Journal of Crystallization Process and Technology
Vol.1 No.3(2011), Article ID:7844,8 pages DOI:10.4236/jcpt.2011.13006
A Statistical Rate Theory of Interface Concentration during Solution Zeolite Crystal Growth
![]()
Department of Mechanical, Materials and Aerospace Engineering, University of Central Florida, Orlando, USA.
Email: *ilegbusi@ucf.edu
Received June 17th, 2011; revised July 30th, 2011; accepted August 9th, 2011.
Keywords: Zeolite, Crystal Growth, Statistical Rate Theory, Boundary Layer, Microgravity
ABSTRACT
A theoretical model is developed using statistical rate theory to determine the rate of molecular transport across the interface of a growing spherical zeolite crystal. The model is expressed in terms of the interface concentration. Two model constants appear in the expression for the equilibrium exchange rate. In order to validate the model, zeolite crystallization is investigated for a system for which experimental data exist. The model constants were first established using the measured growth rates at a specific temperature. Then the model was used to predict the growth rate at other temperatures.
1. Introduction
The synthesis of large zeolite crystals has been the subject of extensive investigation in recent years due to the potential industrial applications of the material in molecular electronics, chemical sensors and zeolite-polymer composites [1-3]. Large crystals are typically synthesized by suppressing nucleation and enhancing growth. However, it is difficult to control these two processes not only because of the complexity of the underlying mechanism of zeolite microstructure formation, but also for the convection-diffusion flow in the boundary layer/depletion zone around the growing crystals. This flow can significantly affect the mass and energy transport across the boundary layer, and thus the surface kinetics of zeolite crystal growth [4]. In order to reduce this density-driven convection, zeolite crystals have been grown in low-earthorbit. Such space experiments [5,6] have demonstrated a substantial increase in crystal size over those grown on ground. Although this size increase has been attributed to the reduction and suspension of primitive nuclei in nutrient-rich pool in the absence of gravitational sedimentation, our understanding of the interaction between the transport phenomena and surface kinetics as the zeolite crystal grows is still rather limited [7-10]. The objective of the present work was to develop a theoretical model to relate the rate of transport of solute species across the boundary layer to the kinetics of crystallization.
Zeolite crystal growth is hypothesized to be solutionmediated process. In such a process, the diffusion and reaction steps such as dissociation and integration of solute species at a terrace or at a step play an important role in determining the quality of the final products [11]. Several models have been proposed [12-14] based on the theory that the crystal surfaces grow in such a way that solute species in a supersaturated solution are first transported by diffusion and convection and then deposited onto the surface of the crystal by integration or an integration reaction with supersaturation being the driving force. Therefore, two points are worthy of note: first, the solution-mediated process is one in which the crystal growth is diffusion-controlled and, second, the surface reaction of soluble species with the crystal surface is rate-determining. We use a theoretical approach (statistical rate theory) to obtain the expression for the rate of solute transport across the boundary layer in terms of the concentration at the interface. The advantage of this approach is that once the model has been established, it could be incorporated into the kinetics equations and used to predict the crystal growth rate [15-17].
The transport of species across the boundary layer depends on a number of factors including species size, viscosity of solution, kinetic energy, and fluid convection. In particular, particle settling under gravity can produce a convection zone which stores strain energy and, in the present case a barrier to the diffusion of solute species to the crystal surface. The rate of settling, typically determined from estimate of the terminal velocity, is a function of three forces: gravity, buoyancy and fluid drag on the particle surface. More specifically, the shape and depth of the boundary layer depend strongly on various parameters such as crystal size, orientation, speed and fluid properties. For example, the motion directed toward the crystal enhances solute collision rate with the solid surface above the normal collision frequency, while the backward motion of the solution from the crystal decreases the collision rate with the solid surface. Nonetheless, it is highly desirable to provide a fundamental understanding of the impact on the transport of solute species, of the flow field in the boundary layer. Such information will guide design strategies for synthesizing large crystals through modification of the flow field.
In this paper, a model is developed that relates crystal growth rate to the flow properties within the boundary layer around the growing crystal. Specifically, a statistical rate theory is used to obtain the rate of solute transport across the interface of a growing crystal. This rate is expressed in terms of the concentration at the interface, and serves as the boundary condition for the solute transport equations.
2. Formulation
2.1. Crystal Growth Rate under Steady State Conditions
The mechanism of zeolite crystal growth from solution is complicated, as are all processes occurring in the boundary layer between two phases. These processes involve the diffusion of solute species (molecules, atoms, and ions) from the bulk gel phase to the interface and the incorporation into the crystal phase. Zeolites are complex inorganics based on an infinitely extended framework of 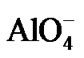 and
and 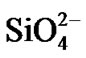 tetrahedra linked to each other by shared oxygen atoms [18]. In order to study the mass transport across the interface between the solid and liquid phases, we assume the following mechanism for zeolite crystal growth:
tetrahedra linked to each other by shared oxygen atoms [18]. In order to study the mass transport across the interface between the solid and liquid phases, we assume the following mechanism for zeolite crystal growth:
 (1)
(1)
where M represents the cation. The diffusion and surface incorporation in the boundary layer of the tetrahedral units, TO4 (T representing Al or Si), dominate the growth rate of zeolite crystals.
In order to express the instantaneous transport rate of the TO4 tetrahedral units across the interface between the solution and the growing crystals under steady state conditions, we assume that the zeolite crystallization occurs in an isolated system. This transport rate can be expressed in terms of: 1) the chemical potential of the TO4 units in the solid phase, 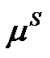 (
(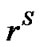 ), where
), where 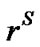 is in the solid but at the interface with the liquid phase; 2) the chemical potential of the TO4 units in the liquid solution
is in the solid but at the interface with the liquid phase; 2) the chemical potential of the TO4 units in the liquid solution  (
(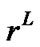 ) at the interface with the solid phase; and 3) the equilibrium exchange rate between the liquid and solid that would exist if the isolated system were allowed to evolve to equilibrium and both phases were present. Thus:
) at the interface with the solid phase; and 3) the equilibrium exchange rate between the liquid and solid that would exist if the isolated system were allowed to evolve to equilibrium and both phases were present. Thus:
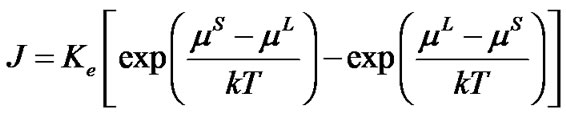 (2)
(2)
where J is the transport rate of TO4 units across the interface, Ke is the equilibrium exchange rate between the liquid and solid, k is the Boltzmann constant and T is the temperature.
The chemical potential of the TO4 units in the solution can be expressed as
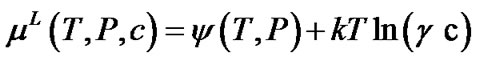 (3)
(3)
where P is the pressure, c is the concentration, g is the activity coefficient and the term y(T, P) can be written in terms of the properties at saturation condition:
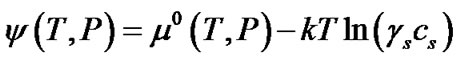 (4)
(4)
where 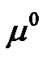 (T,P) is the chemical potential of the pure TO4 units,
(T,P) is the chemical potential of the pure TO4 units, 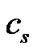 is the concentration of TO4 units in the saturated solution, and
is the concentration of TO4 units in the saturated solution, and  is the activity coefficient in the saturated solution. Substituting Equation (4) into Equation (3), we can obtain the chemical potential in the solution as,
is the activity coefficient in the saturated solution. Substituting Equation (4) into Equation (3), we can obtain the chemical potential in the solution as,
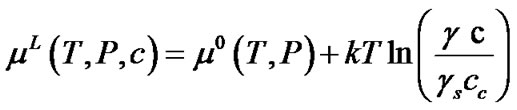 (5)
(5)
Although the activity coefficients are in general a function of the concentration, for simplicity, we assume that the ratio of activity coefficients in the above equation is unity. In reality, this assumption needs to be validated using experimental data for zeolite crystallization. The chemical potential in the solution can thus be rewritten as:
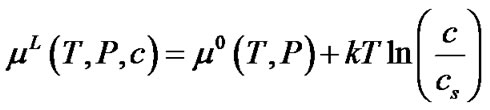 (6)
(6)
Under steady state conditions, the chemical potential in the solid phase must be equal to that in the saturated liquid solution, thus:
 (7)
(7)
Substituting Equations (5) and (7) into Equation (2) and evaluating the concentrations appearing in these expressions at the interface, we can rewrite the equilibrium exchange rate of the TO4 units between solution and crystal as,
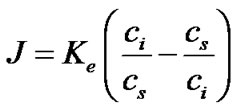 (8)
(8)
where ci is the concentration at the interface between the two phases.
In the statistical rate theory, the equilibrium exchange rate Ke is typically defined as the production of the collision frequency of the solute molecules with the solid surface, and the cross-sectional areas of the available sites for solute transfer from one phase to the other under equilibrium condition [19,20]. However, in the case of zeolite crystal growth, the collision frequency of the TO4 units with the solid surface is significantly influenced by the flow condition resulting from the motion of growing crystals. For example, when the crystal settles down under gravity, the motion of the solution toward the crystal enhances the rate of solute collision with the solid surface above the normal solute collision frequency. On the other hand, the motion of the solution away from the crystal decreases the rate of collision with the solid surface. Therefore, we propose a modified expression for the equilibrium exchange rate of TO4 units in which an additional term is introduced to represent the modification of collision frequency as a result of the flow in the diffusion layer. The equilibrium exchange rate assumes the form:
 (9)
(9)
where  is the collision frequency under equilibrium conditions of the solute molecules with the crystal surface due to their random motions,
is the collision frequency under equilibrium conditions of the solute molecules with the crystal surface due to their random motions, 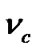 is the enhancment of this rate due to convection, and
is the enhancment of this rate due to convection, and 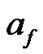 is the fraction of crystal surface that is available for transport of solute molecules from the solution.
is the fraction of crystal surface that is available for transport of solute molecules from the solution.
To represent the collision frequency nr, we invoke the analogy with a gas phase contacting a solid surface. Typically, the gas has a Maxwellian distribution under equilibrium condition. Therefore, the collision frequency of gas molecules with the solid surface under equilibrium conditions, ng, can be expressed as:
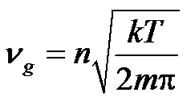 (10)
(10)
where n is the number density of gas molecules, and m is the mass per gas molecule.
We assume a similar expression for the collision frequency for the liquid solution considered here. Under equilibrium conditions the concentration of the solute molecules would be cs and the collision frequency can be expressed as:
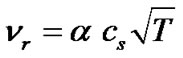 (11)
(11)
where a is a proportionality constant.
In order to obtain the enhancement of collision frequency nc, we denote the radial velocity of solute molecules at the edge of the diffusion layer by 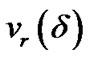 Therefore, under equilibrium conditions,
Therefore, under equilibrium conditions,
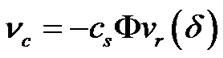 (12)
(12)
where F is the fraction of solute molecules that will strike the crystal surface and d is the thickness of the diffusion layer. Substituting Equations (11) and (12) into Equation (9), we obtain the following expression for the overall transport rate of solute units from the solution to the crystal:
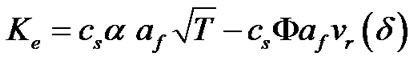 (13)
(13)
Since a, b, and af are constants, Ke can be expressed more simply in terms of two constants Kr and Kc. Therefore, the expression for the transport rate of solute molecules from the solution to the crystal is:
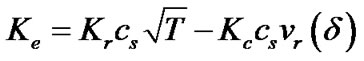 (14)
(14)
The first term in the above expression is the rate that would exist if the solution were static, and the second term results from the convection due to the crystal sedimentation of growing crystals. Therefore, this expression combines the contributions to the crystal growth of both the reaction kinetics and the fluid convection through two coefficients, Kr and Kc. The coefficient Kr can be obtained from the experimental data on zeolite crystallization. However, representing Kc is quite difficult because it involves a number of factors including the size and shape of crystals, solution viscosity and gravity level. In the following section, we will represent Kc from the dynamics of particles immersed in an incompressible fluid.
2.2. Convection-Diffusion Equation
We consider a spherical particle growing in an incompressible solution at low Reynolds number. As shown in Figure 1, due to the density variation, the growing crystal
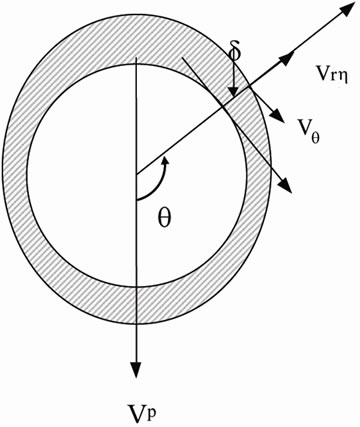
Figure 1. Schematic of the boundary layer (depletion zone) around the crystal.
settles down under gravity, with a boundary layer/depletion zone developing around the particle with different solute concentration and/or temperature from the bulk. In this boundary layer, the stream function y, and the concentration of solute molecules, c(r,q), must satisfy the two-dimensional, steady state convection-diffusion equations [21]:
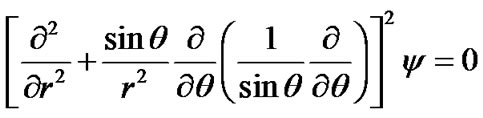 (15)
(15)
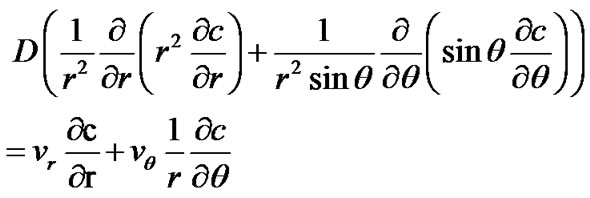 (16)
(16)
where D is the diffusion coefficient of solute molecules, vr and vq are the radial and axial velocity components of the solution, respectively, which can be obtained from the stream function from the expressions:
 (17)
(17)
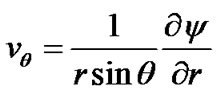 (18)
(18)
This approach amounts to assuming that the bulk fluid acts as a nutrient pool for the crystal. When r approaches infinity, the velocity of solution reaches the terminal velocity of particles and the solute concentration is assumed to be a constant cb. This terminal velocity Vp can be expressed as [22],
 (19)
(19)
where R is the radius of the spherical crystal, g is the gravity, n is the kinematic viscosity of solution, and D is the ratio of crystal density to the solution density.
The boundary condition for the convection-diffusion equation is expressed using the transport rate of solute molecule across the interface between the crystal and solution, thus:
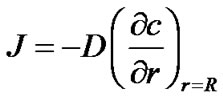 (20)
(20)
Substituting Equation (20) into Equation (2), we can rewrite the equilibrium exchange rate of solute molecules across the interface as,
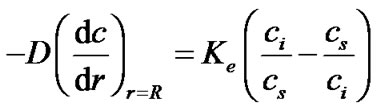 (21)
(21)
We assume the concentration in the bulk is constant throughout the growth process. Thus, the portion of solution outside of the diffusion layer is approximated as a reservoir that has a given concentration. Since the concentration in the bulk is unchanged by the growth process, strictly speaking the system cannot reach equilibrium. This situation will be re-examined subsequently when we consider the equilibrium exchange rate.
To estimate the thickness of the diffusion layer, we may determine the location where c begins to deviate from the bulk value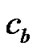 . We define this thickness from the relation:
. We define this thickness from the relation:
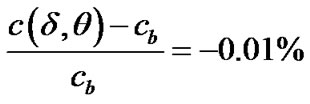 (22)
(22)
In order to characterize the crystal growth, we rewrite Equation (21) in a dimensionless form by introducing a reference length and a reference concentration, thus,
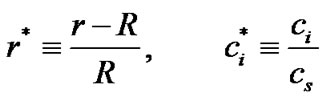 (23)
(23)
Then, we can rewrite Equation (21) as,
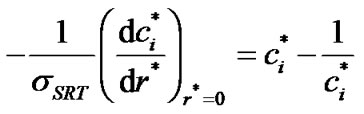 (24)
(24)
where the parameter, 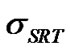 , is the dimensionless equilibrium coefficient defined by,
, is the dimensionless equilibrium coefficient defined by,
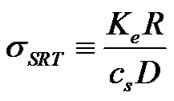 (25)
(25)
It should be noted that if 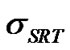 becomes much larger than 1, the left-hand side of Equation (24) is small and
becomes much larger than 1, the left-hand side of Equation (24) is small and  approaches unity. Thus, in this case, equilibrium exists at the interface and the transport rate of solute molecules is diffusion-controlled. On the other hand, if
approaches unity. Thus, in this case, equilibrium exists at the interface and the transport rate of solute molecules is diffusion-controlled. On the other hand, if 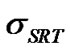 is much smaller than 1, the left-hand side of Equation (24) is large and
is much smaller than 1, the left-hand side of Equation (24) is large and 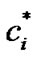 is much larger than 1 at the interface, suggesting a highly non-equilibrium process in which the crystal growth rate is controlled by molecular transport across the interface.
is much larger than 1 at the interface, suggesting a highly non-equilibrium process in which the crystal growth rate is controlled by molecular transport across the interface.
2.3. Crystal Growth under Non-Equilibrium Conditions
In the last section, we developed the expression for the equilibrium exchange rate Ke of solute TO4 units in a solution assumed to have a saturation concentration corresponding to the temperature of the system. However, a real zeolite crystal growth situation cannot reach equilibrium since the bulk region is approximated by one having a constant concentration that is not necessarily equal to the saturation concentration corresponding to this temperature. In the statistical rate theory, Ke is evaluated in the hypothetical equilibrium state in which there is no net molecular transport across the interface between the liquid solution and the solid. The procedure that we have adopted amounts to defining this hypothetical state as being that in which the solution is saturated at the system temperature.
These expressions for the chemical potential of TO4 units in the liquid solution and the solid phase are also valid at the non-equilibrium condition of zeolite crystal growth. Therefore, the instantaneous transport rate of these units across the interface is enhanced above the equilibrium exchange rate in the direction of the phase having the smaller chemical potential by a factor that appears in Equation (2).
3. Computational Details
The equations governing the flow (stream function) and solute transport are solved with the SIMPLEC algorithm embodied in the CFD-ACE software code. This code uses a control volume based finite difference formulation with upwind difference for convective fluxes. We apply the mathematical model developed above to a zeolite crystallization process. Once the equilibrium exchange rate and the velocity field have been calculated for a growing crystal suspended in the solution, it is possible to compare the nonlinear rate of solute molecular transport across the interface to the diffusion rate.
The crystal growth rates measured on ground and at a temperature of 100˚C were used to determine the values of Kr and Kc. The values of the diffusion coefficient, viscosity, and saturation concentration used in these calculations are given in Table 1. The values obtained for Kr and Kc using this procedure are also listed in the table. These values were used in all subsequent calculations
4. Results and Discussion
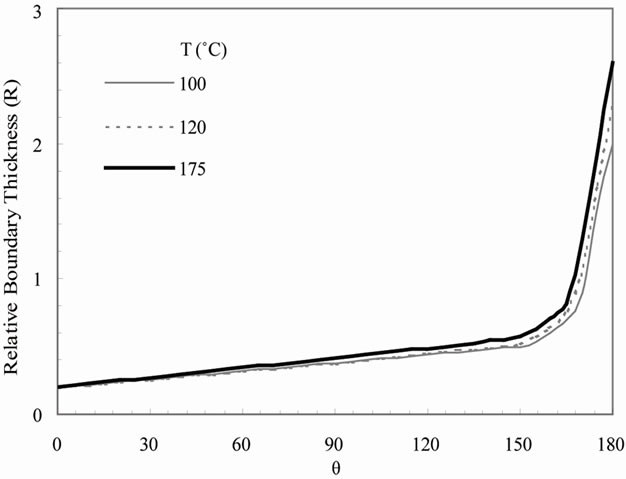
Figure 2(a) shows the computed relative diffusion boundary layer thickness of a particle growing on ground as a function of temperature and position around the particle (angle q). The corresponding results in microgravity are presented in Figure 2(b). The diffusion boundary layer thickness is defined as the location where the solute concentration has been altered by less than 1% of its undisturbed value. Thus, the shape and relative thickness of the diffusion boundary are independent of the particle size. Figures 2(a) and (b) show that the predicted relative thickness is much larger in microgravity than on ground. In addition, it increases slightly with increasing temperature both on ground and low earth orbit. It should be noted that the solute distribution and boundary layer thickness generally depend on the solute incorporation at the interface expressing crystal growth. For zeolites the crystal growth rate is typically determined not solely by the temperature, but also flow condition and solute concentration. In the present situation the effect of flow condition and solute concentration predominate in the range of temperature considered. However, the increase in thickness with temperature is observed to be significantly higher in low earth orbit than on ground. This trend is attributed to the reduction of convection and mass transport in microgravity (i.e. suppressed effect of flow condition), leading to diffusion-controlled solute transport in microgravity. In addition, Figures 2(a) and (b) also show that the relative thickness varies with angle q. Specifically, the thickness is larger in the upper periphery (q = 180˚) of the particle where the motion of solution is outward than the lower surface of the particle (q = 0) where the motion of solution is towards the particle.
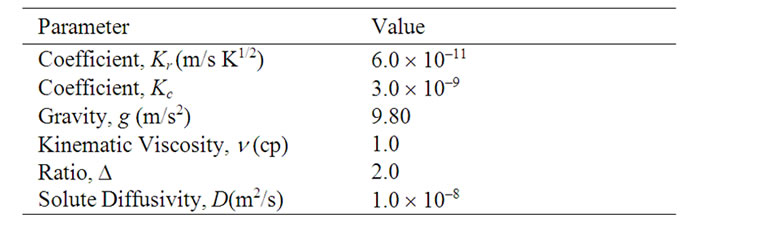
Table 1. Parameter values used in these calculations.
 (a)
(a)  (b)
(b)
Figure 2. Predicted relative diffusion boundary thickness as a function of temperature on (a) ground and (b) low earth orbit.
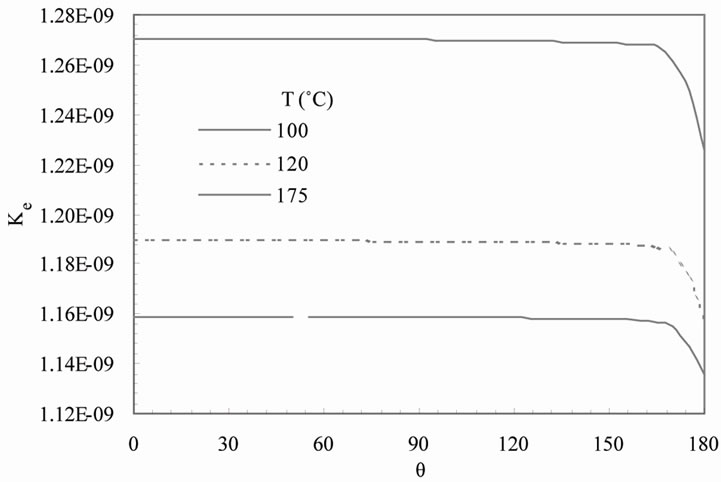
The predicted equilibrium exchange rate Ke as a function of temperature and angle q is presented in Figure 3(a) for crystal growth on ground. The corresponding curves for low earth orbit are presented in Figure 3(b). In both cases, the equilibrium exchange rate increases with increasing temperature. This trend may be attributed to the enhancement of solute incorporation into the growing surface governed by chemical reaction kinetics, which in turn is enhanced at higher temperature. This trend is also consistent with the effect of temperature on boundary layer thickness presented in previous Figures 2(a) and (b). As might be expected, a change in the relative thickness may have a significant effect on the equilibrium exchange rate because the boundary thickness determines the velocity of solutes entering the boundary and thus the exchange rate. Figures 3(a) and (b) also show that the predicted equilibrium exchange rate is nearly independent of q on both ground and low earth orbit. However, there is a slight decrease on the upper surface of the particle (q = 180˚) on ground (Figure 3(a)) where the solution moves away from the crystal. This motion can induce a reduction in crystal growth rate in this zone.
 (a)
(a) (b)
(b)
Figure 3. Predicted equilibrium exchange rate Ke predicted as a function of temperature on (a) ground and (b) low earth orbit.
Figure 4(a) shows the solute mass velocity J as a function of temperature and angle q for growth on ground. The corresponding prediction for low earth orbit is presented in Figure 4(b). The mass velocity increases with temperature both on ground and microgravity. It remains nearly constant around the particle on ground except near the top surface (close to q = 180˚) where there is a sharp decrease. On the other hand, this velocity decreases as q increases for growth in microgravity. The solute mass velocity strongly depends on the diffusion boundary layer thickness due to the boundary-dependence of the exchange rate. Thus the observed trends are consistent with the previous results presented in Figures 2 and 3. It is not clear from Figure 4 if there is any disagreement between the theory and the experiments conducted at the varying temperatures. Since no fitting parameters are used in this paper for the prediction of the crystal growth rates at these temperatures, the results appear to support the statistical rate theory expression developed for the rate of molecular transport across the solid-liquid interface.
Figure 5(a) shows the dimensionless equilibrium co-
 (a)
(a)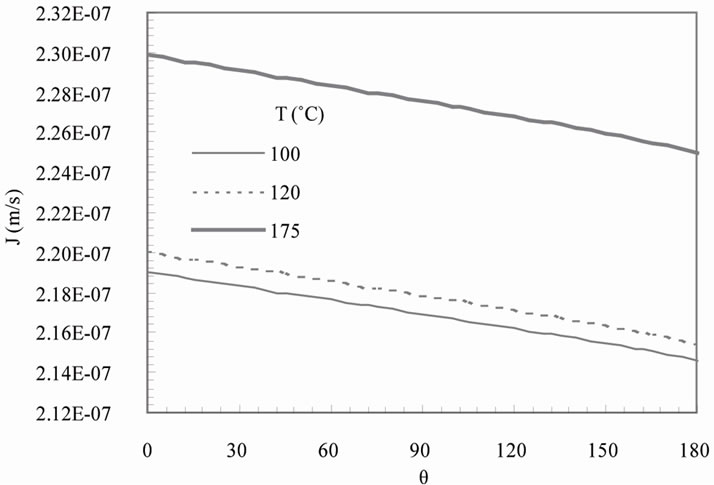 (b)
(b)
Figure 4. Predicted solute flux velocity J predicted as a function of temperature on (a) ground and (b) low earth orbit.
efficient sSRT as a function of temperature and q onground. The corresponding result for low earth orbit is presented in Figure 5(b). In the consensus of the previous results on boundary thickness, the predicted parameter sSRT increases slightly with temperature in both ground and microgravity, and increases from bottom (q = 0) to top (q = 180˚) of particle in both cases but with a slightly higher rate of increase on ground. The parameter sSRT is much less than unity at all the temperatures considered on ground (Figure 5(a)). Thus the crystal growth rate on ground is controlled by molecular transport across the interface. In other words, the interfacial transport rate and the diffusion rate are of the same magnitude and neither can be neglected. The solute concentration at the interface is large, which suggests a strong degree of non-equilibrium. At low earth orbit (Figure 5(b)), sSRT is much larger than unity at all temperatures, suggesting a diffusion-controlled crystal growth mechanism. In addition, the observed increase in sSRT from top to bottom of
 (a)
(a)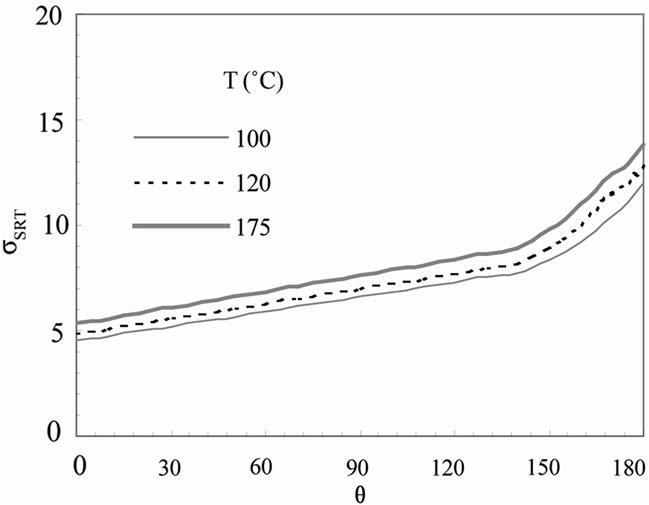 (b)
(b)
Figure 5. Predicted dimensionless parameter, sSRT, predicted as a function of temperature on (a) ground and (b) low earth orbit.
the particle reflects the difference in crystal growth rates between the upper and lower surface of the particle, especially under gravity (Figure 5(a)).
5. Conclusions
The statistical rate theory has been used to develop an expression for the rate of molecular transport across the interface of a growing zeolite crystal. The transport rate is expressed in terms of the concentration in the liquid phase at the crystal surface. This rate expression provides a nonlinear boundary condition for the convectiondiffusion equation. The equation is solved in terms of two coefficients that appear in the equilibrium exchange rate expression. In order to predict the rate of crystal growth, the values of these coefficients are first determined from available experimental data at a single temperature, but at different gravity levels. Then the system of equations is solved to predict the steady state crystal growth rates at the two-phase interface at different temperatures and gravity levels. The results include the concentration at the interface, the diffusion layer thickness, and equilibrium exchange rate at the crystal-solution interface. They indicate that gravity levels significantly affect the shape and thickness of diffusion layer, and thus the growth mechanisms. The present work suggests for zeolite crystallization a surface reaction-controlled growth mechanism on ground and a diffusion-controlled growth mechanism in low earth orbit.
6. Acknowledgements
The authors gratefully acknowledge the useful discussions and support of Dr. Albert Sacco, Jr. of the NASA Center for Advanced Microgravity Materials Processing (CAMMP), Northeastern University.
REFERENCES
- E. R. Geus, A. Mulder, D. J. Vischjager and J. Birlein, “Inclusion Tuning of Nonlinear Optical Materials: Switching the SHG of p-nitroaniline and 2-methyl-p-nitroaniline with Molecular Sieve Hosts,” Journal of the American Chemical Society, Vol. 110, No. 9, 1988, pp. 2986- 2987. doi:10.1021/ja00217a057
- J. Warzywoda, A. G. Dixon, R. W. Thompson and A. Sacco Jr., “Synthesis and Control of the Size of Large Mordenite Crystals Using Porous Silica Substrates,” Journal of Materials Chemistry, Vol. 5, No. 7, 1995, p. 1019. doi:10.1039/jm9950501019
- A. Sacco Jr., “The NASA GAS Program: A Stepping Stone to Education,” IEEE Transactions on Education, Vol. 34, No. 1, 1991, pp. 27-30. doi:10.1109/13.79875
- S. P. Zhdanov, “Molecular Sieve Zeolites,” In: R. F. Gould, Ed., Advances in Chemistry Series, Washington, DC, 1971, p. 20.
- A Sacco Jr., N. Bac, J. Warzywoda, I. Guray, R. W. Thompson and L. A. McCauley, “Zeolite Crystal Growth in Microgravity,” Proceedings of Space Technology and Applications, Albuquerque, 1996, pp. 429- 432,
- A Sacco Jr., N. Bac, J. Warzywoda, I. Guray, M. Marceau, T. L. Sacco and L. M. Whalen, “Zeolite Crystal Growth (ZCG) Flight on USML-2,” Final Report, NASA Contract No: NAS8-40260.
- F. Otalora, M. L. Novella, J. A. Gavira, B. R. Thomas and J. M. G. Ruiz, “Experimental Evidence for the Stability of the Depletion Zone around a Growing Protein Crystal under Microgravity,” Acta Crystallographica, Vol. D57, 2001, pp. 412-417. doi:10.1107/S0907444901000555
- S. Bosnar, T. Antonić, J. Bronić and B. Subotić, “Mechanism and Kinetics of the Growth of Zeolite Microcrystals. Part 2: Influence of Sodium Ions Concentration in the Liquid Phase on the Growth Kinetics of Zeolite A Microcrystals,” Microporous and Mesoporous Materials, Vol. 76, No. 1-3, 2004, pp. 157-165. doi:10.1016/j.micromeso.2004.07.021
- H. W. Song, O. J. Ilegbusi and A. Sacco Jr., “Effects of Gravity on Zeolite Crystallization from Solution,” Journal of Crystal Growth, Vol. 277, No. 1-4, 2005, pp. 623- 630. doi:10.1016/j.jcrysgro.2004.12.161
- H. W. Song, O. J. Ilegbusi and A. Sacco Jr., “Kinetics of Zeolite NaA Crystallization in Microgravity,” Materials Letters, Vol. 59, No. 21, 2005, pp. 2668-2672. doi:10.1016/j.matlet.2005.04.015
- I. J. Kim and H. J. Lee, “Effects of Seeding on the Formation of Large NaX Zeolite Crystals, Materials Science Forum, Functionally Graded Materials VIII,” In: O. Van der Biest, M. Gasik, J. Vleugels, Eds., Vols. 492-493, 2005, pp. 287-292.
- B. M. Lowe, “Innovation in Zeolite Materials Science,” In: P. J. Grobt, W. J. Mortier, E. F. Vansant and G. F. Schulz-Ekloff, Eds., Studies in Surface Science and Catalysis, Elsevier, Amesterdam, 1988.
- A. Berthoud, “Theorie de la Formation des Faces d’un Crystal,” Journal of Chemical Physics, Vol. 10, 1912, p. 624.
- B. Subotić and J. Bronić, “Theoretical and Practical Aspects of Zeolite Crystal Growth,” In: S. M. Auerbach, K. A. Carrado, P. Dutta, Eds., Handbook of Zeolite Science and Technology, K. Marcel Dekker Inc. New York, 2003, pp. 129-203.
- J. J. P. Valeton, “Wachstun and Auflosung der Kristalle,” Z. Kristallogr, Vol. 59, No. 1923, pp. 135-169.
- J. A. W. Elliott and C. A. Ward, “Temperature Programmed Desorption: A Statistical Rate Theory Approach,” Journal of Chemical Physics, Vol. 106, No. 13, 1997, pp. 5677-5685. doi:10.1063/1.473588
- J. A. W. Elliott and C. A. Ward, “Chemical Potential of Adsorbed Molecules from a Quantum Statistical Formulation†,” American Chemical Society, Vol. 13, No. 5, 1997, pp. 951-960. doi:10.1021/la951019w
- R. P. Townsend, “Ion Exchange in Zeolites-Basic Principles,” Chemistry and Industry, Vol. 2, No. 7, 1984, pp. 246-251.
- C. A. Ward, “The Rate of Gas Absorption at a Liquid Interface,” Journal of Chemical Physics, Vol. 67, No. 1, 1977, pp. 229-233. doi:10.1063/1.434547
- C. A. Ward. M. Rizk and A. S. Tucker, “Statistical Rate Theory of Interfacial Transport. II. Rate of Isothermal Bubble Evolution in a Liquid-Gas Solution,” Journal of Chemical Physics, Vol. 76, No. 11, 1983, pp. 5606-5615. doi:10.1063/1.442866
- J. Crank, “The Mathematics of Diffusion,” Oxford University Press, Oxford, 1956.
- M. R. Maxey and J. J. Riley, “Equation of Motion for a Small Rigid Sphere in a Nonuniform Flow,” Physics of Fluids, Vol. 26, No. 4, 1983, pp. 883-890. doi:10.1063/1.864230