Journal of Analytical Sciences, Methods and Instrumentation
Vol.3 No.4(2013), Article ID:38319,5 pages DOI:10.4236/jasmi.2013.34023
Low Level Determination of Genotoxic Impurity in Deferasirox Formulation
![]()
Department of Chemistry, Ramnarian Ruia College, Mumbai, India.
Email: *pravish1981@yahoo.com
Copyright © 2013 Pravish Kumar Tiwari et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 17th, 2013; revised September 28th, 2013; accepted October 11th, 2013
Keywords: Deferasirox; Genotoxic; 4-Hydrazinobenzoic Acid; HPLC
ABSTRACT
4-hydrazinobenzoic acid has been highlighted as one of the potential genotoxic impurities (PGIs). A sensitive HPLC method was developed and validated for the determination of 4-hydrazino benzoic acid in Deferasirox Tablet. HPLC method on Zorbax SB C18 column (250 × 4.6 mm i.d.), 5 μm, with UV Detector was used. The proposed method was cost effective, specific, linear, accurate, rugged and precise. The calibration curves showed good linearity over the concentration range of 0.5 μg/ml to 1.5 μg/ml with respect to the sample. The correlation coefficient was 0.999. Excellent recoveries of 102% were obtained at the level 0.5 μg/ml.
1. Introduction
Deferasirox is an orally administered iron chelator used to reduce chronic overload in patients who are receiving long term blood transfusion for conditions such as beta thalassemia and chronic anaemia. It is the first oral medication approved in USA for this. Two molecules of Deferasirox are capable of binding to one atom of iron which is subsequently eliminated by fecal excretion. Its low molecular weight and high lipophilicity allows the drug to be taken orally.
Synthetic starting materials and intermediates are reactive by design and may occur as impurities in the final API. The nature of this chemical reactivity can often be translated into biological reactivity and these materials can often be mutagens or carcinogens. Many times it has been established that due to high chemical relativities the fate of the several genotoxic agents precluded their retention within the final API especially if their formation was separated from the final API by several synthetic steps.
4-hydrazinobenzoic acids are often used during manufacture of pharmaceuticals, as counter-ions to form salt, as acid catalysts or as the result of protecting group removal during the synthesis. In fact 4-hydrazinobenzoic acid is a known genotoxin and is known to be carcinogenic in rats and mice [1]. The potential presence of these genotoxins has attracted the attention of regulatory authorities, although no official guidelines have yet been issued. Draft guidelines [2] from the European agency and feedback from the US Food Drug Administration (FDA) to the pharmaceuticals industry via responses to drug applications have enabled the industry to establish interim strategies. Generally, it is accepted that genotoxins will be limited to a daily dose of 1.0 μg/day to 1.5 μg/day unless safety studies establish that it is safe to receive a higher dose or that the drug is used for only a short term exposure, e.g. as an antibiotic. As some genotoxicity studies can take up to two years, e.g. carcinogenicity studies, it is preferable for the potential genotoxins to be controlled during the synthesis. In some cases where levels cannot be controlled and no safety data exists, it may be preferable for the pharmaceutical company to change the route of the drug substance or the isolation procedure, though this normally happens only during early development. Although, 4-hydrazinobenzoic acid is a well known carcinogen [3,4], this data would infer that the regulatory authorities may be expected to request levels of 4-hydrazinobenzoic acid to be controlled to 0.7 ppm in the drug substance (assuming a 1 g/day daily dose) or safety data to justify a greater dose.
Due to the increasing concern from the regulatory perspective in relation to the potential hazards, there has been a general renaissance and increased number of analytical techniques, Liquid chromatographic methods utilizing mass spectrometric detectors is reported in literature for the determination of 4-hydrazinobenzoic acid impurities [5]. But this method is quite expensive as it requires MS detector for low level determination.
The proposed HPLC method for determination of 4- hydrazinobenzoic acid is sensitive and robust involving no laborious sample preparation steps. This method has many advantages over the method reported in the literature in terms of cost, specificity, accuracy and reproducibility. Matrix interference is dealt in the method by utilizing two diluents. First diluent in which high concentration of Deferasirox and 4-hydrazinobenzoic acid is soluble and then high concentration of Deferasirox is precipitated out by addition of Second Diluent and filtrate contains 4-hydrazinobenzoic acid.
Synthesis of Deferasirox
Deferasirox can be prepared from simple commercially available starting materials (salicylic acid, salicylamide and 4-hydrazinobenzoic acid) in the following two-step synthetic sequence (Figure 1). The condensation of salicyloyl chloride (formed in situ from salicylic acid and thionyl chloride) with salicylamide under dehydrating reaction conditions results in formation of 2-(2-hydroxyphenyl)-1,3(4H)-benzoxazin-4-one. This intermediate is isolated and reacted with 4-hydrazino benzoic acid in the presence of base to give 4-(3,5-bis(2-hydroxyphenyl)-1, 2,4-triazol-1-yl)benzoic acid (Deferasirox) [6].
2. Material and methods
2.1. Reagents and Chemicals
HPLC-grade acetonitrile was purchased from J T Baker (Phillipsburg, USA); Perchloric acid was purchased from Sigma-Aldrich (Milwaukee, WI), purified water collected through Milli-Q water purification system (Millipore, Bedford, MA, USA). Reference substances 4-hydrazinobenzoic acid was purchased from Sigma Aldrich (Milwaukee, WI) and Deferasirox Tablet (Exjade) samples were obtained from Market.
2.2. Column Selection and Mobile Phase Optimization
For adequate retention of 4-hydrazinobenzoic acid on different columns like Hypersil BDS C18, Kromasil C18 and Zorbax C18 of different dimensions were evaluated. On Hypersil BDS C18 and Kromasil C18 early elution, blank interference and inadequate separation of analytes was observed. However, on Zorbax C18 column the separation and the response for 4-hydrazinobenzoic acid were found to be suitable.
Different composition of mobile phase using 0.1% triethylamine adjusted pH 2 with perchloric acid and acetonitrile were studied. Good separation and responses were observed using gradient composition of 0.1% triethylamine (v/v in water) adjusted pH 2 with percholoric acid and acetonitrile was optimized with column oven temperature of 30˚C at a flow rate of 1.0 mL per minute. Under these conditions the retention time of 4-hydrazinobenzoic acid were observed to be about 4.0 minutes.
2.3. Instrumentation
Chromatography
The Liquid Chromatography system used was an Agilent 1100 series LC system (Agilent Technologies) consisting of an 1100 series pump with a degasser, a temperature controlled micro-well plate, auto sampler and a column compartment. The analytical column was a Zorbax SB C18 (250 × 4.6 mm) 5 μm. The mobile phase consisted of mobile phase-A (0.2% Triethylamine adjusted to pH 2.0 with perchloric acid) and mobile phase-B (Acetonitrile). The flow rate was 1.0 ml/min and the run time was 45 minutes. Column oven temperature was maintained at 30˚C. Injection volume was 100 μL. The gradient optimized (Table 1) was as follows. The diluents are -DiluentA: Diluted 6.5 ml of triethylamine in 100 ml water; Diluent B-70% Perchloric acid; Diluent C-Diluent A:Diluent B:Water (6:2:2).
2.4 Standard and Sample Preparation
2.4.1. Standard Preparation
Stock standard solution of 4-hydrazinobenzoic acid was prepared in diluents C to obtain the concentration as 100.0 μg/ml. Final standard solution was prepared in Diluent C having 0.05 μg/ml (which is equal to 1 μg/ml with respect to sample concentration).
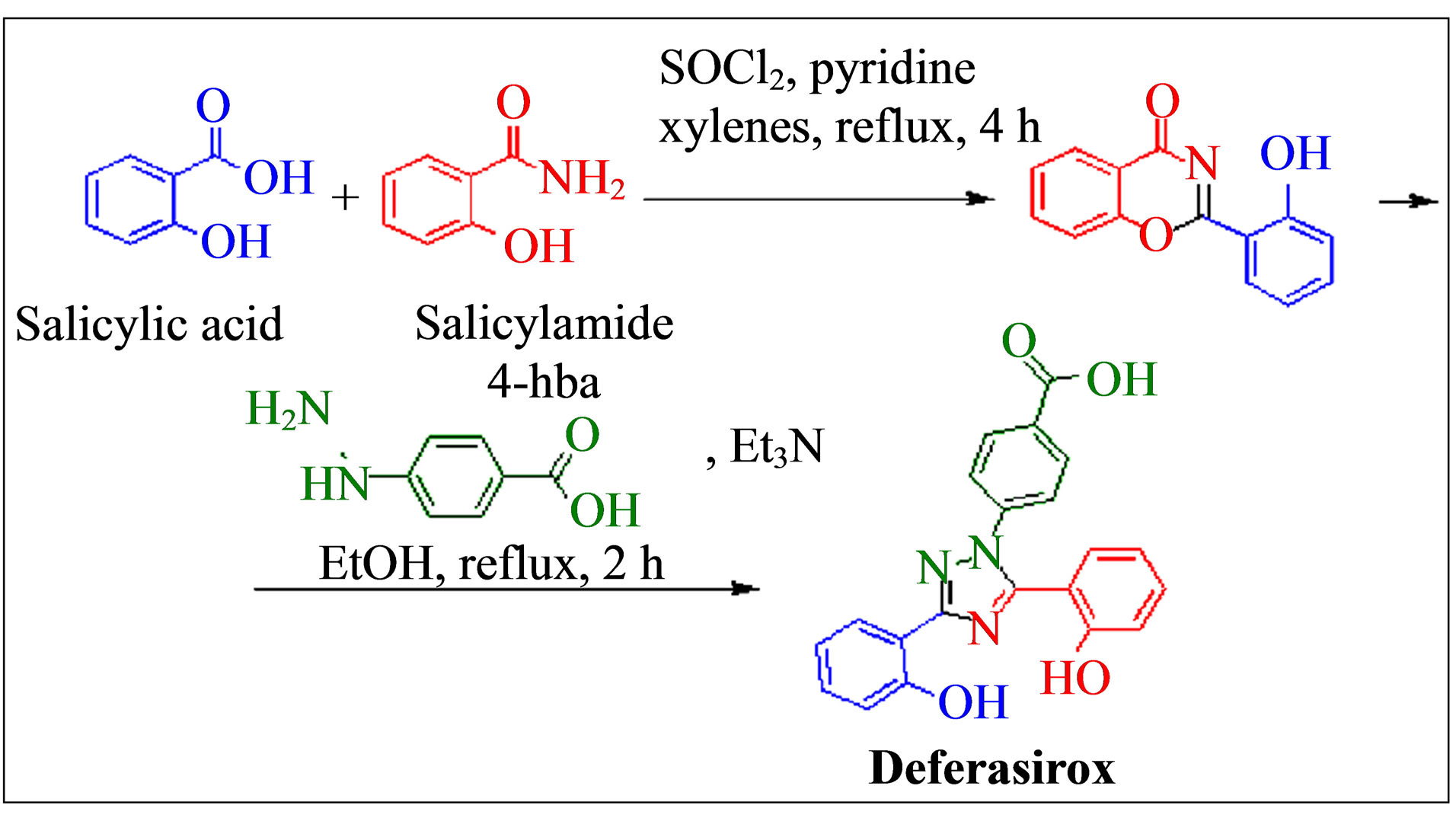
Figure 1. Synthesis of deferasirox.
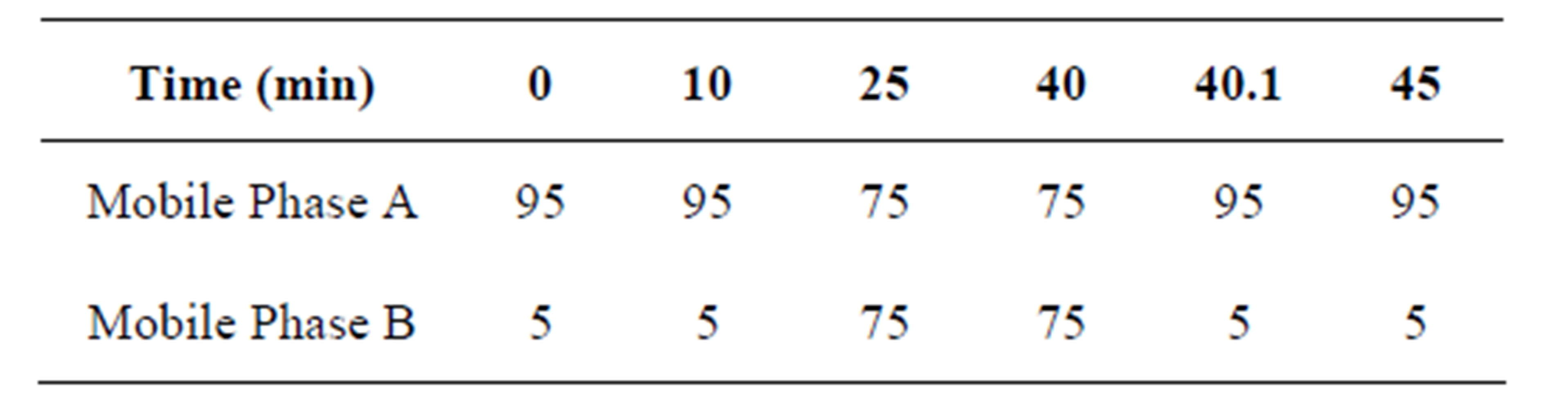
Table 1. Gradient composition.
2.4.2. Sample Preparation
10 tablets of Deferasirox were crushed and weighed sample of equivalent weight to 500 mg Deferasirox in 25 ml volumetric flask, added 6 ml of diluent A. After sonication for 15 minutes, 2 ml of diluent B was added. Sonicate again for 15 minutes and cool the solution. Add 2 ml of water and filter this sample solution through 0.45 μ polytertafluoroethylene (PTFE) filter. This filtrate is used for further analysis.
2.4.3. Optimization of Sample Preparation
Sample preparation is an important part of the genotoxic impurity analysis. This is important as the matrix effects in trace analysis get magnified resulting in the loss of sensitivity, abnormal recovery. Matrix effect also primarily causes analyte’s instability. As the quantitation was required to be done at very trace levels, therefore, the sample concentration was required to be increased in order to reach accurate detection at trace levels. Sample preparations were carried out in first diluent followed by addition of second diluents to precipitate. Diluent selection was accomplished in such a way that in diluent A, Deferasirox and 4-hydrazinobenzoic acid had high solubility and the addition of second diluent B caused precipitation of Deferasirox at the same time 4-hydrazinobenzoic acid was completely extractable from the matrix.
The deduction of selecting basic additives in the diluents A and B that is first and second diluent is given ahead. First diluent-Diluent A brought about Deferasirox and 4-hydrazino benzoic acid to dissolve. As mobile phase contained triethylamine hence 6.5% triethylamine used as basic medium in first diluent. The role of acidic additive in second diluent diluent B was to precipitate large quantity of Deferasirox by neutralizing triethylamine. As perchloric acid is strong acid hence it is required in very less quantity to neutralize triethylamine without diluting the sample. Therefore, 70% perchloric acid was selected as second diluent.
In second diluent, Deferasirox was insoluble and the analyte 4-hydrazinobenzoic acid had excellent solubility. This ensued in removal of the matrix and at the same time optimum extraction of the analyte, resulting in proper peak shapes and good recovery in the range of 100.67% to 107% for 4-hydrazinobenzoic acid.
3. Results and Discussion
The developed HPLC method for the determination of 4- hydrazinobenzoic acid in Deferasirox tablet was validated. The linearity was assessed by preparing and analyzing six calibrators of 4-hydrazinobenzoic acid in the concentration range of 0.5 - 1.5 μg/ml. The slope, intercept and regression coefficient were determined by the least squares linear regression analysis. System precision was done by six replicated injections of the standard preparation.
The limit of quantitation (LOQ) was calculated on the basis of the lowest concentration of 4-hydrazinobenzoic acid that gives signal to noise ratio not less than (NLT) 10. The precision and accuracy were evaluated by spiking 4-hydrazinobenzoic acid and determining the % RSD < 5%. Stability of analytical solution was evaluated at 25˚C for 24 hours. Ruggedness study was performed by different scientist using different column by spiking analytes and determining the % RSD < 10%.
Validation was necessary before the application of the developed mass procedure to the commercial products. The developed method for the determination of 4-hydrazinobenzoic acid in Deferasirox Formulation was validated. The linearity was established by plotting the peak area counts of analytes versus concentration of analytes in the concentration range of 0.5 μg/ml to 1.5 μg/ml. Data is summarized in Table 2. The slope, intercept and regression coefficient were determined by the least squares linear regression analysis. Linearity correlations of the peak area counts and concentration of the analytes was achieved r2 = 0.999. This is represented graphically in Figure 2.
The LOQ was calculated on the basis of the lowest concentration of analytes that gives Signal to Noise ratio NLT 10 and for LOD NLT 3. LOD was found 0.16 μg/ml and LOQ was found 0.5 μg/ml. System precision was 2.2% for 4-hydrazinobenzoic acid. 4-hydrazinobenzoic acid was not found in the Deferasirox sample hence method precision and accuracy experiments were performed by spiking 4-hydrazinobenzoic acid and deter-
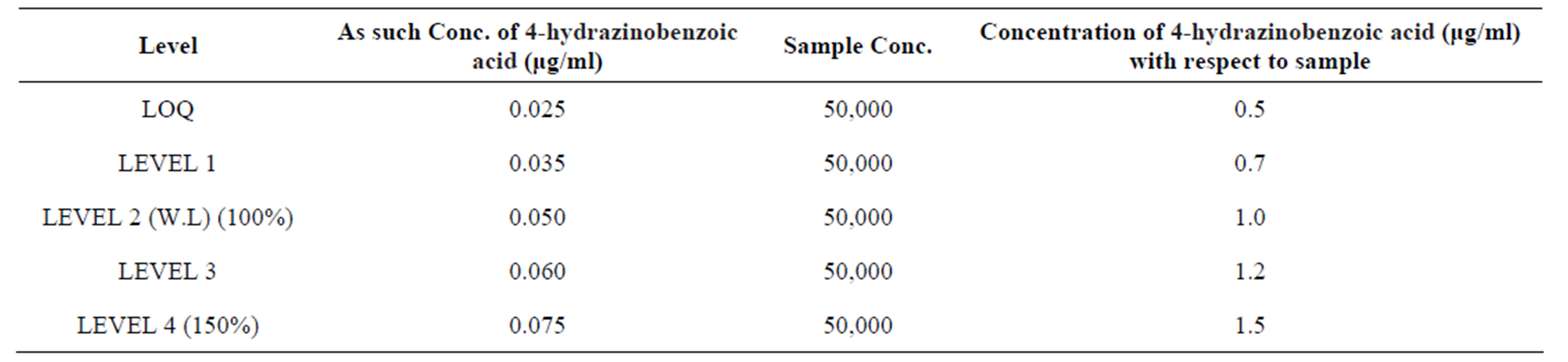
Table 2. Linearity of 4-hydrazino benzoic acid at different level.
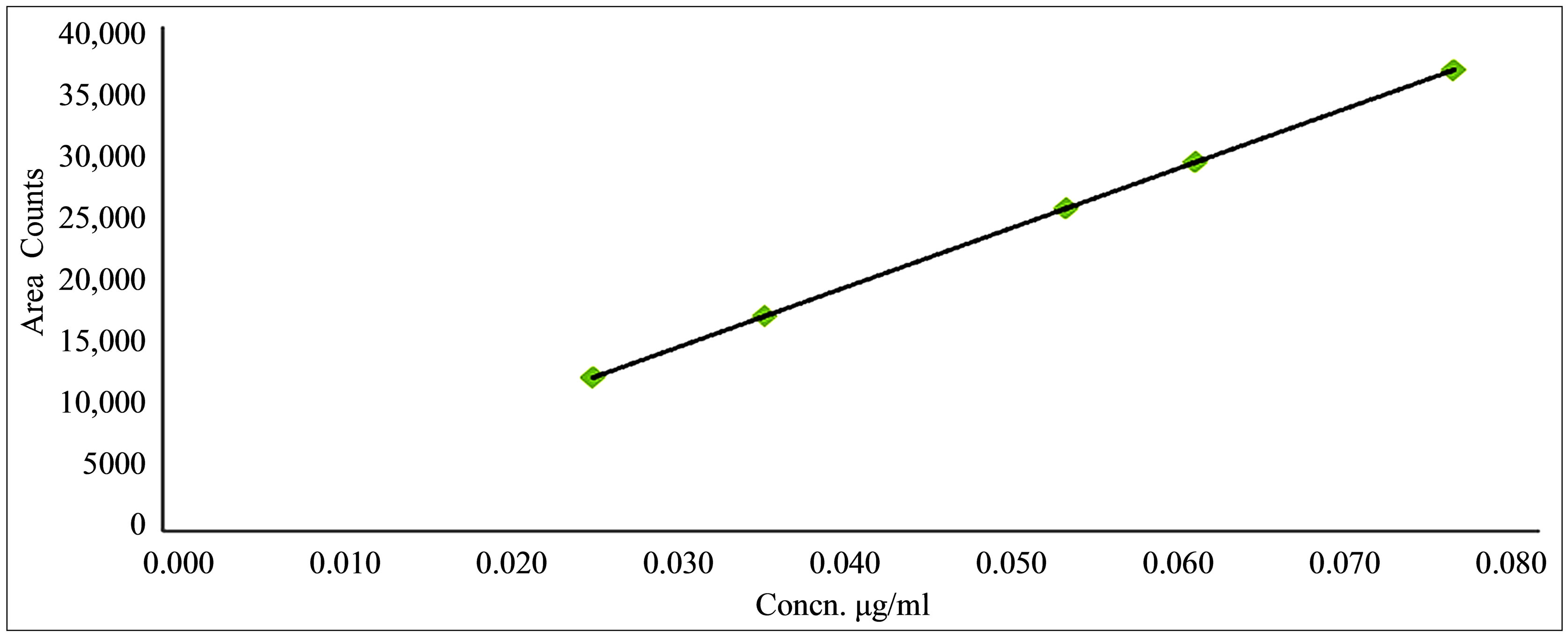
Figure 2. Linearity plot of 4-hydrazinobenzoic acid in the concentration range of 0.025 μg/ml to 0.075 μg/ml.
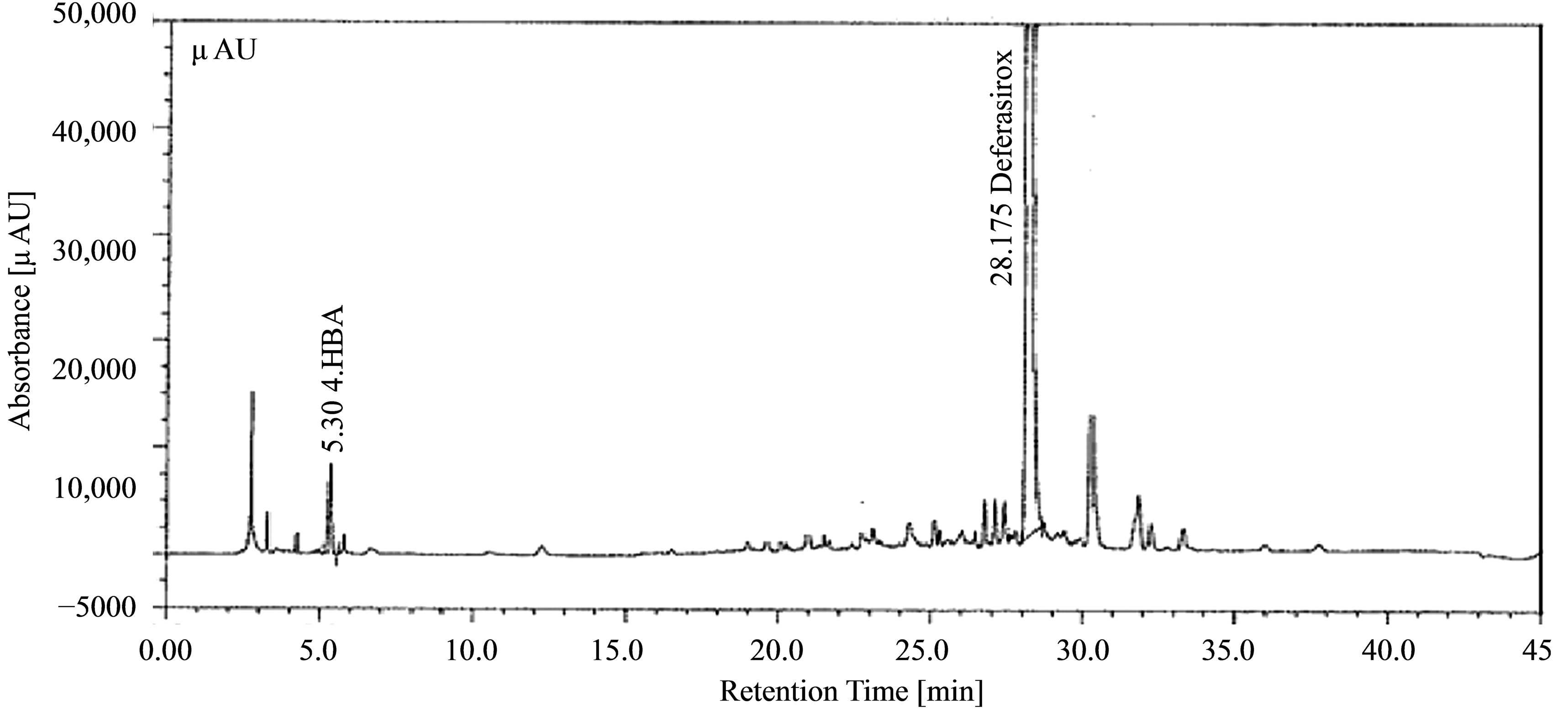
Figure 3. Accuracy of 4-hydrazinobenzoic acid in Deferasirox Formulation at Working Level.
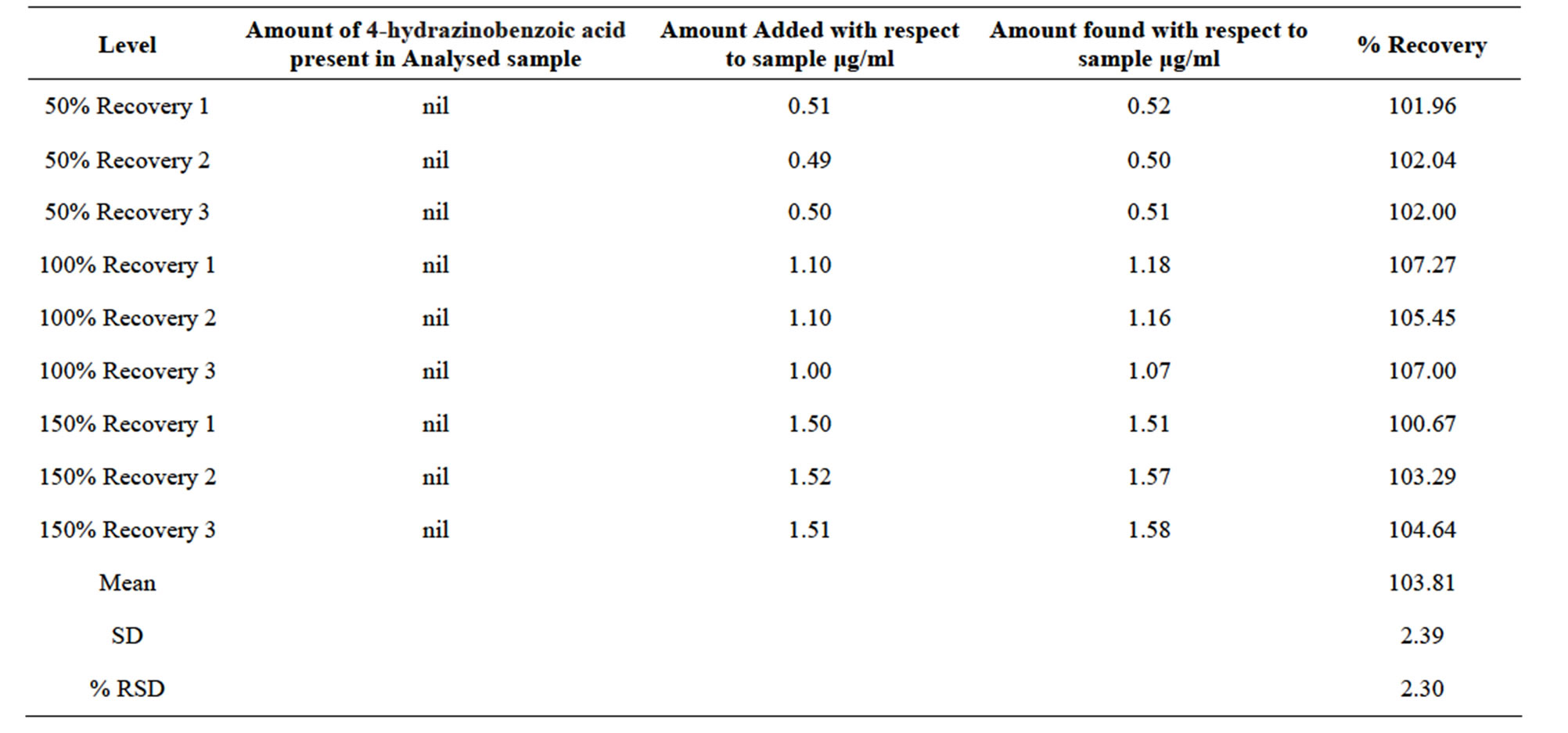
Table 3. Accuracy of spiking 4-hydrazinobenzoic acid at different level.
mining the % RSD. Recovery of the spiked amounts of analytes were calculated (Figure 3), the mean recovery percentages were observed to be in the range of 102% - 107% for 4-hydrazinobenzoic acid. Data is summarized in Table 3. Stability of analytical solution was evaluated at 25˚C for 24 hours and the solution was observed stable during this period.
Ruggedness study was performed by different scientist using different column. The % RSD for ruggedness study was observed as 2.5% for 4-hydrazinobenzoic acid. The derived values indicated good reproducibility and sensitivity of the method.
4. Conclusion
A method on HPLC was fabricated for screening and quantification of 4-hydrazinobenzoic acid in the Deferasirox Formulation. This HPLC method was cost effective, sensitive and specific for the detection of 4-hydrazinobenzoic acid in the Deferasirox Formulation. Furthermore, the method was accurate and reproducible for measurement of 4-hydrazinobenzoic acid. The high levels of 4-hydrazinobenzoic acid in the formulation might be dangerous if this product is not properly tested by drug quality control laboratories. The described method presents a highly reliable technique for rapid detection of genotoxic impurities in this formulation with accuracy and precision.
REFERENCES
- S. Glowienke, W. Frieauff, T. Allmendinger, H. Martus, W. Suter and L. Mueller, “Structure-Activity Considerations and in Vitro Approaches to Assess the Genotoxicity of 19 Methane-, Benzeneand Toluenesulfonic Acid Esters,” Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 581, No. 1-2, 2005, pp. 23- 24. http://dx.doi.org/10.1016/j.mrgentox.2004.10.004
- “Guidelines on the Limits of Genotoxic Impurities CPMP/ SWP/ 5199/02,” The European Medicines Agency [EMEA], Committee for Medical Products for Human Use [CHMP].
- I. Colon and S. M. Richoll, “Determination of Methyl and Ethyl Esters of Methanesulfonic, Benzenesulfonic and PToluenesulfonic Acids in Active Pharmaceutical Ingredients by Solid-Phase Microextraction (SPME) Coupled to GC/SIM-MS,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 39, No. 3-4, 2005, pp. 477-485. http://dx.doi.org/10.1016/j.jpba.2005.04.037
- H. G. Ramjit, M. M. Singh and A. B. Coddington, “Gas Chromatographic/Mass Spectrometric Analysis of Methyl Methanesulphonate and Ethyl Methanesulphonate in the Bismesylate Salt of DPI 201-106, a Positive Inotropic Agent for the Treatment of Heart Failure,” Journal of Mass Spectrometry, Vol. 31, No. 8, 1996, pp. 867-872. http://dx.doi.org/10.1002/(SICI)1096-9888(199608)31:8<867::AID-JMS362>3.0.CO;2-Q
- S. Harikrishna, K. Nagabhusana Reddy, Y. Suresh, M. Ramkrishna, M. Sarat, K. Kishore and C. Rambabu, “Low-level Determination of 4-Hydrazino BenzoicAcid in Drug Substance by High Performance Liquid Chromatography/Mass Spectrometry,” E-Journal of Chemistry, Vol. 7, No. 2, 2010, pp. 403-408. http://dx.doi.org/10.1155/2010/616494
- S. Steinhauser, U. Heinz, M. Bartholomä, T. Weyhermüller, H. Nick and K. Hegetschweiler, “Complex Formation of ICL670 and Related Ligands with FeIII and FeII,” European Journal of Inorganic Chemistry, Vol. 2004, No. 21, 2004, pp. 4177-4192. http://dx.doi.org/10.1002/ejic.200400363
NOTES
*Corresponding author.