International Journal of Clean Coal and Energy
Vol.1 No.1(2012), Article ID:17622,11 pages DOI:10.4236/ijcce.2012.11001
Efficient Theoretical Screening of Solid Sorbents for CO2 Capture Applications*
National Energy Technology Laboratory, United States Department of Energy, Pittsburgh, USA
Email: yuhua.duan@netl.doe.gov
Received December 15, 2011; revised January 28, 2012; accepted February 13, 2012
Keywords: ab initio thermodynamics; CO2 sorbent and capture technology; DFT and phonon lattice dynamics
ABSTRACT
By combining thermodynamic database mining with first principles density functional theory and phonon lattice dynamics calculations, a theoretical screening methodology to identify the most promising CO2 sorbent candidates from the vast array of possible solid materials has been proposed and validated. The ab initio thermodynamic technique has the advantage of allowing identification of thermodynamic properties of CO2 capture reactions without any experimental input beyond crystallographic structural information of the solid phases involved. For a given solid, the first step is to attempt to extract thermodynamic properties from thermodynamic databases and the available literatures. If the thermodynamic properties of the compound of interest are unknown, an ab initio thermodynamic approach is used to calculate them. These properties expressed conveniently as chemical potentials and heat of reactions, which obtained either from databases or from calculations, are further used for computing the thermodynamic reaction equilibrium properties of the CO2 absorption/desorption cycles. Only those solid materials for which lower capture energy costs are predicted at the desired process conditions are selected as CO2 sorbent candidates and are further considered for experimental validations. Solid sorbents containing alkali and alkaline earth metals have been reported in several previous studies to be good candidates for CO2 sorbent applications due to their high CO2 absorption capacity at moderate working temperatures. In addition to introducing our computational screening procedure, in this presentation we will summarize our results for solid systems composed by alkali and alkaline earth metal oxides, hydroxides, and carbonates/bicarbonates to validate our methodology. Additionally, applications of our computational method to mixed solid systems of Li2O with SiO2/ZrO2 with different mixing ratios, our preliminary results showed that increasing the Li2O/SiO2 ratio in lithium silicates increases their corresponding turnover temperatures for CO2 capture reactions. Overall these theoretical predictions are found to be in good agreement with available experimental findings.
1. Introduction
Carbon dioxide is one of the major combustion products which once released into the air can contribute to the global climate warming effects [1-3]. In order to mitigate the global climate change, we must stop emitting CO2 into the atmosphere by separating and capturing CO2 from coal combustion and gasification plants and sequestering the CO2 underground. Current technologies for capturing CO2 including solvent-based (amines) and CaO-based materials are still too energy intensive. Hence, there is critical need for new materials that can capture and release CO2 reversibly with acceptable energy costs. Accordingly, solid sorbent materials have been proposed for capturing CO2 through a reversible chemical transformation and most of them result in the formation of carbonate products. Solid sorbents containing alkali and alkaline earth metals have been reported in several previous studies to be good candidates for CO2 sorbent applications due to their high CO2 absorption capacity at moderate working temperatures [4-6].
To achieve such goals, one of these new methods considered at National Energy Technology Laboratory (NETL) is based on the use of regenerable solid sorbents. In this case sorbents such as alkaline earth metal oxides or hydroxides are used to absorb CO2 at warm temperatures typically ranging from 100˚C - 300˚C [7,8]. The key phenomenon used in these processes is transformation of the oxide or hydroxide materials to a carbonate upon CO2 absorption. Regeneration of the sorbent can be obtained, if necessary, in a subsequent step represented by the reverse transformation from the carbonate phase to the oxide or hydroxide phases. The efficiencies of these processes are highly dependent on identification of the optimum temperature and pressure conditions at which absorption, respectively regeneration are performed. In the case of high-performance sorbents, both these two mechanistic steps are optimized in order to achieve minimal energetic and operational costs.
Optimization of the sorbent material can be obtained starting from the analysis of their intrinsic atomistic structure and of their transformations upon interaction with CO2. Particularly important is to identify the corresponding thermodynamic and kinetic characteristics of the sorbent material of interest. For this purpose scientists at NETL have developed a multi-step computational methodology based on combined use of first principles calculations combined with lattice phonon dynamics to describe the thermodynamic properties of CO2 capture reactions by solid sorbents [4,9-15]. This methodology has been used to screen different classes of solid compounds and has as major objective identification of the optimum candidate materials that can be further subjected to experimental testing. The advantage of this proposed method is that it allows identification of the thermodynamic properties of the CO2 capture reaction as a function of temperature and pressure conditions without any experimental input, excepting the crystallographic structural information of the solid phases involved. Such thermodynamics information is essential to guide experimental groups at NETL in development of highly optimized CO2 sorbents. For a given database of solid materials, our screening scheme allows identification of a short list of promising candidates of CO2 sorbents with optimal energy usages, which can be further evaluated by our experimental research groups.
In this work, we summarize our progress on development of novel screening scheme to indentify most promising candidates for CO2 sorbents. The remainder of this report is organized as follows: In the second section we briefly describe the screening method we developed. In the third section, we provide validation results of our computational method for the case of alkali and alkaline metal compounds. Then, we present the preliminary results on CO2 capture reactions by lithium related salts. The main conclusions are summarized in the last section.
2. Screening Methodology
2.1. Ab Initio Thermodynamics Approach
The complete description of the computational methodology can be found in our previous papers [4,9-15]. Here, we limit ourselves to provide only the main aspects relevant for the current study. The CO2 capture reactions by solids in the presence of water vapors can be expressed generically in the form
 (1)
(1)
where the terms given in [∙∙∙] are optional and n1 and n2 are the numbers of moles of CO2 and H2O involved in the capture reactions. We treat the gas phase species CO2 and H2O as ideal gases. By assuming that the difference between the chemical potentials (∆μº) of the solid phases of A, B (and C) can be approximated by the difference in their electronic energies (∆Eº), obtained directly from first-principles DFT calculations, and the vibrational free energy of the phonons and by ignoring the PV contribution terms for solids, the variation of the chemical potential (∆μ) for capture reaction with temperature and pressure can be written as
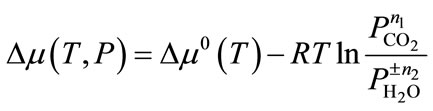 (2)
(2)
where ∆μ0(T) is the standard chemical potential changes between reactants and products. If these thermodynamiccal data are available in the thermodynamic database or literature, we can direct apply them into above equation. If these data are not available, they can be calculated using the ab initio thermodynamic approach based on the following approximation.
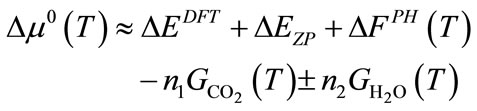 (3)
(3)
Here, ∆EZP is the zero point energy difference between the reactants and products and can be obtained directly from phonon calculations. The ∆FPH is the phonon free energy change between the solids of products and reactants. If the capture reaction does not involve H2O, then the PH2O in above equations is set to P0, which is the standard state reference pressure of 1 bar, and the GH2O term is not present. The “+” and “−” signs correspond to the cases when H2O is a product, respectively a reactant, in the general reaction. The free energies of CO2 (GCO2) and H2O (GH2O) can be obtained from standard statistical mechanics. The enthalpy change for the reaction (1), ∆Hcal(T), can be derived from above equations as
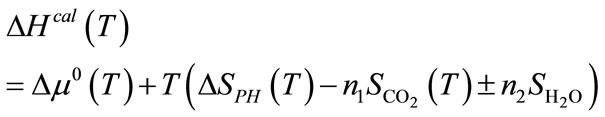 (4)
(4)
In Equation (3), ∆EDFT is the total energy change of the reactants and products calculated by DFT. In this work, the Vienna Ab-initio Simulation Package (VASP) [16,17] was employed to calculate the electronic structures of the solid materials involved in this study. All calculations have been done using the projector augmented wave (PAW) pseudo-potentials and the PW91 exchange-correlation functional [18]. This computational level was shown to provide an accurate description of oxide systems [13-14,19]. Plane wave basis sets were used with a cutoff energy of 500 eV and a kinetic energy cutoff for augmentation charges of 605.4 eV. The k-point sampling grids of n1 × n2 × n3, obtained using the Monkhorst-Pack method [20], were used for these bulk calculations, where n1, n2, and n3 were determined consistent to a spacing of about 0.028 Å−1 along the axes of the reciprocal unit cells. In Equations (3) and (4), the zero pointenergies (EZP), entropies (SPH), and harmonic free energies (FPH, excluding zero-point energy which was already counted into the term ∆EZP) of solids were calculated by the PHONON software package [21] in which the direct method is applied following the formula derived by Parlinski et al. [22] to combine ab initio DFT with lattice phonon dynamics calculations.
As an optimal CO2 solid sorbent, it should not only be easy to absorb CO2 in the capture cycle but also be easy to release the CO2 during regeneration cycle. The operating conditions for absorption/desorption processes depend on their use as in a preor a post-combustion application. The US Department of Energy (DOE) programmatic goal for post-combustion and oxy-combustion CO2 capture is to capture at least 90% of the CO2 produced by a plant with the cost in electricity increasing no more than 35%, whereas the goal in the case of pre-combustion CO2 capture is to capture at least 90% of the CO2 produced with the cost in electricity increasing no more than 10% [23-24]. Under pre-combustion conditions, after the water-gas shift reactor, the gas stream mainly contains CO2, H2O and H2. The partial CO2 pressure could be as high as 20 to 30 bar and the temperature
(T1) is around 313 - 573 K. To minimize the energy consumption, the ideal sorbents should work in these ranges of pressure and temperature in order to separate CO2 from H2. For post-combustion conditions, the gas stream mainly contains CO2 and N2, the partial pressure of CO2 is in the range 0.1 to 0.2 bar, and the temperature range (T2) is quite different. Currently, in post-combustion CO2 capture technology, the amine-based solvents, carbonand zeolite-based solid sorbents (including metal organic framework) capture CO2 within a lower temperature range (<200˚C) [25], while oxides (such as CaO, Na2O, etc.) and salts (such as Li4SiO4, Li2ZrO3, etc.) capture CO2 usually within a higher temperature range (>400˚C) [9- 13]. Based on Equation (2), the working conditions of each solid capturing CO2 can be evaluated and used for determining its suitability as CO2 sorbent.
In this study, the thermodynamic database HSC Chemistry [26] and Factsage [27] packages were employed to search for the available thermodynamic properties of solids.
2.2. Screening Scheme
Figure 1 shows the schematic of our screening methodology. For a given solid databank, this methodology includes four main screening steps (or filters) which allow identification of the most promising candidates [13].
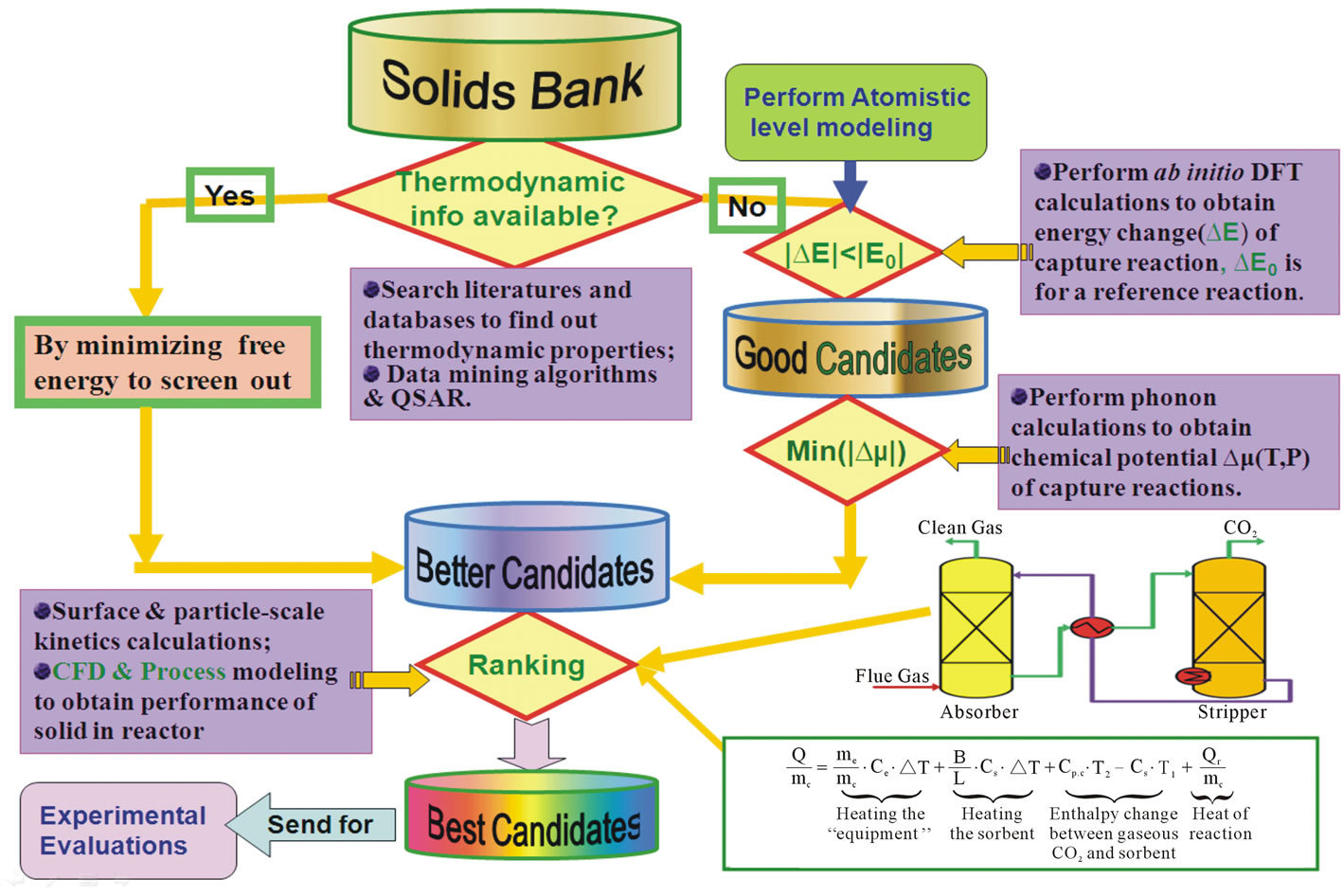
Figure 1. Schematic of our screening methodology.
Step 1: For each solid in the data bank, we first conduct basic screening based on acquisition of general data, such as the wt% of absorbed CO2 in the assumption of a complete reaction, the materials safety and cost, etc. We also include where available the thermodynamic data from literature and from general thermodynamic databases, such as HSC Chemistry, Factsage, etc. If the necessary data for evaluation of the thermodynamic properties exists, then the use of DFT calculations is not necessary and the optimal candidates can be obtained by minimizing their known free energies based on the operating conditions. Otherwise, if the material passes basic screening, but no thermodynamic data are available, then continue to the next step.
Step 2: Perform DFT calculations for all compounds in the candidate reaction with this solid. If |∆EDFT − ∆Eref|/n1 < 20 kJ/mol, where n1 is CO2 molar number in capture reaction, and ∆Eref is the DFT energy change for the reference capture reaction (e.g. CaO + CO2 = CaCO3), we add this compound to the list of good candidates. Otherwise, we go back to step 1 and pick another solid.
Step 3: Perform phonon calculations for reactant and product solids to obtain the corresponding zero point energies and the phonon free energies for the list of good candidates. Specify the target operating conditions (temperature, partial pressures of CO2 and H2O) and compute the change in chemical potential for the reaction, namely ∆m(T, P) from above equations. If ∆m(T, P) is close to zero (e.g. |∆m(T, P)| < 5 kJ/mol) at the operating conditions, then we select this reaction as a member of the “better” list. Only a short list of compounds will likely be left after application of step 3.
Step 4: Additional modeling could be performed to rank the remaining short list of better candidates both obtained from database searching and ab initio thermodynamic calculations as shown in figure 1. One is the kinetics of the capture reactions, which could be done by transport and diffusion calculations as well as using experimental measurements. Another necessary and doable modeling task is the behavior of the solid in the reactor, which can be done by computational fluid dynamics (CFD) methods based on finite element method (FEM) approach and process modeling to estimate the overall costs [28]. These simulations are currently underway. Application of these screening filters will ensure that only the most promising candidates will be identified for the final experimental testing.
This screening methodology provides a path for evaluating materials for which experimental thermodynamic data are unavailable. One area where this approach could be used to great advantage is in evaluating mixtures and doped materials, where thermodynamic data are generally not available but for which the crystallographic structure is known or can be easily determined. Based on the above screening methodology, we have screened hundreds of solid compounds and found some promising candidates for CO2 sorbents. Here, in this work we summarize the results obtained by applying the screening methodology to several classes of solid materials.
3. Results and Discussions
3.1. Applications to Alkali and Alkaline Earth Metal Oxides and Hydroxides [4,12-14]
The thermodynamic data for these oxides, hydroxides and corresponding carbonates and bicarbonates are available in thermodynamic databases, in order to validate our theoretical approach, we also made the ab initio thermodynamic calculations for these known crystals. Table 1 shows the calculated thermodynamic properties of these reactions accompanying with experimental data obtained from HSC Chemistry database [27].
As an example, Figure 2 shows the heats of reactions for alkali and alkaline earth metal oxides capture CO2. From it, one can see that, except for BeO + CO2 à BeCO3 reaction, overall, the calculated results are in good agreement with HSC experimental data. These findings indicate that our theoretical approach can predict the right thermodynamic properties of various solid reacting with CO2 if the right crystal structure of solids is known or is easy to be determined. The larger discrepancy observed for BeO/BeCO3 system is due to lack of the crystal structure information of BeCO3. As the only one input property of the solid in the ab initio thermodynamics calculations, this indicates that in order to obtain reliable results the crystal structure must be known or can be easily predicted correctly.
Table 1 listed the calculated DFT energy changes and

Figure 2. The calculated (solid line) and HSC data (dot line) heat of reaction for alkali and alkaline earth oxides reacting with CO2 to form carbonates [13,14].


Table 1. The calculated and experimental thermodynamic properties of reactions of CO2 captured by alkali and alkalineearth oxides and hydroxides [13,14].
thermodynamic properties of reactions of these oxides and hydroxides capture CO2. Among the 25 CO2 capture reactions indicated in this table, after applying the first filter (steps 1 and 2), only 10 reactions satisfied our selection criteria and are worth to be considered for third screening step (filter two). After applying the second filter on these 10 reactions, as summarized in Figure 3, we found that only MgO(Mg(OH)2)/MgCO3, Na2CO3/ NaHCO3, K2CO3/KHCO3 are promising candidates for CO2 sorbents in either post-combustion or pre-combustion CO2 capture technologies [7,8,10]. These results are in good agreement with the experimental facts, which means our screening methodology is reliable and could be used to identify promising solid CO2 sorbents by predicting the thermodynamic properties of solids reacting with CO2 [14].
Based on Equation (2), Figure 4 gives the calculated relationships of the chemical potential ∆m(T,P) with temperature and CO2 pressure for reactions M2CO3 + CO2 + H2O = 2MHCO3 (M = Na, K), MgO + CO2 = MgCO3, and Mg(OH)2 + CO2 = MgCO3 + H2O. From Figure 4, one can see that Na2CO3/NaHCO3 and K2CO3/KHCO3

Figure 3. Schematic screening results of alkali and alkaline metal oxides, hydroxides and bicarbonates.

Figure 4. The calculated chemical potentials (∆μ) versus CO2 pressure PCO2/P0 and temperatures for the reactions of MgO, Mg(OH)2, and alkali metal carbonates capturing CO2 at fixed PH2O = 1.0 bar and 0.1 bar [13,14]. Only the curve with ∆μ = 0 for each reaction is shown explicitly.
can capture CO2 at low temperature range (400 - 500 K) when CO2 pressure is around 0.1 bar (post-combustion) or 20 - 30 bar (pre-combustion) [10, 14]. We have examined the effect of H2O on the reaction thermodynamics and have found that our modeling approach can be used to account for partial pressures of CO2 and H2O and the temperature. We found that formation of bicarbonates from the alkali metal oxides results in a lower sorbent regeneration temperature and that formation of bicarbonate from the carbonates, by addition of CO2 and H2O, reduces the CO2 capturing temperature even further. Indeed, as shown in Figure 4, we predict that Na2CO3 and K2CO3 have turnover temperatures for CO2 capture through bicarbonate formation that are suitable for operation under both preand post-combustion conditions. When the steam pressure (PH2O) increases as shown in Figure 4, at the same temperature, the PCO2 is decreased because both CO2 and H2O are on the reactant sides.
As one can see from Figure 4, our results show that MgO could be used for both preand post-combustion capture technologies due to its low regenerating temperature (T2 = 540 K for post-combustion conditions and T1 = 690 K for pre-combustion conditions) which are close to experimental findings. However, Mg(OH)2 can only be used for post-combustion capture technologies with a turnover T2 = 600 K because its turnover temperature (T1) is very high, outside the temperature range of interest for pre-combustion applications.
Among the list of alkaline-earth metal oxides and hydroxides analyzed in Table 1, comparing with CaO, only MgO and Mg(OH)2 are found to be good sorbents for CO2 capture. Upon absorption of CO2 both of MgO and Mg(OH)2 can form MgCO3. However, the regeneration conditions of the original systems can take place at different conditions as indicated in Figure 4. In this case we present the calculated phase diagram of MgO-Mg(OH)2- MgCO3 system at different CO2 pressures and at two fixed PH2O values (0.1 and 1.0 bar). From Figure 4 it can be seen that when H2O is present and at low temperatures, MgCO3 can release CO2 to form Mg(OH)2 instead of forming MgO. For example, at PH2O = 0.1 bar, only for temperatures under the transition temperature (Ttr) 460 K, MgCO3 can be regenerated to form Mg(OH)2. By the increase in the H2O pressure, the transition temperature is increased. As shown in Figure 4, when PH2O is increased to 1.0 bar from 0.1 bar, the corresponding Ttr = 520 K. Above Ttr, MgCO3 is regenerated to MgO. Therefore, when water is present in the sorption/desorption cycle, no matter whether the initial sorbent is MgO or Mg(OH)2, and for temperatures below Ttr, the CO2 capture reaction is dominated by the process Mg(OH)2 + CO2 ↔ MgCO3 + H2O(g), whereas above Ttr the CO2 capture reaction is given by MgO + CO2 ↔ MgCO3. The reason is that between MgO and Mg(OH)2, there is a phase transition reaction MgO + H2O(g) = Mg(OH)2 happening at the transition temperature Ttr. Obviously, by controlling the H2O pressure as shown in Figure 4, the CO2 capture temperature (T swing) can be adjusted because the CO2 is a reactant while H2O is a product. However, adding more water in the sorbent system will require more energy due to its sensible heat. These results are in good agreement with the experimental measurements [8].
3.2. Applications to Mixture of Solids [9-11,15]
Lithium silicate (Li4SiO4) and zirconate (Li2ZrO3) have been proposed experimentally as promising high-temperature CO2 sorbents [29-35]. And our previous theoretical studies confirmed these findings [9-11]. However, with different ratios of Li2O/SiO2 and Li2O/ZrO2, one can get different lithium salt compounds as shown in Table 2. We performed first filter (steps 1 and 2) on these lithium salts. The absorbed CO2 molar and weight
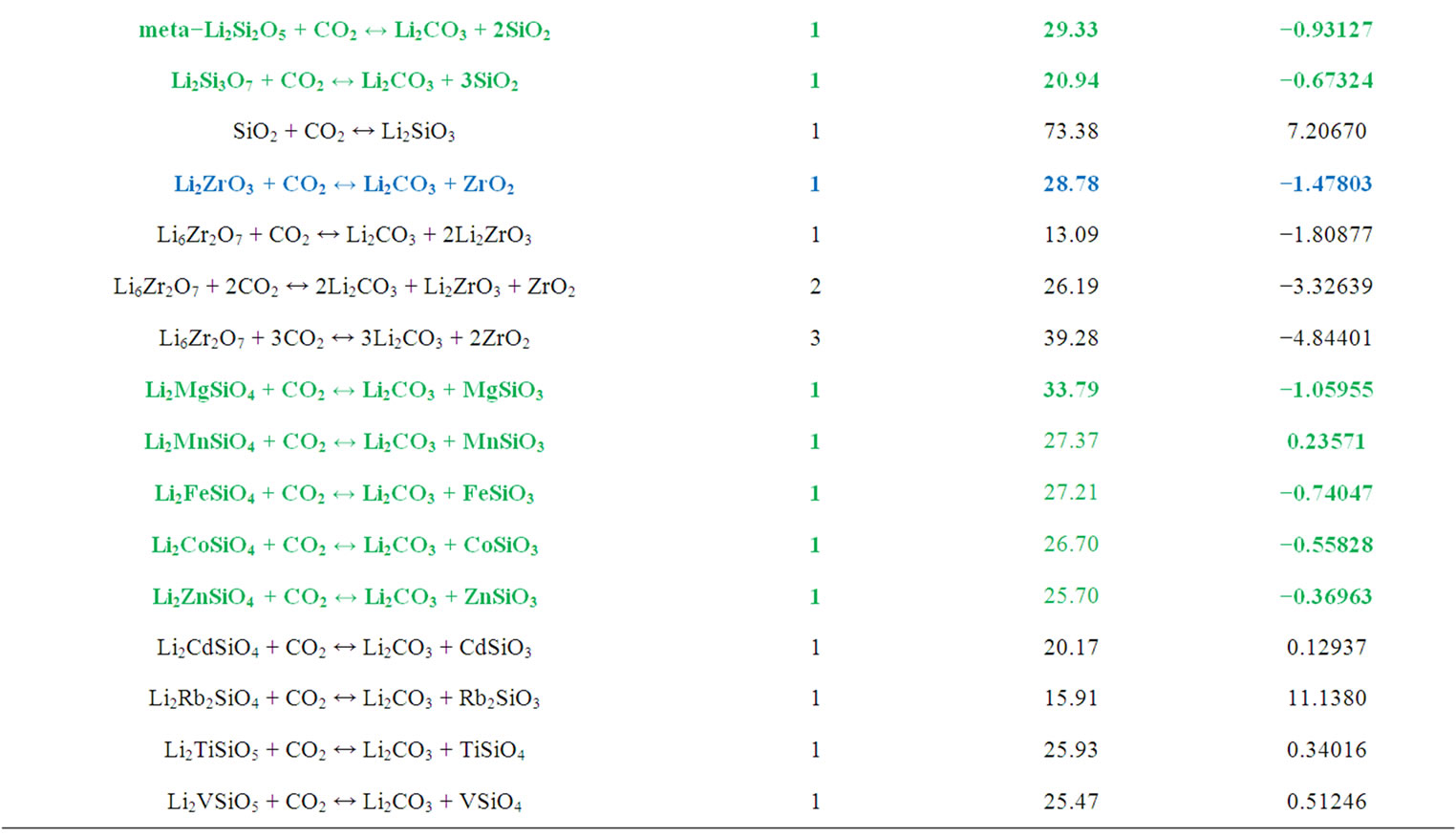

Table 2. The mole and weight percentages of CO2 capture by lithium silicates and zirconates, and the calculated energy change (∆EDFT) of the absorption reactions.
percentages as well as the calculated DFT energy differrences for the capture reactions are also listed in Table 2.
Figure 5 shows the free energy changes of CO2 capture reactions by some lithium silicates as obtained from HSC Chemistry database. From Table 2 and Figure 5, one can see that comparing with Li2O, Li4SiO4, and Li2ZrO3, the Li2SiO3, Li2Si2O5, and Li2Si2O7 are better CO2 solid sorbent candidates because they require less free energy to reverse the CO2 capture reactions and have lower regenerating temperatures. Our calculations show

Figure 5. The Gibbs free energy changes of some lithium silicates capture CO2 reactions from HSC Chemistry database [26].
that although pure Li2O can absorb CO2 efficiently, it is not a good solid sorbent for CO2 capture because the reverse reaction, corresponding to Li2CO3 releasing CO2, can only occur at very low CO2 pressure and/or at very high temperature [12]. SiO2 does not interact with CO2 at normal conditions. Therefore, it can be concluded that when a lithium silicate compound with the ratio of Li2O/SiO2 is less or equal to 1.0, it could have better CO2 capture performance than Li4SiO4, because its regeneration can occur at low temperature and hence require less regeneration heat. Further calculations (steps 3 and 4) and analysis on these lithium silicates capture CO2 properties are underway.
Figure 6(a) summarizes our calculated heats of reactions (ΔH) for four alkali metal silicate and zirconates [9-11]. From Figure 6(a) and Table 2, one can see that the K2ZrO3 capture CO2 has a larger ΔH than the other three solids. Li4SiO4 has a relative small ΔH while along a large temperature range the Li2ZrO3 and Na2ZrO3 have similar ΔH. Therefore, K2ZrO3 is not a good candidate as CO2 sorbent because it needs more heat to regenerate. Among these four solids, Li4SiO4 is the best choice. These results are in good agreement with available experimental measurements [29-35].
According to Equation (2), the calculated relationships of Δµ with CO2 pressure and temperature for these four solids are shown in Figure 6(b). The line in Figure 6(b) indicates that for each reaction, ∆µ(T, P) is approaching zero. The region close to the line is favorable for the absorption and desorption because of the minimal energy costs at a given temperature and pressure. Above the line, the solid (Li4SiO4, M2ZrO3 (M = Li, Na, K)) is favorable to absorb CO2 and to form Li2CO3, while below the line the Li2CO3 is favorable to release CO2 and to regenerate lithium silicate solids. The calculated thermodynamic
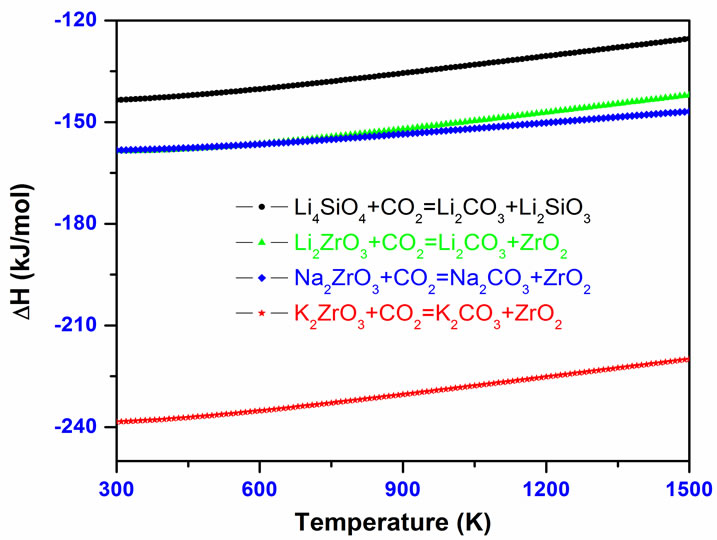 (a)
(a)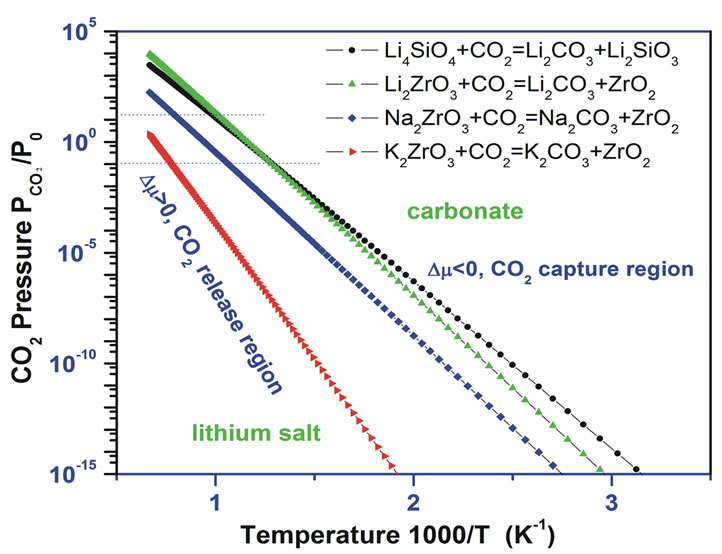 (b)
(b)
Figure 6. The calculated thermodynamic properties of some alkali metal silicate and zirconates capture CO2 [9-11]. (a) The heat of reactions; (b) the contour plotting of calculated chemical potentials (∆μ) versus CO2 pressures and temperatures of the sorbents capture CO2 reactions. Y-axis plotted in logarithm scale. Only ∆μ = 0 curve is shown explicitly. For each reaction, above its ∆μ = 0 curve, their ∆μ < 0, which means the sorbents absorb CO2 and the reaction goes forward, whereas below the ∆μ = 0 curve, their ∆μ > 0, which means the CO2 start to release and the reaction goes backward to regenerate the sorbents.
properties of these solids are also summarized in Table 3.
From Figure 6(b) and Table 3 one can see that these solids capture CO2 up to higher temperatures (T1 > 1000 K) compared with desired pre-combustion condition (313 - 573 K). Therefore, they are not good sorbents for capturing CO2 in pre-combustion technology. However, some of them could be used for high-temperature postcombustion CO2 capture technology with T2 = 1285 K, 925 K, 780 K, 880 K, and 770 K for K2ZrO3, Na2ZrO3, Li2ZrO3 and Li4SiO4 respectively. Obviously, compared
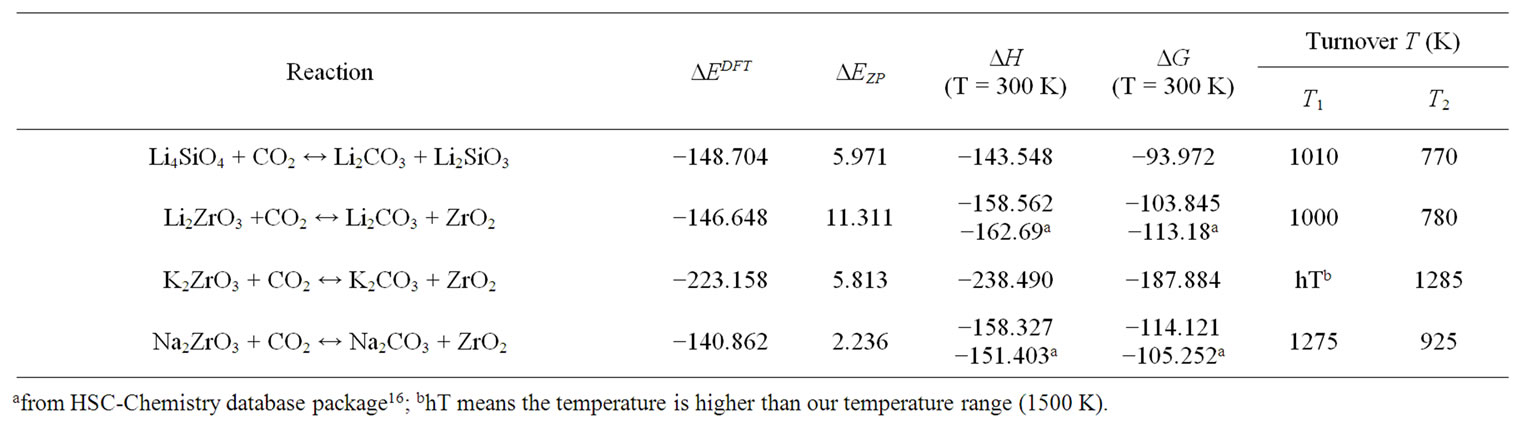
Table 3. The summary of the calculated energy change ∆EDFT, the zero-point energy changes ∆EZP and the thermodynamic properties (∆H, ∆G) of the CO2 capture reactions by alkali metal silicates and zirconates. (unit: kJ/mol) [9,11-13]. The turnover temperatures (T1 and T2) of the reactions of CO2 capture by solids under the conditions of pre-combustion (PCO2 = 20 bar) and post-combustion (PCO2 = 0.1 bar) are also listed.
to CaO, the T2 of K2ZrO3 is still too high to be used for post-combustion technology. This may be part of the reason that there is no experimental work found in the literature for pure K2ZrO3 capturing CO2. Therefore, Li4SiO4, Na2ZrO3, and Li2ZrO3 are good candidates for CO2 sorbents working at high temperature.
Although Li4SiO4 and Li2ZrO3 have similar turnover temperature T2 as shown in Table 3, from Figure 6(a) one can see that the reaction heat of Li2ZrO3 capture CO2 is about 20 kJ/mol lower than that of Li4SiO4. This indicates that more heat is needed for regenerating Li2ZrO3 from Li2CO3 and ZrO2. Therefore, as a CO2 sorbent, the Li4SiO4 is thermodynamically better than Li2ZrO3 despite they may have different kinetics behaviours [36].
4. Conclusions
By combining thermodynamic database searching with first principles density functional theory and phonon lattice dynamics calculations, from vast of solid materials, we proposed a theoretical screening methodology to identify most promising candidates for CO2 sorbents. The thermodynamic properties of solid materials are obtained and used for computing the thermodynamic reaction equilibrium properties of CO2 absorption/desorption cycle based on the chemical potential and heat of reaction analysis. According to the preand post-combustion technologies and conditions in power-plants, based on our calculated thermodynamic properties of reactions for each solid capturing CO2 varying with temperatures and pressures, only those solid materials, which result in lower energy cost in the capture and regeneration process and could work at desired conditions of CO2 pressure and temperature, will be selected as promised candidates of CO2 sorbents and further be considered for experimental validations. Compared to experimental thermodynamic data for known systems, our results show that this screening methodology can predict the thermodynamic properties for sorbents capture CO2 reactions and therefore can be used for screening out good CO2 solid sorbents from vast of solid materials which thermodynamic data are unknown.
5. Acknowledgements
One of us (YD) thanks Drs. D. C. Sorescu, J. K. Johnson, B. Zhang, Y. Soong, R. Siriwardane and G. Richards for fruitful discussions.
REFERENCES
- D. Aaron and C. Tsouris, “Separation of CO2 from flue gas: A review,” Separation Science and Technology, Vol. 40, No. 1-3, 2005, pp. 321-348. doi:10.1081/SS-200042244
- M. R. Allen, D. J. Frame, C. Huntingford, C. D. Jones, J. A. Lowe, M. Meinshausen and N. Meinshausen, “Warming caused by cumulative carbon emissions towards the trillionth tonne,” Nature, Vol. 458, No. 7242, 2009, pp. 1163-1166. doi:10.1038/nature08019
- R. S. Haszeldine, “Carbon Capture and Storage: How Green can Black Be?” Science, Vol. 325, No. 5948, 2009, pp. 1647-1652. doi:10.1126/science.1172246
- Y. Duan, “Computational Screening of Solid Materials for CO2 Capture,” 2011. http://www.netl.doe.gov/publications/proceedings/11/co2cture/Posters/Poster-Duan-NETL-ComputationalScreening.pdf
- C. M. White, B. R. Strazisar, E. J. Granite, J. S. Hoffman and H. W. Pennline, “Separation and capture of CO2 from large stationary sources and sequestration in geological formations-coalbeds and deep saline aquifers,” Journal of the Air & Waste Management Association, Vol. 53, 2003, pp. 645-715.
- J. C. Abanades, E. J. Anthony, J. Wang and J. E. Oakey, “Fluidized bed combustion systems integrating CO2 capture with CaO,” Environmental Science & Technology, Vol. 39, No. 8, 2005, pp. 2861-2866. doi:10.1021/es0496221
- R. Siriwardane, J. Poston, K. Chaudhari, A. Zinn, T. Simonyi and C. Robinson, “Chemical-looping combustion of simulated synthesis gas using nickel oxide oxygen carrier supported on bentonite,” Energy & Fuels, Vol. 21, No. 3, 2007, pp. 1582-1591. doi:10.1021/ef0604947
- R. V. Siriwardane and R. W. Stevens, “Novel Regenerable Magnesium Hydroxide Sorbents for CO2 Capture at Warm Gas Temperatures,” Industrial & Engineering Chemistry Research, Vol. 48, No. 4, 2009, pp. 2135-2141. doi:10.1021/ie8011598
- Y. Duan, “Electronic structural and phonon properties of lithium zirconates and their capabilities of CO2 capture: A first-principle density functional approach,” Journal of Renewable and Sustainable Energy, Vol. 3, No. 1, 2011, Article ID: 013102. doi:10.1063/1.3529427
- Y. Duan, “A first-principles density functional theory study of the electronic structural and thermodynamic properties of M2ZrO3 and M2CO3 (M=Na, K) and their capabilities of CO2 capture,” Journal of Renewable and Sustainable Energy, Vol. 4, No. 1, 2012, Article ID: 013109. doi:10.1063/1.3683519
- Y. Duan and K. Parlinski, “Density functional theory study of the structural, electronic, lattice dynamical, and thermodynamic properties of Li4SiO4 and its capability for CO2 capture,” Physical Review B, Vol. 84, No. 10, 2011, Article ID: 104113. doi:10.1103/PhysRevB.84.104113
- Y. Duan and D. C. Sorescu, “Density functional theory studies of the structural, electronic, and phonon properties of Li2O and Li2CO3: Application to CO2 capture reaction,” Physical Review B, Vol. 79, No. 1, 2009, Article ID: 014301. doi:10.1103/PhysRevB.79.014301
- Y. Duan and D. C. Sorescu, “CO2 capture properties of alkaline earth metal oxides and hydroxides: A combined density functional theory and lattice phonon dynamics study,” Journal of Chemical Physics, Vol. 133, No. 7, 2010, Article ID: 074508. doi:10.1063/1.3473043
- Y. Duan, B. Zhang, D. C. Sorescu and J. K. Johnson, “CO2 Capture Properties of M-C-O-H (M = Li, Na, K) Systems: a Combined Density Functional Theory and Lattice Phonon Dynamics Study,” Journal of Solid State Chemistry, Vol. 184, No. 2, 2011, pp. 304-311. doi:10.1016/j.jssc.2010.12.005
- B. Zhang, Y. Duan and J. K. Johnson, “First-principles density functional theory study of CO2 capture with transition metal oxides and hydroxides,” Journal of Chemical Physics, Vol. 136, No. 6, 2012, Article ID: 064516. doi:10.1063/1.3684901
- G. Kresse and J. Hafner, “Ab initio Molecular-Dynamics for Liquid-Metals,” Physical Review B, Vol. 47, No. 1, 1993, pp. 558-561. doi:10.1103/PhysRevB.47.558
- G. Kresse and J. Furthmuller, “Efficient iterative schemes for ab initio total-energy calculations using a planewave basis set,” Physical Review B, Vol. 54, No. 16, 1996, pp. 11169-11186. doi:10.1103/PhysRevB.54.11169
- J. P. Perdew and Y. Wang, “Accurate and Simple Analytic Representation of the Electron-Gas Correlation-Energy,” Physical Review B, Vol. 45, No. 23, 1992, pp. 13244-13249. doi:10.1103/PhysRevB.45.13244
- Y. Duan, “Electronic properties and stabilities of bulk and low-index surfaces of SnO in comparison with SnO2: A first-principle density functional approach with an empirical correction of van der Waals interactions,” Physical Review B, Vol. 77, No. 4, 2008, Article ID: 045332. doi:10.1103/PhysRevB.77.045332
- H. J. Monkhorst and J. D. Pack, “Special Points for Brillouin-Zone Integrations,” Physical Review B, Vol. 13, No. 12, 1976, pp. 5188-5192. doi:10.1103/PhysRevB.13.5188
- K. Parlinski, “PHONON Software,” 2006. http://wolf.ifj.edu.pl/phonon/
- K. Parlinski, Z. Q. Li and Y. Kawazoe, “First-principles determination of the soft mode in cubic ZrO2,” Physical Review Letters, Vol. 78, No. 21, 1997, pp. 4063-4066. doi:10.1103/PhysRevLett.78.4063
- National Energy Technilogy Laboratory, “Cost and Performance Baseline for Fossil Energy Plants,” 2007. http://www.netl.doe.gov/energy-analyses/baseline_studies. html
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, “Advancesn in CO2 capture technology— The US Department of Energy’s Carbon Sequestration Program,” International Journal of Greenhouse Gas Control, Vol. 2, No. 1, 2008, pp. 9-20. doi:10.1016/S1750-5836(07)00094-1
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, “CO2 capture by solid adsorbents and their applications: current status and new trends,” Energy & Environmental Science, Vol. 4, No. 1, 2011, pp. 42-55.
- software HSC Chemistry, “Pori: Outotec Research Oy,” 2006. www.outotec.com/hsc
- Factsage, www.factsage.com
- H. W. Pennline, J. S. Hoffman, M. L. Gray, R. V. Siriwardane, D. J. Fauth and G. A. Richards, “NETL in-house postcombustion sorbent-based carbon dioxide capture research,” Annual NETL CO2 capture technology for existing plants R&D meeting, Pittsburgh, 24-26 March 2009.
- K. Essaki, K. Nakagawa, M. Kato and H. Uemoto, “CO2 absorption by lithium silicate at room temperature,” Journal of Chemical Engineering of Japan, Vol. 37, No. 6, 2004, pp. 772-777. doi:10.1252/jcej.37.772
- M. Kato and K. Nakagawa, “New series of lithium containing complex oxides, lithium silicates, for application as a high temperature CO2 absorbent,” Journal of the Ceramic Society of Japan, Vol. 109, No. 11, 2001, pp. 911-914. doi:10.2109/jcersj.109.1275_911
- K. Nakagawa and T. Ohashi, “A novel method of CO2 capture from high temperature gases,” Journal of the Electrochemical Society, Vol. 145, No. 4, 1998, pp. 1344- 1346. doi:10.1149/1.1838462
- K. Nakagawa and T. Ohashi, “A reversible change between lithium zirconate and zirconia in molten carbonate,” Electrochemistry, Vol. 67, No. 6, 1999, pp. 618-621.
- M. Olivares-Marin, T. C. Drage and M. M. Maroto-Valer, “Novel lithium-based sorbents from fly ashes for CO2 capture at high temperatures,” International Journal of Greenhouse Gas Control, Vol. 4, No. 4, 2010, pp. 623- 629.
- R. Rodriguez-Mosqueda and H. Pfeiffer, “Thermokinetic Analysis of the CO2 Chemisorption on Li4SiO4 by Using Different Gas Flow Rates and Particle Sizes,” Journal of Physical Chemistry A, Vol. 114, No. 13, 2010, pp. 4535- 4541. doi:10.1021/jp911491t
- R. Xiong, J. Ida and Y. S. Lin, “Kinetics of carbon dioxide sorption on potassium-doped lithium zirconate,” Chemical Engineering Science, Vol. 58, No. 19, 2003, pp. 4377-4385. doi:10.1016/S0009-2509(03)00319-1
- A. Lopez-Ortiz, N. G. P. Rivera, A. R. Rojas and D. L. Gutierrez, “Novel carbon dioxide solid acceptors using sodium containing oxides,” Separation Science and Technology, Vol. 39, No. 15, 2004, pp. 3559-3572. doi:10.1081/SS-200036766
NOTES
*The original manuscript of this paper was initially included in the Proceedings of 28th Annual International Pittsburgh Coal Conference, September 12-15, 2011, Pittsburgh, PA.