Journal of Analytical Sciences, Methods and Instrumentation
Vol.3 No.3A(2013), Article ID:37131,10 pages DOI:10.4236/jasmi.2013.33A001
A Rapid Method for Determination of Some Phenolic Acids in Brazilian Tropical Fruits of Mangaba (Hancornia speciosa Gomes) and Umbu (Spondias tuberosa Arruda Camara) by UPLC
![]()
1Laboratory of Flavor and Chromatographic Analysis, Federal University of Sergipe, São Cristóvão, Brazil; 2Instituto Federal de Educação, Ciência e Tecnologia da Bahia, Canela, Brazil; 3Department of Chemistry, R. D. National College, Bandra, Mumbai, India.
Email: *nishanigam@gmail.com
Copyright © 2013 Edelvio de Barros Gomes et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received July 7th, 2013; revised August 25th, 2013; accepted September 8th, 2013
Keywords: Antioxidants; Bioactive Compounds; Tropical Fruits; SPE; UPLC-DAD
ABSTRACT
A rapid chromatographic method for the determination of six phenolic acids (chlorogenic, ferulic, gallic, p-coumaric, protocatechuic and vanillic acids) by ultra performance liquid chromatography (UPLC), was developed and applied for Brazilian tropical fruits mangaba (Hancornia speciosa Gomes) and umbu (Spondias tuberosa Arruda Camara). A multivariate statistical experimental design was employed to optimize analytical conditions (solvent A, solvent B concentrations and flow rates). Samples were cleaned-up by solid-phase extraction (SPE) with different solvents (methanol and acetone) employing SPE cartridges (amine and octadecyl-silane). The method using dihydrogen potassium phosphate 5 mmoL as solvent A and 8% acetonitrile as solvent B presented limits of detection varying from 14 to 94 ng∙mL−1, limits of quantification from 39 to 277 ng∙mL−1 with 2 µL of injection volume while total run time for all six compounds was only 9.6 minutes. Higher recovery was obtained by extraction with methanol-acetone of 69.51% to 72.59% for protocatechuic acid and 69.58% to 126.31% for the chlorogenic acid. The concentrations of chlorogenic, p-coumaric and ferulic acids in mangaba extracts were 113.4, 32.1 and 1.5 µg∙g−1, respectively while concentrations of chlorogenic, protocatechuic, gallic, vanillic, ferulic and p-coumaric acids present in umbu fruit were 118.9, 141.3, 3.5, 2.5, 2.2 and 1.8 µg∙g−1, respectively.
1. Introduction
Evidences suggest that a diet rich in fruits and vegetables may decrease the risk of chronic diseases, due to the presence of high concentrations of antioxidant compounds, such as phenolic acids [1]. These compounds are widely spread throughout the plant kingdom, occurring in the forms of esters, ethers or free form, and are wellknown for their bioactive properties such as antimutagenic, anticarcinogenic, anti-inflammatory and antimicrobial. In particular, some typical low molecular weight phenolic acids such as gallic, chlorogenic, protocatechuic (3,4 dihidroxy benzoic), p-coumaric, vanillic and ferulic acids, have been reported to have some of these properties [2].
There are extensive studies describing techniques for determination of phenolic acids in food, including UV spectrophotometry [3], gas chromatography [4], thinlayer chromatography [5] and high-performance liquid chromatography [6,7]. The last, carried out in reversedphase mode in conventional high pressure liquid chromatographic system (RP-HPLC) is the most common technique employed and in which the octadecyl-silane (C18) is used as the stationary phase, and diode array as the detector [8]. Mobile-phase, stationary phase and elution system (gradient or isocratic mode) conditions were optimized in developing a RP-HPLC method [9]. The elution systems are usually binary with an aqueous acidified polar solvent (the solvent “A”—responsible for the minimization of peak tailing) such as aqueous acetic acid, perchloric acid, trichloroacetic acid, trifluoracetic acid, phosphoric acid, dihydrogen-potassium phosphate or formic acid; and a less polar organic solvent (the solvent “B”) such as methanol or acetonitrile [10].
In general, the RP-HPLC determinations are timeconsuming and this is attributed largely due to the gradient elution mode. Isocratic elution mode, on the contrary, is advantageous, since it tends to be accomplished in a single chromatographic run, with economy of solvent. Additionally, in the RP-HPLC systems, acetonitrile has been preferentially employed as the solvent “B” due to its low viscosity and excellent mixing capacity with water. Nevertheless, the recent worldwide scarcity of acetonitrile caused dramatic price increases, requiring for minimized usage of this solvent [11]. Moreover analysis in RP-HPLC conventional system requires more time when compared to analysis performed in reversed-phase ultra performance liquid chromatographic (RP-UPLC) system.
Few reports using isocratic RP-HPLC for the determination of phenolic acids in fruits have been published [12,13], and there is just one report describing the development of isocratic method using RP-UPLC system for the simultaneous determination of phenolic acids (gallic, 3,5 dihydroxybenzoic, protocatechuic, chlorogenic, gentisic, 4-hydroxybenzoic, caffeic, vanillic, syringic, 3-hydroxybenzoic, coumaric, sinapic, ferulic, salicylic and trans-cinnamic acids) in beverages using massspectrometer as the detector [14]. However to the best of our knowledge, an isocratic RP-UPLC method using diode array as detector for determining phenolic acids in fruits has not been reported.
In this paper our objective was to develop an isocratic method for rapid determination of phenolic acids such as chlorogenic, ferulic, gallic, p-coumaric, protocatechuic and vanillic acids using RP-UPLC system and to analyze their concentrations in Brazilian tropical fruits of mangaba and umbu. Differents mobile phases at varying flow-rates were used and evaluated to optimize analytical conditions. The method was validated by evaluating selectivity, linear range and limits of detection, quantification and recovery of the phenolic acids.
2. Experimental
2.1. Fruits
Ripe fruits used in this study were purchased from the Central Market in the city of Aracaju, Sergipe state, Brazil. Two tropical fruits, largely cultivated in this region, were selected: mangaba (Harcornia speciosa Gomes) and umbu (Spondia stuberosa Arruda Camara).
2.2. Standards, Solvents and Reagents
All standards of phenolic acids as well as the uracil standard were purchased from Sigma-Aldrich. (St Louis, MO). Phenolic acids standards were: chlorogenic, ferulic, gallic, p-coumaric, protocatechuic and vanillic acid. Reagents (hydrochloric acid, dihydrogen-potassium phosphate, trichloroacetic acid and trifluoroacetic acid) and solvents (acetonitrile, acetone and methanol) were of HPLC grade and these were purchased from Vetec Ltd (Duque de Caxias, Brazil). Dihydrogen-potassium phosphate was used at 5 mmoL∙L−1, and both trichloroacetic and trifluoroacetic acids were used at 1%v∙v−1 concentration. All standards were dissolved in methanol. The LC grade water was obtained from milliQ water purification system (Millipore, Bedford, MA, USA).
2.3. UPLC Analysis
Analyses were performed using a Prominence Ultra Performance Liquid Chromatographic system (Shimadzu Inc., Japan) which comprised of a binary pump (model LC-20AD), diode array detector (model SPD-M20A), auto-sampler (model SIL-20A HT), oven (model CTO- 20A), controller (model CBM-20A), degasser (model DGU-20A3). The LC-Solution software version 1.2 was used to control the auto-sampler, detector, data acquisition and run settings. A C18 reverse-phase column, (Shimpack, Shimadzu Inc. Japan; 50 mm × 3.0 mm, 2.2 µm particle size) was employed for all analysis at a temperature of 40˚C. Peak areas were determined at 254 nm for the gallic, vanillic and protocatechuic acid and 310 nm for the chlorogenic, ferulic and p-coumaric acids.
2.4. Parameters Optimization
A three-factor full factorial design (FFD) was used to describe the relationship between the response function and the process variables, as well as to determine the conditions that influenced the retention time. The independent variables studied were: solvent “A” (dihydrogen-potassium phosphate, trichloroacetic acid and trifluoroacetic acid); flow rates of mobile phase (X1: 0.4; 0.5 and 0.6 mL∙min−1) and solvent “B” (X2: 8%, 10% e 12%v∙v−1 of acetonitrile) while response variables were retention time of the six phenolic acids (gallic, chlorogenic, protocatechuic, p-coumaric, vanillic and ferulic: Y1, Y2, Y3, Y4, Y5, Y6, respectively). Twenty-seven randomized experiments were performed, based on FFD parameters as cited in Table 1. Solvent “A” solutions were prepared using deionized water and pH was adjusted to 2.4.

Table 1. Conditions of full factorial design of 33 detailing independent variables, their codes and values.
2.5. Data Interpretation and Statistical Analysis
Results of statistical factorial design experiments were computed using Statistica software version 7.0 (Microsoft, Tulsa-Oklahoma, USA). Statistical models generated were re-parameterized considering only statistically significant effects (p < 0.05). The influence of the three independent variables, were primarily based on the statistical analysis of retention time (Y) as response-variable at (p < 0.05). Additionally, parameters such as number of efficient theoretical plates (Nef) [15] resolution between critical adjacent peaks (RS) and minimal usage of acetonitrile were also considered in optimizing the analytical conditions.
2.6. Sample Preparation and Clean-Up
For sample preparation, pulp weight of about 2 g was used. The SPE cartridges used for clean-up of samples were a Strata C18-E cartridge 500 mg-6 mL and a Strata NH2 cartridge 500 mg - 6 mL (PhenomenexTM, TorranceCalifornia, USA). Two sets of samples were prepared. The first one, non-fortified sample—without any addition of phenolic acids, while the second set was fortified (addition of 0.66 µg of each standard of phenolic acids before elution). The cartridges were conditioned with 10 mL of methanol, followed by 10 mL of distilled water. Both sets of samples were directly loaded onto the cartridges, washed and followed by elution. Both cartridges were eluted with 2 mL of 0.1 mol∙L−1 of HCl, and then continuously eluted with 10 mL of methanol. Additionally, another sequence of solvents was used being 10 mL of methanol and 2 mL of acetone. These eluates were collected in a flask and then evaporated under vacuum in a rotary evaporator at 40˚C to dryness. Each dried extract of sample was re-suspended in 2 mL of methanol.
2.7. Identification and Quantification of Phenolic Acids
A volume of 2 µL of the extract prepared as described in item 2.6 was injected in UPLC System (item 2.3). The identification of phenolic acids in the samples was carried out by comparing their retention times and the spectra obtained from those of the standards injected at identical analytical conditions. The quantification of all phenolic acids was done based on the peak area of the compounds related to the calibration curves prepared from injecting the standard solutions.
2.8. Method Validation
2.8.1. Selectivity, Sensitivity and Linearity
Linear least-squares regression was employed to calculate the slope, intercept and correlation coefficient. The calibration graphs were constructed by plotting each chromatographic peak area against its corresponding concentration. Selectivity was evaluated by comparing the chromatograms of sample extracts to those of standards and possible interferences were investigated considering shapes of peaks, retention time and absorption spectra. Additionally, blank chromatogram at selected wavelengths (254 nm for gallic, vanillic and protocatechuic acid and 310 nm for chlorogenic, p-coumaric and ferulic acid), was used to confirm nonexistence of these compounds or any other interferences. The method sensitivity was determined by the angular coefficients (slope) of the calibration graphs for each phenolic acid. The linearity of the detector responses for the studied compounds was represented by the linear regression coefficient (r2).
2.8.2. Limit of Detection (LOD) and Limit of Quantification (LOQ)
Peaks which produced signals between 3 and 10 times the amplitude of noise were selected and LOD calculated by using the equation: 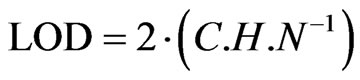 where, C is the amount of analyte which produced such signal; H is the height of the peak; and N is the amplitude of noise. The quantification limits (LOQ) were stated as the 3 times the limit of detection (LOQ = 3 × LOD) and these were also determined experimentally by using varying concentrations of phenolic acids [16].
where, C is the amount of analyte which produced such signal; H is the height of the peak; and N is the amplitude of noise. The quantification limits (LOQ) were stated as the 3 times the limit of detection (LOQ = 3 × LOD) and these were also determined experimentally by using varying concentrations of phenolic acids [16].
2.8.3. Recovery
The extracts were spiked with known amounts of each standard in triplicate and analyzed. The recovery was determined as the quotient of the determined concentration of phenolic acids (after chromatographic analysis) per concentration added of individual standards. Results were expressed in percentage.
3. Results and Discussion
The variables such as flow rate (X1) and the usage of solvent B (X2) were selected based on preliminary studies, which indicated that flow rates higher than 0.6 mL∙min−1 implied in lower resolution between all contiguous pairs of peaks (Rs < 1.3). On the other hand, flow rates lower than 0.4 mL∙min−1 extended total retention time to more than 15 minutes which was an undesirable feature in this work as it aimed for shorter retention times. Furthermore based on preliminary studies, concentrations of solvent B (acetonitrile) lower than 8% also extended total retention time to more than 15 minutes, and values higher than 12% decreased resolution to undesirable values (Rs < 1.3). The effect of independent variables (flow rate and percentage of solvent B) was evaluated by retention time data for all six phenolic acids at a significant level of p < 0.05 and statistical data analysis results are presented in Table 2.
3.1. Optimal Analytical Conditions
The statistical models reported the influence of the variables on response-factor by the second-order polynomial regression equation. For retention time (Y), effect of flow rate (X1) and percentage of solvent “B” (X2) were significant (p < 0.05) in first-order linear effect and second-order quadratic effect (Table 2).
Based on the conditions predicted by the statistical model, Figure 1 shows flow rate and solvent “B” (acetonitrile) range that promoted lower retention times for chlorogenic acid. Dark-green colour indicates the optimal region representing flow rate of 0.6 mL∙min−1 and the usage of 12% acetonitrile. Further experiments were performed to determine resolution, using 8%, 10% and 12% of acetonitrile.
Differences among three solvents “A” (KH2PO4, TCA and TFA) as well as, interactive effects of all independent variables were not statistically significant (p < 0.05). Moreover the present study reveals that the retention time was not affected by using different solvents “A” (Tables 3 - 5).
Since the retention time response was almost similar for the three solvents A (KH2PO4, TCA and TFA), KH2PO4 was selected as solvent A over other acids tried in this study.

Table 2. Polynomial equations and statistical parameters (p < 0.05) calculated from full factorial experimental design experiments.

Figure 1. Contour surface plot showing the combined effect of flow rate (X1) and percentage of acetonitrile (X2) on the retention time of chlorogenic acid (Y2).
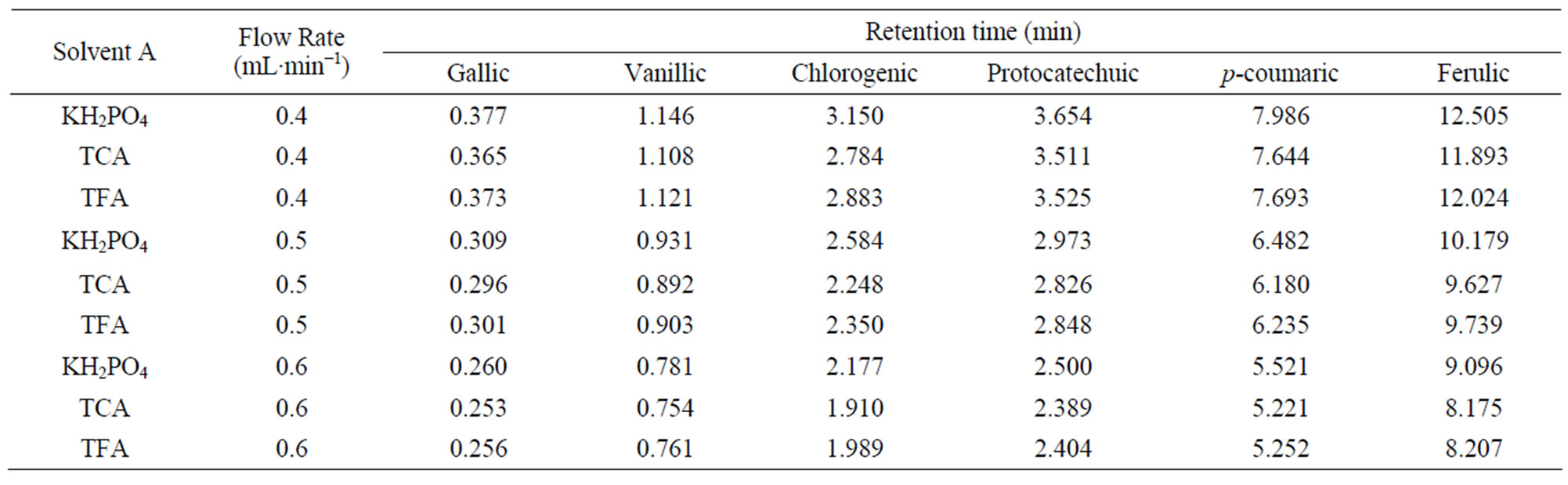
Table 3. Retention time data of phenolic acids for three solvents “A” (KH2PO4, TCA and TFA) and solvent “B” (8% acetonitrile) at different flow rates.
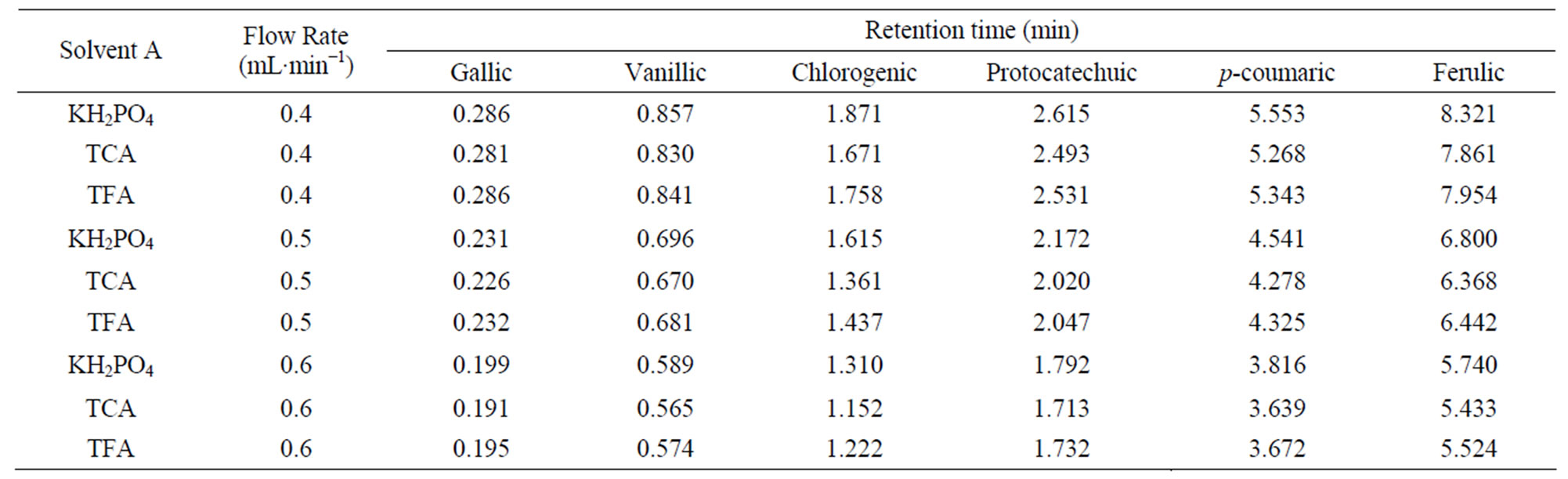
Table 4. Retention time data of phenolic acids for three solvents “A” (KH2PO4, TCA and TFA) and solvent “B” (10% acetonitrile) at different flow rates.
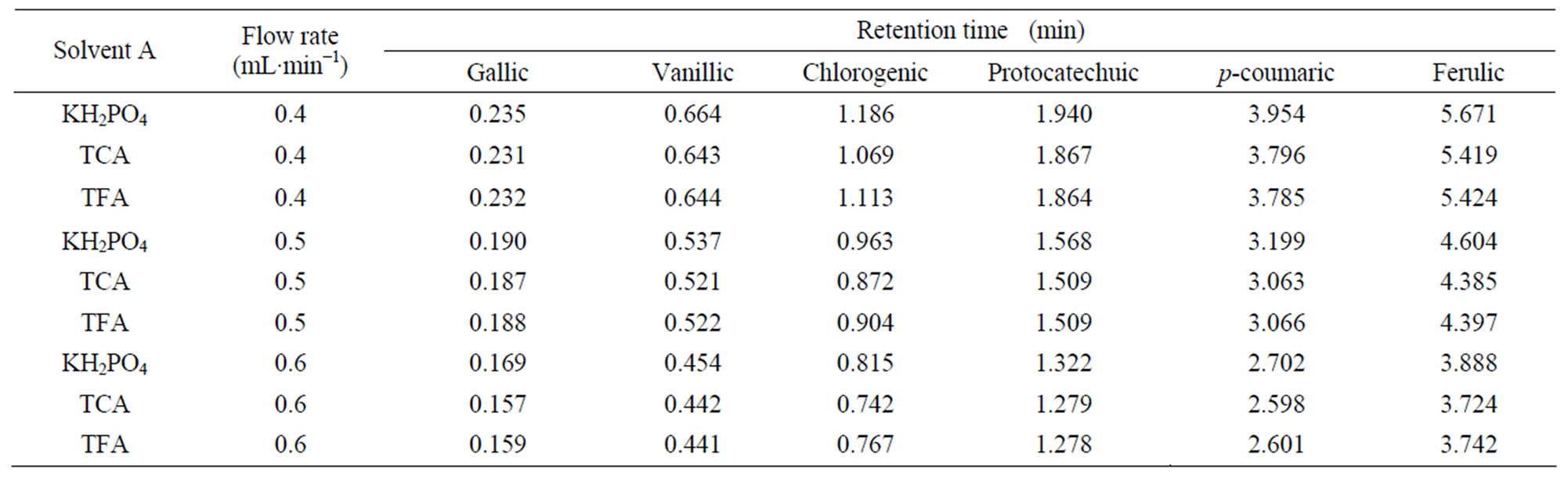
Table 5. Retention time data of phenolic acids for three solvents “A” (KH2PO4, TCA and TFA) and solvent “B” (12% acetonitrile) at different flow rates.
3.2. Usage of Lower Concentration of Acetonitrile
The lower concentration of 8% acetonitrile promoted minimal satisfactory resolution (RS) for a critical pair of peaks (RS = 1.3 for the pair chlorogenic/protocatechuic acid) and high Nef for the analytes (Tables 6 and 7).
Acetonitrile as solvent “B” (X2) had a greater influence on retention time of all phenolic acids. Increases in less-polar component of mobile phase in RP-HPLC/ UPLC systems, tend to dislocate the analyte toward mobile phase, resulting in decreased retention time of compounds. There were sharp and well separated peaks for all phenolic acids even when acetonitrile concentration was 8%. Figure 2 shows these chromatograms along with those obtained with the usage of 12% acetonitrile. It
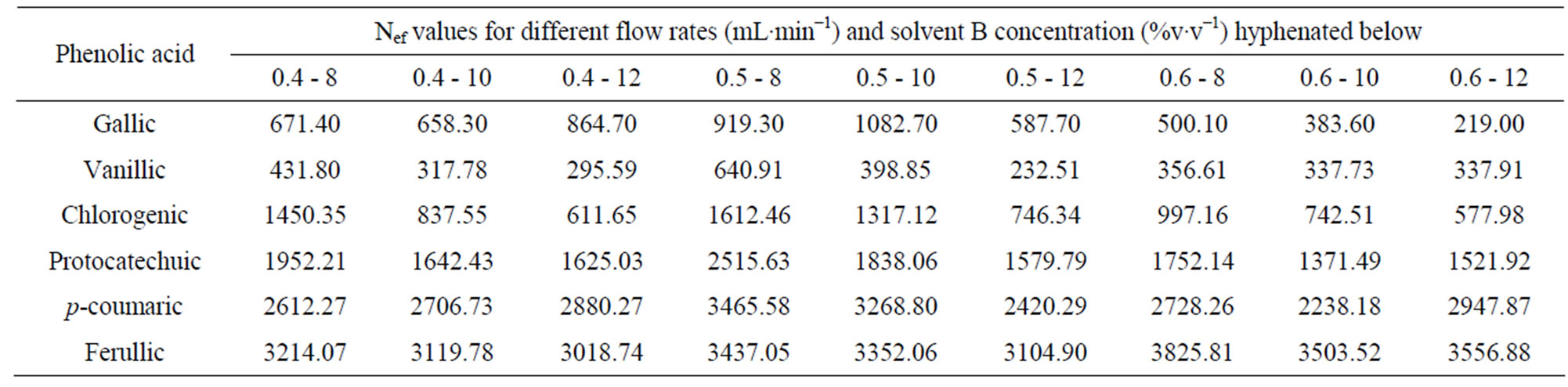
Table 6. Number of efficient theoretical plates (Nef) of phenolic acids in different combinations of flow rate and percentage of solvent B (acetonitrile) using KH2PO4 as the solvent A.
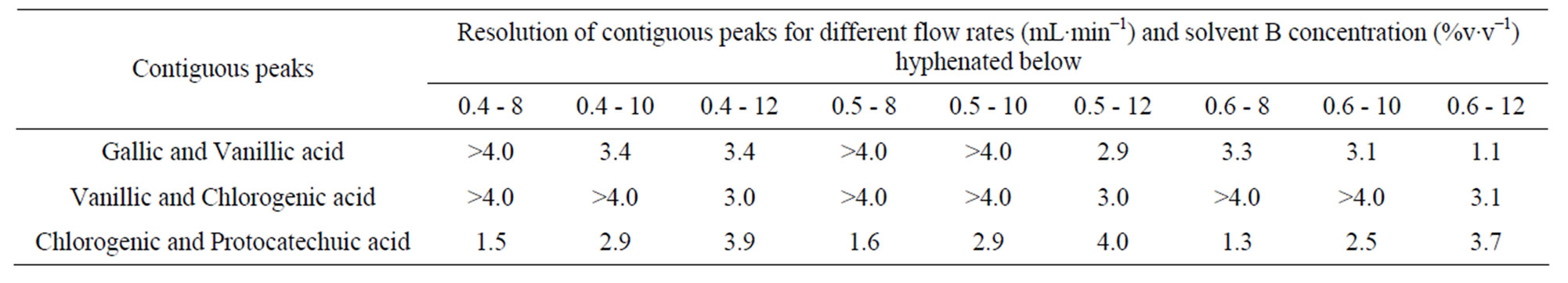
Table 7. Resolution of contiguous peaks of phenolic acids in different combinations of flow rate and percentage of solvent B (acetonitrile) using KH2PO4 as the solvent A.
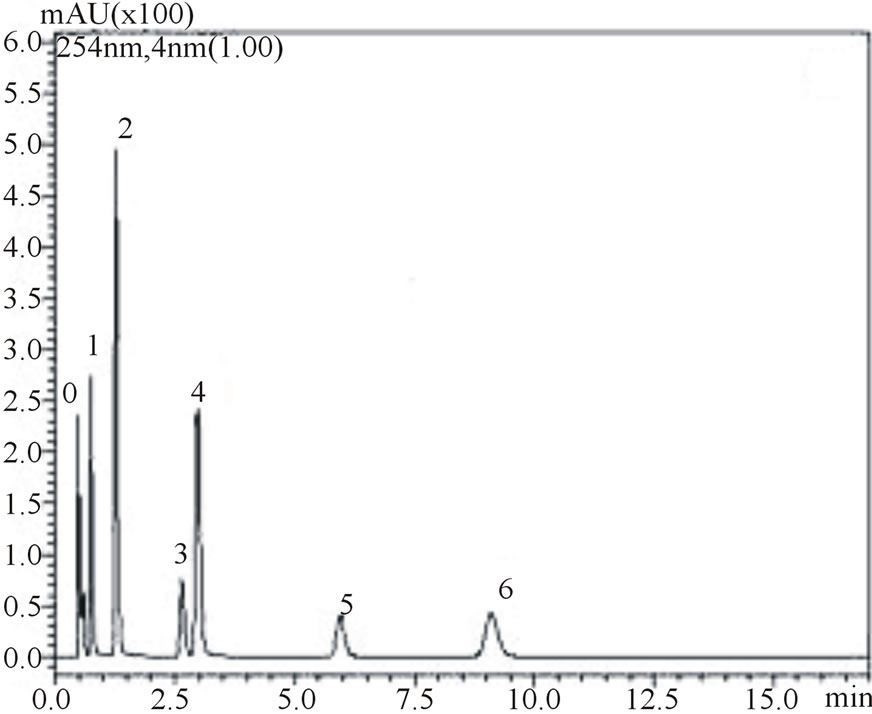
is noteworthy from the figures that gallic, vanilic and protocatechuic acids (peaks 1, 2 and 4, respectively) are larger (Figure 2(a)) when measured at 254 nm while chlorogenic, ferulic and p-coumaric acids (peaks 3, 5 and 6, respectively) possess larger areas at 310 nm measurement (Figure 2(b)). Similar behavior was observed in the flow rate variable. Flow rate of about 0.6 mL∙min−1 promoted decrease in retention times while values in the flow rate such as 0.4 mL∙min−1 resulted in higher retention times of phenolic acids. Based on these results the conditions for the identification of all six phenolic acids were optimized: Usage of KH2PO4 5 mmol∙L−1 as solvent A; flow rate of 0.6 mL∙min−1, 8% acetonitrile as usage of solvent B.
Amakura et al [12] used 15% to 20% of acetonitrile as solvent B for the separation of five phenolic acids (gallic, chlorogenic, caffeic, ellagic and ferulic acids) in grape, apple, pomegranate and prune fruit juices. However, in this work in order to reduce the cost of using acetonitrile, and at the same time get satisfactory results, acetonitrile concentration was reduced from 12% to 8%v∙v−1, and even at this concentration, the peaks were distinct and quite well-separated.
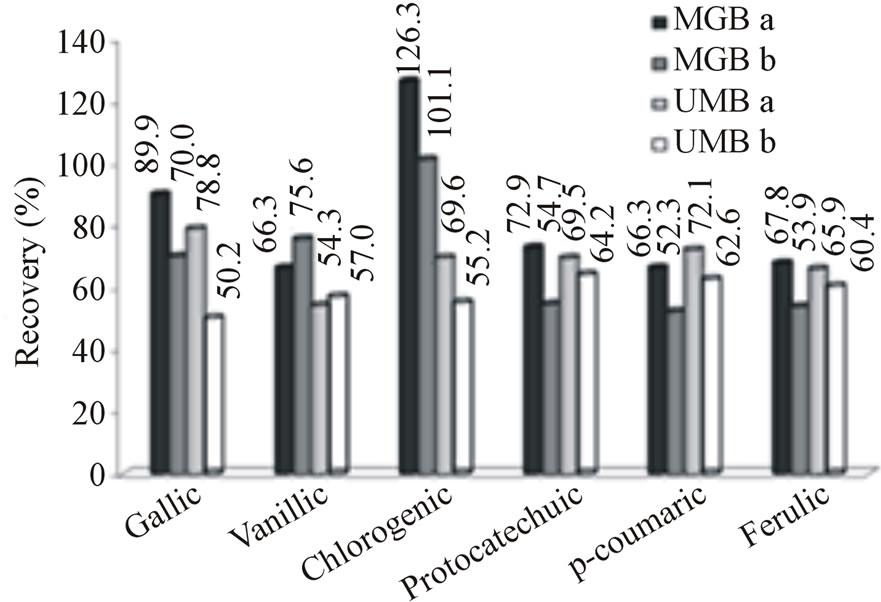
3.3. Clean-Up
All phenolic acids except gallic acid, presented recovery percentage higher or equal to 82.3% with Strata C18-E cartridge (data not shown). A Strata NH2 cartridge was employed to select gallic acid after elution of sample with Strata C18-E cartridge and, as a result, best recovery (89.9%) of gallic acid was observed after using this procedure. Gallic acid recovery using cartridge containing amino group (Strata NH2), was 89.9% and 70.0% for the mangaba pulp with and without the usage of acetone in extraction step, respectively, and for the umbu pulp it was 78.9% and 50.2%, respectively (Figure 3(a)). These results are in agreement with those obtained by Amakura et al [12] who utilized combined Sep-Pak Plus C18 and Bond Elute (NH2) cartridges. Phenolic acids investigated in current study belong to two main subgroups: hydroxybenzoic acids (gallic, protocatechuic and vanillic acids); and hydroxycinnamic acids or its esters (ferulic, p-coumaric and chlorogenic acids) [17]. Cartridges containing linked amino groups are best suited to separate polar compounds in organic mixtures and the gallic acid molecule is the most polar compound among all the hydroxybenzoic acids tested in current work.
3.4. UPLC Method Validation
The standardized method by optimizing analytical conditions was evaluated for the determination of 6 phenolic acids in mangaba and umbu fruit pulps. The chromatograms were evaluated for selectivity, linearity, limits of quantification and recovery of each of the phenolic acids under study. The selectivity was assessed by comparing the chromatograms of the extracts from fruit pulps to those of the standards phenolic acids. Peak shapes, retention time and spectral purity of the peaks was considered for evaluation of possible interferences. No interfering peaks were found in the blank chromatograms at wave-
 (a)
(a) (b)
(b)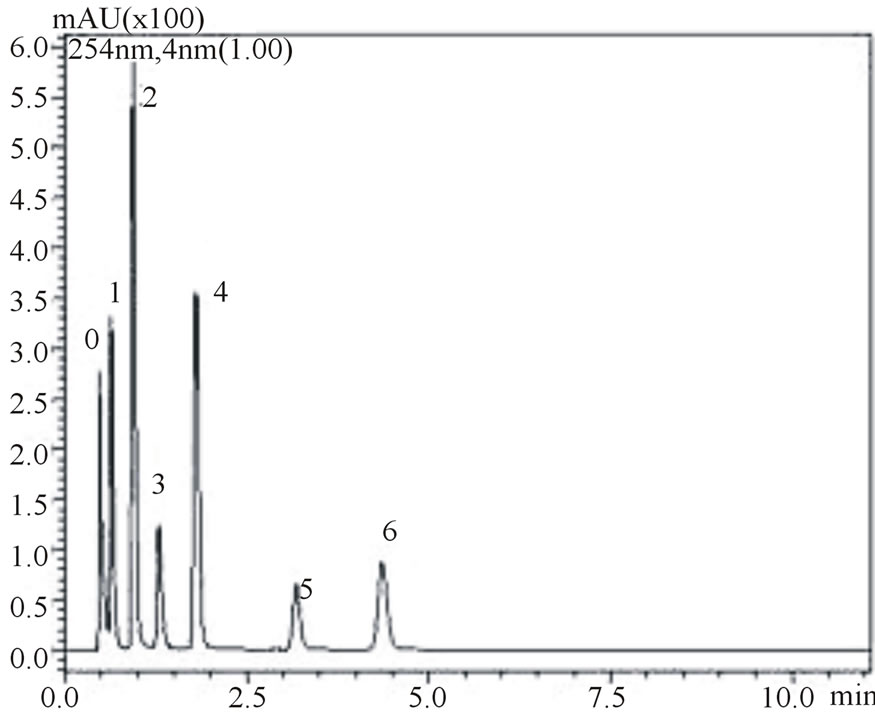 (c)
(c)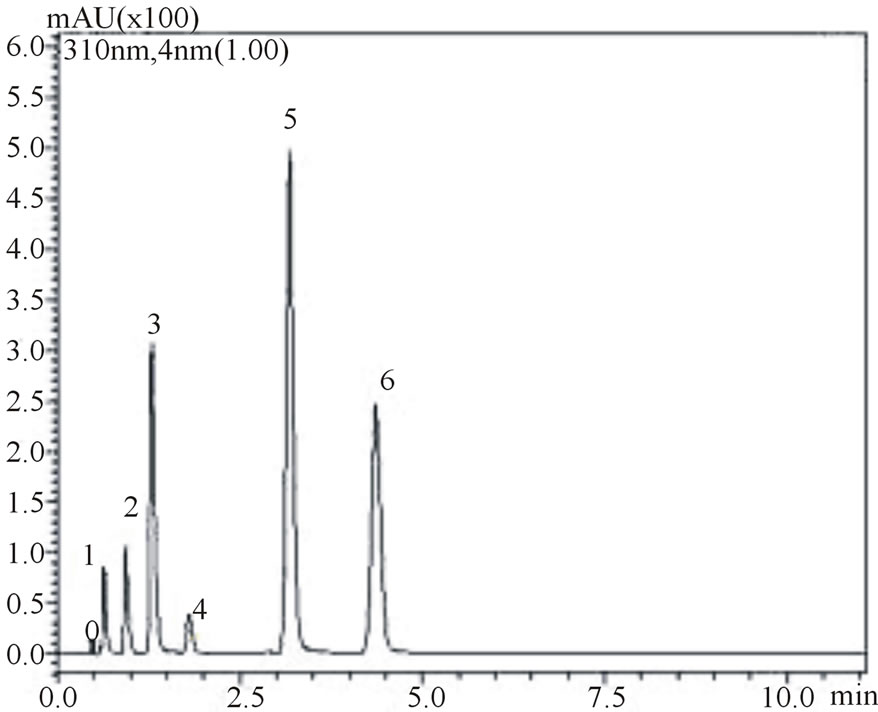 (d)
(d)
Figure 2. Chromatograms demonstrating the effect of acetonitrile concentration on phenolic acids retention times, at 0.6 mL∙min−1 flow rate: 8% acetonitrile at 254 nm (a) and 310 nm (b) wave lengths; 12% acetonitrile at 254 nm (c) and 310 nm (d) wavelengths. The peak numbers: 0—uracil, 1—gallic acid, 2—vanillic acid, 3—chlorogenic acid, 4—protocatechuic acid, 5—ferulic acid and 6—p-coumaric acid.
 (a)
(a) (b)
(b)
Figure 3. (a) Recovery percentage of phenolic acids in fortified pulps of mangaba and umbu with and without usage of acetone at the extraction step, respectively. (b) Concentration of phenolic acids in non-fortified pulps of mangaba and umbu using optimal extraction condictions. Codes MGBa and MGBb represent mangaba fruit pulp extracted with and without acetone, respectively; UMBa and UMBb represent umbu fruit pulp extracted with and without acetone, respectively.
lengths used for measurement of phenolic compounds.
3.4.1. Linearity
The data on linearity and sensitivity for phenolic acids are presented in Table 8. Linearity did not differ statistically (p < 0.05) for gallic, protocatechuic and chlorogenicacids in which correlation coefficients were higher than 0.997 for all phenolic acids. The p-coumaric acid curve showed highest value of sensitivity among all phenolic acids (Table 8). The concentration of all standards varied in the range 0.5 - 2.8 µg∙mL−1 and good linearity was achieved for all phenolic acids in this range.
3.4.2. Limits of Detection (LOD) and Quantification (LOQ)
Detection limits for various phenolic acids varied from 14 to 94 ng∙mL−1 (Table 8). These values are in accordance, and comparable with the results obtained by Shahrzad and Bitsch [13] and Amakura et al [12] who developed an isocratic method to quantify five phenolic acids (gallic, chlorogenic, caffeic, ellagic and ferulic acid) out of which three phenolic acids being gallic, chlorogenic and ferulic acids were subject of study in this work. However, they did not analyze protocatechuic, p-coumaric and vanillic acids in fruit juices (apple, pomegranate and prune). The results obtained for the quantification limits show that the optimized method is sensitive enough for determination of the phenolic acids in mangaba and umbu fruit pulps.
3.4.3. Recovery
Addition of acetone in extraction step enhanced the recovery of chlorogenic acid in both fruit pulps (Figure 4). This augmentation is thought to be due to the affinity of carbonyl group of acetone with the carboxyl group from chlorogenic acid. When the acetone was added after elution with methanol, chlorogenic acid recovery increased from 101.1% to 126.3% for mangaba, and from 55.2% to 69.6% for umbu (Figure 3(a)). Percentages of recovery for various phenolic acids ranged from 66.3% to 126.3% for mangaba pulp when it was extracted with 2 solvents (methanol and acetone) and relatively lower values (52.3% to 101.1%) were obtained when pulp was extracted only with methanol. For the umbu extract recoveries ranged from 65.9% to 78.8% when methanol and acetone was employed in extraction and from 54.2% to 64.2% with methanol only (Figure 3(a)).
The presence of different forms of phenolic acids requires suitable extraction conditions, and among all other aspects, the most important is being the polarity of extracting solvents. As a consequence, combined solvents are more effective for screening different classes of phenolic acids than the usage of only one solvent alone [18,19].
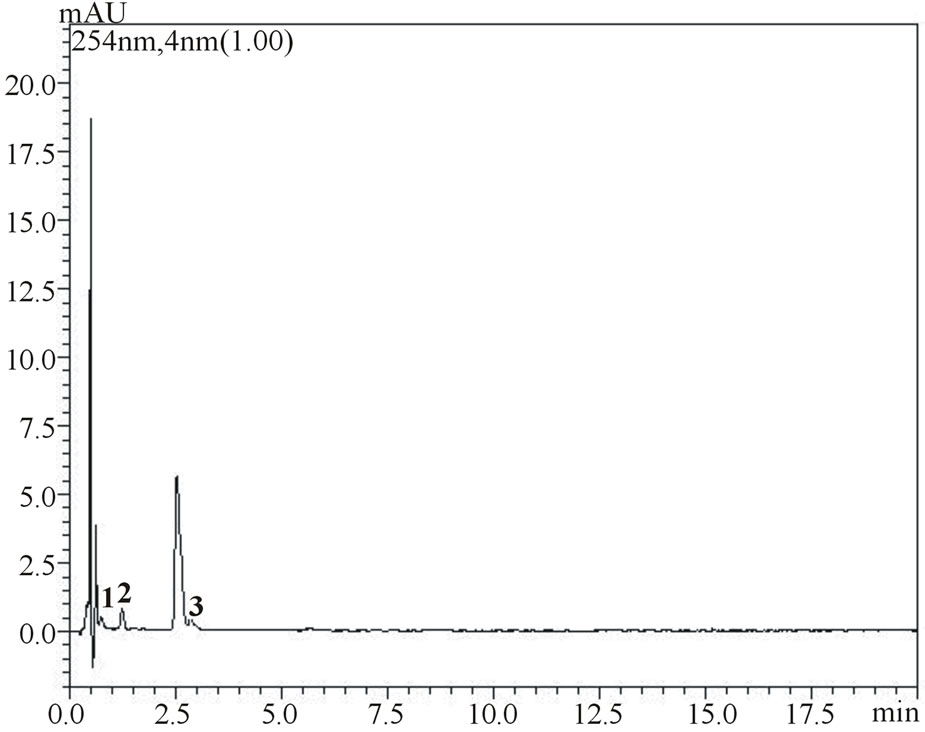
Figure 3(b) shows the concentrations of phenolic acids in non-fortified pulps of both fruits obtained under optimized extraction conditions, Figure 4 shows the chromatograms for phenolic acids measured at different wave lengths (254 nm for gallic, vanillic and protocatechuic acids and 310 nm for chlorogenic, p-coumaric and ferulic acids). Higher concentrations of protocatechuic and chlorogenic acid were obtained for umbu (141.3 and 118.9 µg∙g−1 of pulp, respectively) and higher concentrations of chlorogenic and p-coumaric acids concentrations for mangaba (113.4 and 32.1 µg∙g−1, respectively). Gallic, vanillic and protochatecuic acids were not detected in mangaba pulp and ferulic acid was present in low concentration (1.5 µg∙g−1). In umbu pulp, gallic, vanillic, p-coumaric and ferulic acids were found in lower concentrations being 3.5, 2.5, 1.8 and 2.2 µg∙g−1, respectively.
These experimental data indicate that the phenolic acid standards behaved in a similar way as to the sample extracts. Based on these results on various analytical parameters studied in the method development, the optimized extraction (usage of methanol-acetone) and analytical conditions at 0.6 mL∙min−1 flow rate and 8% of acetonitrile as solvent B could be considered suitable and adequate for the analysis of six phenolic acids in mangaba and umbu tropical fruit juices.
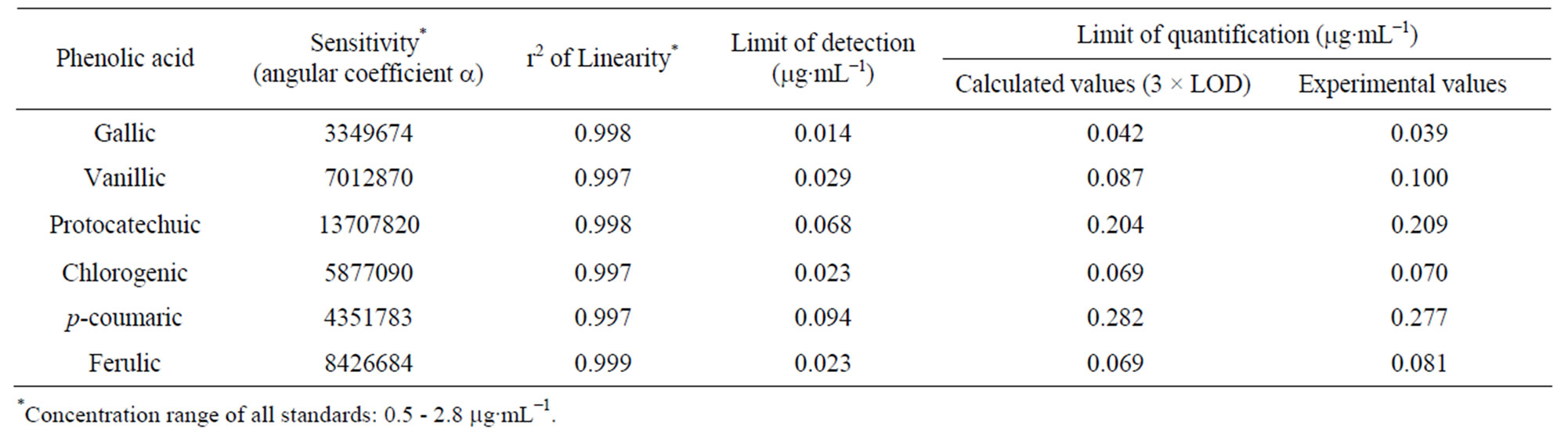
Table 8. Sensitivity, linearity, limits of detection and quantification of phenolic acids under optimized analytical conditions.
 (a)
(a)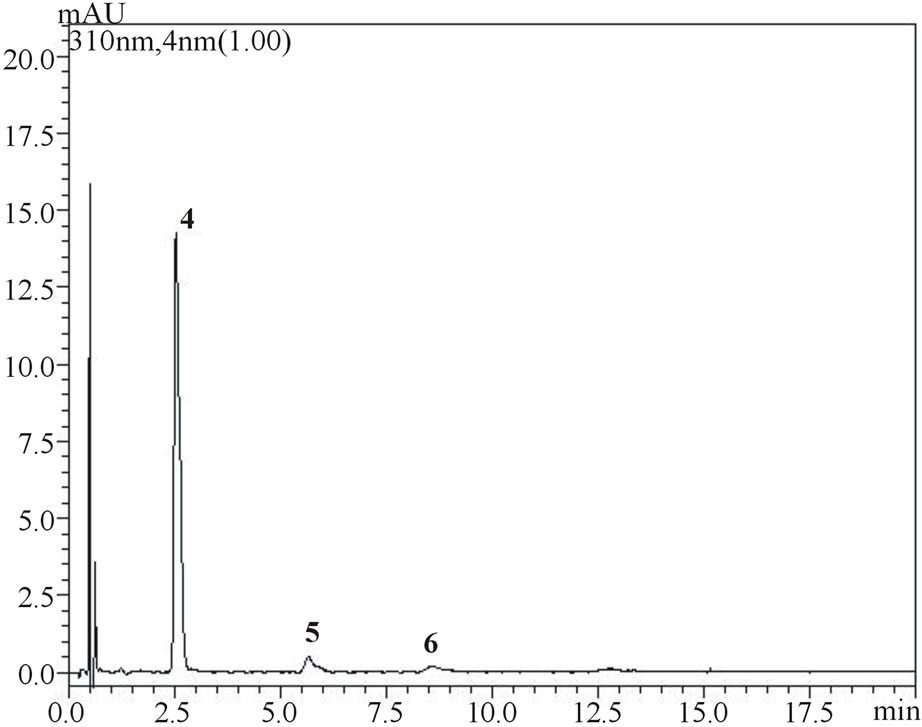 (b)
(b) (c)
(c)
Figure 4. Chromatograms showing separation of gallic, vanillic and protocatechuic acids (1, 2 and 3, respectively) and chlorogenic, p-coumaric and ferulic acids (4, 5 and 6, respectively) in samples of umbu (figure a measured at 254 nm and figure b measured at 310 nm) and mangaba (figure c measured at 310 nm) fruit pulps.
4. Conclusion
A rapid UPLC method was developed for separation, identification and quantification of six phenolic acids viz. gallic, chlorogenic, protocatechuic, p-coumaric, vanillic and ferulic acids in tropical fruits of mangaba and umbu. The experimental results indicate that mangaba pulp contains higher concentrations of chlorogenic, and p-coumaric acids which were 113.4 and 32.1 µg∙g−1, respetively while chlorogenic acid concentration in umbu pulp was equivalent, being 118.9 µg∙g−1. However, protocatechuic acid concentration was higher (141.3 µg∙g−1) in umbu pulp.
5. Acknowledgements
All authors thank the financial support provided from the project, National Institute of Science and Technology (INCT) for Tropical Fruits from CNPq, Brazil offered for promoting research work undertaken in this study. The co-authors (Edelvio and Rita) thank CAPES (Ministry of Education, Brazil) for providing fellowship to them.
REFERENCES
- WHO—World Health Organization, “Diet, Nutrition and the Prevention of Chronic Diseases: Report of a Joint WHO/FAO Expert Consultation,” WHO Reports, Rome, 2003. http://www.who.int/en/
- M. G. L. Hertog, P. C. H. Hollman and B. Van de Putte, “Content of Potentially Anticarcinogenic Flavonoids of Tea Infusions, Wines and Fruit Juices,” Journal of the Agriculture and Food Chemistry, Vol. 41, No. 8, 1993, pp. 1242-1246. http://dx.doi.org/10.1021/jf00032a015
- B. Lapornik, M. Prosek and A. G. Wondra, “Comparison of Extracts Prepared from Plant By-Products Using Different Solvents and Extraction Time,” Journal of Food Engineering, Vol. 71, No. 2, 2005, pp. 214-222. http://dx.doi.org/10.1016/j.jfoodeng.2004.10.036
- T. Y. Chu, C. H. Chang, Y. C. Liao and Y. C. Chen, “Microwave-accelerated Derivatization Processes for the Determination of Phenolic Acids by Gas ChromatographyMass Spectrometry,” Talanta, Vol. 54, No. 6, 2001, pp. 1163-1171. http://dx.doi.org/10.1016/S0039-9140(01)00392-7
- M. Nardini and A. Ghisell, “Determination of Free and Bound Phenolic Acids in Beer,” Food Chemistry, Vol. 84, No. 1, 2004, pp. 137-143. http://dx.doi.org/10.1016/S0308-8146(03)00257-7
- R. J. Robbins and S. R. Bean, “Development of a Quantitative High-Performance Liquid Chromatography-Photodiode Array Detection Measurement System for Phenolic Acids,” Journal of Chromatography A, Vol. 1038, No. 1-2, 2004, pp. 97-105. http://dx.doi.org/10.1016/j.chroma.2004.03.009
- R. Tsao and R. Yang, “Optimization of a New Mobile Phase to Know the Complex and Real Polyphenolic Composition: Towards a Total Phenolic Index Using HighPerformance Liquid Chromatography,” Journal of Chromatography A, Vol. 1018, No. 1, 2003, pp. 29-40. http://dx.doi.org/10.1016/j.chroma.2003.08.034
- R. J. Robbins, “Phenolic Acids in Foods: An Overview of Analytical Methodology,” Journal of the Agriculture and Food Chemistry, Vol. 51, No. 10, 2003, pp. 2866-2887. http://dx.doi.org/10.1021/jf026182t
- H. S. Lee, “HPLC Analysis of Phenolic Compounds,” In: L. M. L. Nollet, Ed., Food Analysis by HPLC, 2nd Edition, Marcel Dekker, Inc., New York, 2000.
- H. M. Merken and G. R. Beecher, “Measurement of Food Flavonoids by High Performance Liquid Chromatography: A Review,” Journal of the Agriculture and Food Chemistry, Vol. 48, No. 3, 2000, pp. 577-599. http://dx.doi.org/10.1021/jf990872o
- J. G. Bordonaba, P. Crespo and L. A. Terry, “A New Acetonitrile-Free Mobile Phase for HPLC-DAD Determination of Individual Anthocyanins in Blackcurrant and Strawberry Fruits: A Comparison and Validation Study,” Food Chemistry, Vol. 129, No. 3, 2011, pp. 1265-1273. http://dx.doi.org/10.1016/j.foodchem.2010.09.114
- Y. Amakura, M. Okada, S. Tsuji and Y. Tonogai, “Determination of Phenolic Acids in Fruit Juices by Isocratic Column Liquid Chromatography,” Journal of Chromatography A, Vol. 891, No. 1, 2000, pp. 183-188. http://dx.doi.org/10.1016/S0021-9673(00)00625-7
- S. Shahrzad and I. Bitsch, “Determination of Some Pharmacologically Active Phenolic Acids in Juices by HighPerformance Liquid Chromatography,” Journal of Chromatography A, Vol. 741, No. 2, 1996, pp. 223-231. http://dx.doi.org/10.1016/0021-9673(96)00169-0
- J. Gruz, O. Novak and M. Strnad, “Rapid Analysis of Phenolic Acids in Beverages by UPLC-MS/MS,” Food Chemistry, Vol. 111, No. 3, 2008, pp. 789-794. http://dx.doi.org/10.1016/j.foodchem.2008.05.014
- A. J. P. Martin and R. L. M. Synge, “A New Form of Chromatogram Employing Two Liquid Phases,” Biochemical Journal, Vol. 35, No. 12, 1941, pp. 1358-1368.
- S. M. Muñoz, M. Montserrat, B. Olga and J. Guasch, “Determination of Some Flavan-3-ols and Anthocyanins in Red Grape Seed and Skin Extracts by HPLC-DAD: Validation Study and Response Comparison of Different Standards,” Analytical Chimica Acta, Vol. 628, No. 1, 2008, pp. 104-110. http://dx.doi.org/10.1016/j.aca.2008.08.045
- L. Bravo, “Polyphenols: Chemistry, Dietary Sources, Metabolism and Nutritional Significance,” Nutrition Review, Vol. 56, No. 11, 1998, pp. 317-333. http://dx.doi.org/10.1111/j.1753-4887.1998.tb01670.x
- M. Markom, M. Hasan, R. W. D. Wan, H. Singh and J. M. Jahim, “Extraction of Hydrostable Tannins from Phyllanthus niruri Linn.: Effect of Solvents and Extraction Methods,” Separation and Purification Technology, Vol. 52, No. 3, 2007, pp. 487-496. http://dx.doi.org/10.1016/j.seppur.2006.06.003
- K. A. Ross, T. Beta and S. D. Arntfield, “A Comparative Study on the Phenolic Acids Identified and Quantified in Dry Beans Using HPLC as Affected by Different Extraction and Hydrolysis Methods,” Food Chemistry, Vol. 113, No. 1, 2009, pp. 336-344. http://dx.doi.org/10.1016/j.foodchem.2008.07.064
NOTES
*Corresponding author.