbons, R. A. Hughes, M. Charalambous, M. Christodoulou, A. Szeto, A. E. Aulabaugh, P. Mascagni and I. Toth, “Lipidic Peptides 1. Synthesis, Resolution and Structural Elucidation of Lipidic Amino-Acids and Their Homo-Oligomers and Heterooligomers,” Liebigs Annalen der Chemie, No. 12, 1990, pp. 1175-1183. doi:10.1002/jlac.1990199001215
- P. Alewood, D. Alewood, L. Miranda, S. Love, W. Meutermans and D. Wilson, “Rapid in Situ Neutralization Protocols for Boc and Fmoc Solid-Phase Chemistries,” Methods in Enzymology, Vol. 289, 1997, pp. 14-29. doi:10.1016/S0076-6879(97)89041-6