Advances in Bioscience and Biotechnology
Vol.4 No.6A(2013), Article ID:33402,11 pages DOI:10.4236/abb.2013.46A009
Purkinje-neuron-specific down-regulation of p38 protects motoric function from the repeated use of benzodiazepine
![]()
Departments of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, USA
Email: Marianna.Jung@unthsc.edu
Copyright © 2013 Marianna Jung, Daniel Metzger. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 25 April 2013; revised 27 May 2013; accepted 5 June 2013
Keywords: Benzodiazepine; Motoric deficit; p38; Purkinje neurons; Mitochondria
ABSTRACT
Benzodiazepine (BZD) is the most prescribed CNS depressant in America to treat hyper-excitatory disorders such as anxiety and insomnia. However, the chronic use of BZD often creates adverse effects including psychomotor deficit. In this study, we investigated a novel mechanism by which chronic BZD impedes motoric function in female mice. We used female mice because BZD use is much more prevalent in female than male populations. We tested the hypothesis that the accumulation of p38 (stress-activated protein) in cerebellar Purkinje neurons mediates motoric deficit induced by chronic BZD. To test this hypothesis, we generated transgenic mice that lack p38 in cerebellar Purkinje neurons by crossing Pcp2 (Purkinje cell protein 2)-Cre mice with p38loxP/loxP mice. p38-knockdown mice and wild-type mice received BZD (lorazepam, 0.5 mg/kg) for 14 days. During this period, they were tested for motoric performance using Rotarod assay in which a quicker fall from rotating rod indicates poorer motoric performance. Cerebellum was then collected to detect p38 in Purkinje neurons and to measure mitochondrial respiration using immunohistochemistry and real-time XF respirometry, respectively. Compared to vehicletreated mice, BZD-treated mice showed poorer motoric performance, a higher number of Purkinje neurons containing p38, and lower mitochondrial respiration. These effects of BZD were much smaller in p38-knockdown mice. These results suggest that the excessive accumulation of p38 in cerebellar Purkinje neurons contributes to motoric deficit associated with chronic BZD. They also suggest that Purkinje neuronal p38 mediates BZD-induced mitochondrial respiratory inhibition in cerebellum. Our findings may provide a new mechanistic insight into chronic BZD-induced motoric deficit.
1. INTRODUCTION
Benzodiazepines (BZD)s, inhibitory neurotransmitter enhancers, are by far the most frequently prescribed CNS depressants in Americans [1]. Although BZDs are powerfully effective in treating hyper-excitatory CNS disorders, many patients encounter the non-therapeutic effects of BZDs [2]. One common problem with BZD therapy is that patients often experience motor incoordination and movement disorders. For example, BZDs increase the risk for falls [3-5], automobile accidents [6], slow motor reaction, and the inaccuracy of motor tasks [7-9]. Animal studies have also shown that diazepam and lorazepam provoke motoric deficit [10,11]. The motor-impairing effect of BZD occurs at a therapeutic dose [12], at an acute and chronic dose, across species [12,13], and across genders, limiting the drugs’ clinical utility [14]. Importantly, the adverse impact of BZD is greater on the population of women than men because they significantly outnumber male BZD users [15,16] even when several factors are normalized [17-22]. This is a clinically important issue because women outlive men, and thus, extending the period of such problems. Therefore, there is a critical need to develop a protective strategy to prevent this problem. As a step toward the development of a protective strategy, we intend to identify a direct mechanism by which chronic BZD impairs motoric functions and this effect of BZD can be minimized.
p38 is a signaling protein kinase whose aberrant activation is implicated in many pathological conditions [23-25]. The known members of the p38 family include p38α [26], p38β [27,28], p38γ [29,30] and p38δ [31]. Among these isozymes, p38α and p38β are highly expressed in brain areas such as cerebellum and cortex [32-34]. p38 is activated upon phosphorylation [35], so phosphorylated p38 (pp38) is often measured as an indicator of p38 activation. p38 is also known as a stressactivated protein kinase because p38 is phosphorylated by stress signals, such as inflammatory cytokines, heat shock, or ischemia [36]. The pathological activation of p38 has been shown in the brains of Alzheimer’s disease patients [37] and in the livers of aged rats after challenged with a prooxidant, H2O2 [38]. Purkinje neurons are the major type of cerebellar neurons, responsible for movement control. Our recent study has shown that p38-containing Purkinje neurons are more populated in rats under the stress of abrupt ethanol withdrawal than healthy rats [39]. The p38 accumulation in this neuronal population is accompanied by poor motoric performance. As such, studies on p38 or BZD have revealed that both compounds (p38 and BZD) are associated with motoric impairment. This led us to the hypothesis that the up-regulation of Purkinje neuronal p38 mediates motoric deficit induced by chronic BZD. p38 is a cytosolic protein, but studies have demonstrated a link between p38 and mitochondria. For example, mitochondrial reactive O2 species contribute to the phosphorylation of p38 during hypoxia in cardiomyocytes [40]. When cells are treated with a p38 inhibitor (SB203580), their mitochondria are less damaged by oxidative stress than vehicle-treated cells [39]. Based on these studies, we also tested whether chronic BZD treatment impairs mitochondrial respiration through Purkinje neuronal p38. To test these hypotheses, we have generated transgenic mice that lack p38 in Purkinje neurons. Here, we report that these p38-knockdown mice show resistance to motoric and mitochondrial impairment induced by chronic BZD.
2. MATERIALS AND METHODS
2.1. Chemicals
Analytic grade reagents were purchased from IDT Company (San Jose, CA), the Jackson Laboratory (Bar Harbor, Maine), Sigma Aldrich (St. Louis, MO), Cellsignaling Technology (Danvers, MA), Seahorse Bioscience (North Billerica, MA), Invitrogen (Grand Island, NY), and Abcam (Cambridge, MA).
2.2. BZD Injection and Motoric Test
All mice were two months old in the beginning of this study. Among BZDs, we selected lorazepam with an intermediate half-life in human. Lorazepam is currently one of the most frequently used BZDs in clinical settings [41]. Clinicians prefer BZD with an intermediate halflife to extensively sedating long half-life BZDs [42,43]. Lorazepam has been reported to impair motoric function in humans [8] and mice [11]. In the current study, mice were injected with lorazepam (0.5 mg/kg) in the afternoon, and next morning they were tested for motoric function using Rotarod apparatus. This procedure was repeated for 14 days. The dose (0.5 mg/kg) of lorazepam was chosen based on our pilot study and previous studies in which lorazepam was used for anxiolytic effects [44-46]. Rotarod is a motor driven treadmill (Omnitech Electronics, Columbus, OH) that measures running coordination and motor performance, such that a shorter latency to fall from an accelerating rod indicates poorer motor performance. The rotor consists of four cylinders that are mounted 35.5 cm above a padded surface. Mice were placed on the cylinder and a timer switch was simultaneously activated to rotate the cylinders. Acceleration continued until 44 rpm for maximum 90 seconds or animals fell to the padded surface, which simultaneously stopped the timer. Mice were tested for 3 sessions/day for 14 days with a 20 minutes resting period between sessions [47].
2.3. Generation of Purkinje-Neuron-Specific p38-Knockdown Mice
Among p38 isoforms, we selected p38α because it is the most abundant isozyme in the brain and the best characterized isoform [48,49]. To avoid lethality, we employed a conditional transgenic mouse system to down-regulate Purkinje p38 genes using the Cre/loxP system and Pcp2 promoter (Purkinje neuron-specific marker) [50,51]. Transgenic mice (Pcp2-Cre mice) that express Cre recombinase under the control of the Pcp2 (Jackson Laboratory) were cross-mated with floxed-p38α mice to generate the Pcp2-Cre+/−/p38loxP/loxP mice. The mice with floxed-p38 were kindly provided by Boehringer Ingelheim Inc. The p38 floxed allele was generated by homologous recombination of embryonic stem cells in which two sites of ATG containing Purkinje p38 sequence were flanked by loxP [52,53] and excised in the presence of Pcp2-Cre. When pups were 21 days old, the tips of the tail were collected for genotype identification.
2.4. Genotyping Procedure for Pcp2 and p38
DNA was isolated by incubating tail samples overnight at 55˚C in proteinase K buffer. Primer sequences were as follows: for Pcp2-Cre transgene forward, 5’-GCGGTCTGGCAGTAAAAACTATC-3’; for Pcp2-Cre transgene reverse, 5’-GTGAAACAGCAT TGCTGTCACTT-3’; for Pcp2-Cre internal positive control forward, 5’-CTAGGCCACAGAATTGAA AGA TCT-3’; for Pcp2-Cre internal positive control reverse, 5’-GTAGGTGGAAATTCTAGCATCATCC-3’; loxP-flanked p38α allele: 5’-TCCTACGAGCGTC GGCAAGGTG-3’ and 5’-ACTCCCCGAGAGTTCC TGCCTC-3’. Sequential denaturing (96˚C, 30 sec), annealing (52˚C, 1 minute) and extension (72˚C, 1 minute) were repeated 35 times for genotyping the Pcp2-Cre transgene. The program of 30 cycles of denaturing (94℃ for 30 sec), annealing (58˚C, 30 sec), and extension (72˚C, 45 sec) was used to genotype the p38α alleles using the polymerase chain reaction method [51].
2.5. Immunohistochemical Detection of p38
Mice were anesthetized with isoflorane and perfused with 0.9% saline. The formalin-fixed and paraffin-embedded left hemispheres were cut into 8 μm-thick slices on a microtome. The slices were deparaffinized in xylene, rehydrated through a series of graded ethanol solutions, and washed with PBS. The slices were subsequently moisturized at 95˚C and incubated with primary antibody, polyclonal rabbit anti-phosphorylated p38α (pp38, an active form of p38) overnight at 4˚C. The slices were then incubated with broad spectrum poly HRP conjugate for 40 minutes at room temperature. The antigen-antibody bindings were visualized with a diaminobenzidine color reaction for 10 minutes. The slides were further rinsed, dehydrated through a series of graded ethanol and xylene, and mounted with Permount. All photographs were taken with a compatible Zeiss digital camera. A 20- fold magnification was used to take pictures.
2.6. Semi-Quantitative Analysis of p38-Positive Purkinje Neurons
Brain slice samples were evaluated using the Carl Zeiss microscope, the image analysis program AxioVision 4 (Carl Zeiss, Thornwood, NY) and a previous method [54] that was modified for our purpose. Three mice per treatment group were evaluated. Six microscopic fields per mouse were selected, such that two microscopic fields were randomly selected from each of anterior (lobes I-V), medius (VI-VII), and posterior (VIII-X) regions of the cerebellar cortex [39,55]. All Purkinje cells with visible p38-positive deposits were individually counted per microscopic field, and the length of the Purkinje layer per field was measured using a software program, Image Pro Plus (Media Cybernetics, Silver Spring, MD) to normalize the cell counts. Data were presented as the average number of p38-positive Purkinje neurons/Purkinje layer (mm) from the 18 data points (3 mice/group, 6 fields/mouse).
2.7. Mitochondrial Respiration
Mitochondrial respiration was assessed by measuring mitochondrial O2 consumption rate (pmoles/minutes) according to a method provided by the XF respirometer manufacturer (Seahorse Bioscience). XF sensor cartridge was hydrated overnight in XF calibration buffer (at 37˚C, no CO2). Isolated mitochondria were diluted with mitochondrial assay solution (Seahorse Bioscience, North Billerica, MA) to yield a final concentration of 200 µg/ml. Diluted mitochondria (50 µl) were transferred into each well of XF microplate and spun down at 4˚C for 10 - 20 minutes at 2000 - 3600 g. A consistent monolayer of mitochondrial adhesion to well bottom was visually ensured. A volume of 450 µl of succinate (5.5 mM) and rotenone (2.2 µM) was then added to each well. The XF microplate was warmed at 37˚C for 8 - 10 minutes and placed in XF respirometer. Real-time (data are obtained while mitochondria respire) mitochondrial respiration was subsequently recorded every 5 - 7 minutes.
2.8. Statistical Analysis
All numerical data are expressed as mean ± standard error of mean (SEM). The data were analyzed by oneway ANOVA as a factor of treatment or two-way ANOVA as a factor of treatment and days of testing. When a significant difference was observed, post-hoc Tukey’s test was conducted to detect an individual group difference. p value was set less than 0.05 to indicate statistical significance.
3. RESULTS
3.1. Chronic BZD Inhibits Motoric Function
Figure 1 illustrates the motoric performance of mice that were injected with BZD or vehicle (methyl cellulose) for 14 days. Two-way ANOVA was conducted as a factor of treatment and days of testing. Compared to vehicle-injected mice, BZD-injected mice fell quicker (a shorter latency, poor performance) from Rotarod from day 9 to the end of the test (day 14) (*p < 0.01). There was no
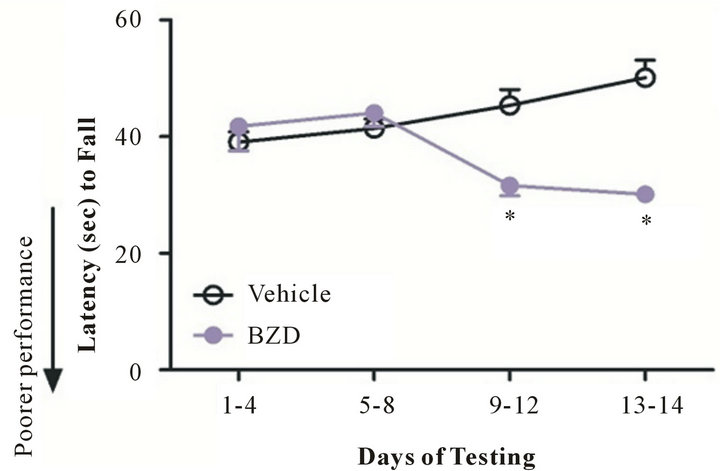
Figure 1. Chronic BZD inhibits motoric function. Mice were injected with lorazepam (0.5 mg/kg, I.P.) at 2 PM and tested on Rotarod (3 sessions/day) the next morning (10 AM). This procedure was repeated daily for 14 days. Compared to vehicle-injected mice, BZD-injected mice show poorer motoric performance from day 9 of testing. *p < 0.01 vs. vehicleinjected mice. Data are presented as mean ± SEM out of all data collected during the time period indicated in X axis. N = 4 - 7 mice/group.
significant difference in Rotarod performance between day 1 and 8 of the test. These data indicate that chronic rather than acute BZD results in motoric deficit.
3.2. Chronic BZD Increases the Number of p38-Containing Purkinje Neurons
We tested whether chronic BZD affects p38 expression in Purkinje neurons. Although p38 immunoreactivity was found in other types of neurons, such as granular neurons, we focused on Purkinje neurons because Purkinje neurons are the sole output of the cerebellar cortex and play a major role in motor function [56]. The immunohistochemical photographs revealed that p38 (as a form of pp38) immunoreactivity (dark deposits) was more distinctively visualized in the Purkinje neurons along the Purkinje layer of BZD-injected mice than vehicle-injected mice (Figure 2).
When the number of p38-positive Purkinje neurons were computed, the number was higher in BZD-injected mice than vehicle-injected mice (*p < 0.001). These data indicate that chronic BZD use results in p38 accumulation in cerebellar major neurons.
3.3. p38-Knockdown Mice Lack p38 in Purkinje Neurons
We generated transgenic mice that were designed to lack p38 in Purkinje neurons to test the role of Purkinje neuronal p38 in BZD-induced motoric deficit. Our recent study reported the DNA results from genotyping, such as the DNA images of Pcp2-Cre at 100 base pair, homozygous p38loxP/loxP at 414 base pair, and an internal positive control (DNA quality control) at 324 base pair [51]. Immunohistochemistry results (Figure 3) reveal that Purkinje p38-knockdown mice (Pcp2-Cre−/+/p38loxP/loxP) show much less p38-positive stains (dark brown deposits) in the Purkinje neurons and a significantly smaller number of p38-positive Purkinje neurons along the Purkinje layer compared to wild-type mice (C57BL/6 mice) (*p < 0.01). These results prove that p38 has been successfully knocked down in Purkinje neurons.
3.4. p38-Knockdown Mice Show Better Motoric Performance than Wild-Type Mice after Chronic BZD Injection
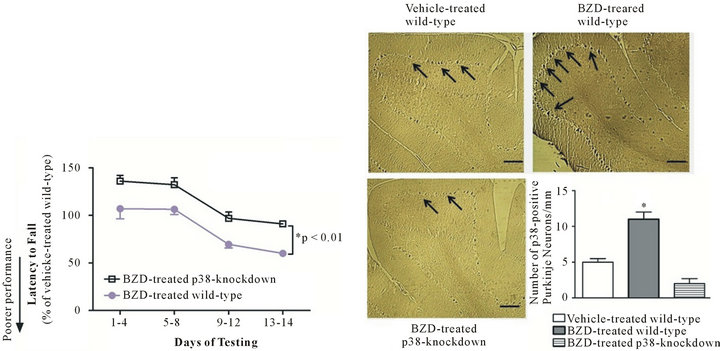
If Purkinje-neuron-specific p38 contributes to BZD-induced motoric deficit, the down-regulation of the p38 would attenuate such effect of BZD. To test this hypothesis, p38-knockdown and wild-type mice were injected with BZD and tested on Rotarod, and this procedure was repeated for 14 days. BZD-injected p38- knockdown mice were able to run on Rotarod longer than BZD-injected wild-type mice throughout the test period (Figure 4(a), *p < 0.01), an indicator of better motoric performance. There was no significant difference in motoric performance between BZD-injected and vehicle-injected p38-knockdown mice (data not shown). When p38-containing Purkinje neurons were examined, chronic BZD treatment failed to increase the number of p38-positive Purkinje neurons in p38-knockdown mice compared to wild-type mice (Figure 4(b)). These data indicate that Purkinje neuronal p38 mediates BZD-induced motoric deficit.
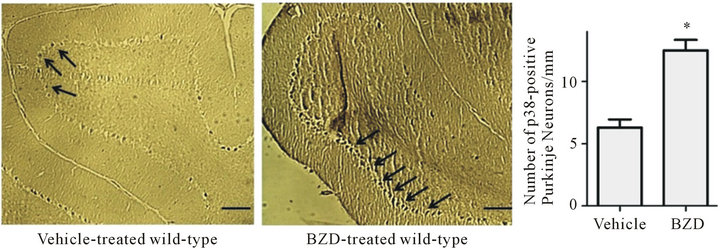
Figure 2. Chronic BZD increases the number of p38-containing Purkinje neurons. Mice were injected with lorazepam (0.5 mg/kg, I.P.) for 14 days. Next morning, they were deeply anesthetized and perfused intracardially with saline followed by paraformaldehyde. Left hemisphere containing the cerebellar vermis was processed for immunohistochemical analysis. All photographs were taken of the cerebellar cortex area containing Purkinje layers that showed a clear image across all treatment groups. Dark deposits marked with arrows indicate p38 immunoreactivity in Purkinje neurons along the Purkinje layer, and they more distinctively appeared in BZD-injected mice than vehicle-injected mice. p38-containing Purkinje neurons were counted using a software program Image Pro Plus, revealing that BZD-injected mice had a higher number of p38-positive Purkinje neurons/Purkinje layer (mm) than vehicle-injected mice. *p < 0.001 vs. vehicle-injected mice. The scale bar indicates an actual length of 100 µm. Depicted are mean ± SEM for 6 microscopic fields/mouse for 3 mice/group.
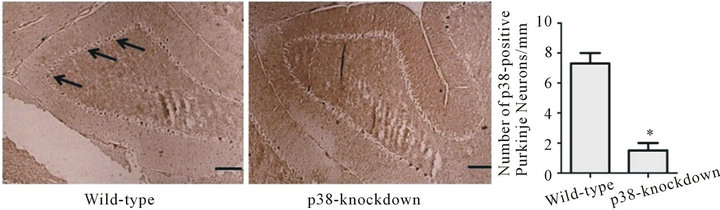
Figure 3. p38-knockdown mice lack p38 in Purkinje neurons. Transgenic mice with Purkinje neuron-specific down-regulation of p38 were generated by cross-mating Pcp2-Cre mice with floxed-p38α mice. When the mice were two months old, the cerebellar sections were immunostained using pp38α antibody. C57BL/6 mice were used as wild-type mice that showed p38-positive stains (dark brown deposits) in the Purkinje neurons along the Purkinje layer. The number of Purkinje neurons (per 1 mm of Purkinje layer) was counted using a software program, ImageProPlus. p38-positive markers and Purkinje neurons were significantly smaller in Purkinje p38- knockdown mice than wild-type mice. *p < 0.01 vs. wild-type mice. The scale bar indicates an actual length of 100 µm. Depicted is mean ± SEM for 6 microscopic fields/mouse for 3 mice/group.
 (a) (b)
(a) (b)
Figure 4. p38-knockdown mice show better motoric performance than wild-type mice after chronic BZD injection. p38-knockdown mice and wild-type mice were injected with lorazepam (0.5 mg/kg, I.P.) and tested on Rotarod (3 sessions/day) the next morning. This procedure was repeated daily for 14 days (a). Data are relative to those of vehicle-injected wild-type mice and presented as mean ± SEM out of all data collected during the time period indicated in X axis. BZD-injected p38-knockdown mice showed a higher latency to fall from Rotarod throughout the test period, an indicator of better motoric performance than BZD-injected wild-type mice (*p < 0.01). Next morning after the completion of behavioral tests, cerebellar area was processed for the immunohistochemical analysis of p38-positive Purkinje neurons (b). All photographs were taken of the cerebellar cortex area containing Purkinje layers. Dark deposits marked with arrows indicate p38 immunoreactivity in Purkinje neurons along the Purkinje layer. BZD-injected wild-type mice showed more distinct p38 immunoreactivity and a higher number of p38-positive Purkinje neurons/Purkinje layer (mm) than vehicle-injected wild-type mice or BZD-injected p38-knockdown mice (*p < 0.002). In (b), depicted are mean ± SEM for 6 microscopic fields/mouse for 3 mice/group.
3.5. Chronic BZD Suppresses Mitochondrial Respiration in a Manner that Is Prevented by p38-Knockdown
The effect of chronic BZD on mitochondrial respiration was tested in p38-knockdown and wile-type mice. As seen in Figure 5, mice injected with chronic BZD show significantly lower mitochondrial respiration than vehicle-injected mice (*p < 0.01). This difference was almost
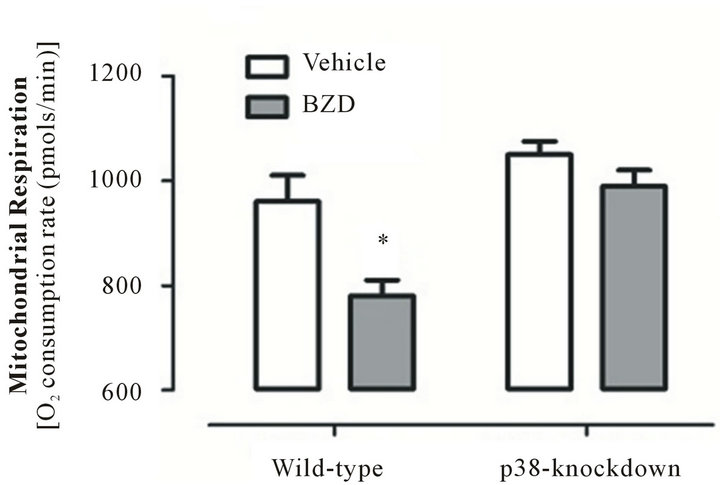
Figure 5. Chronic BZD suppresses mitochondrial respiration in a manner that is prevented by p38-knockdown. p38-knockdown mice and wild-type mice were injected with lorazepam (0.5 mg/kg, I.P.) for 14 days. Next morning, these mice were euthanized under anesthetics and mitochondria were isolated from cerebellum to assess mitochondrial respiration using XF respirometer. Among mice groups, mitochondrial respiration was decreased only in BZD-injected wild-type mice. *p < 0.01 vs. three other groups. Data are presented as mean ± SEM out of all data collected at four time points of measurements with 5 - 7 minutes interval between time points for 5 or 6 mice/group.
completely abolished in p38-knockdown mice injected with BZD or vehicle. These data indicate that Purkinje neuronal p38 mediates BZD-induced mitochondrial respiratory suppression.
4. DISCUSSION
The key finding of the current study is that chronic BZD treatment impedes motoric function and mitochondrial respiration in a manner that is protected by the downregulation of Purkinje neuron-specific p38. These results suggest that chronic BZD acts as a stressor, provoking the excessive accumulation of p38 in vulnerable neurons accompanied by behavioral deficit.
The most studied mechanism of BZDs or BZD analogues is the enhancement of inhibitory GABA neurotransmission through GABA-A receptors, thereby exerting therapeutic effects such as anxiolytic, sedative, or anti-seizure effects [57]. While BZD is powerfully effective and largely safe, evidence indicates that the prolonged use of BZDs often creates adverse effects including motoric deficit [2,7,9,58]. Nearly all the disadvantages of BZDs result from long-term use [59], but most mechanistic studies on motor deficit have focused on BZD’s acute effect. Savic et al. [60] have studied the effects of a single dose of BZD analogues on the motor performance of rats and reported that GABA-A receptor α1 subunit mediates the ataxic potential of BZDs. Similarly, Korpi et al. [10] have reported that GABA-A receptor α6 subunit mediates motoric deficit associated with an acute dose of diazepam. In a study done by Stanley et al. [11], the motor incoordination induced by a single dose of diazepam and lorazepam required a high occupancy of a GABA-A receptor. In comparison with these studies that employed an acute dose regimen of BZD, we studied the effect of a prolonged BZD use on motoric function. The majority of patients who receive BZD therapy continue to take BZD for an extended period of time. This phenomenon is more prevalent in women than men [9], which increases their risk of motoric deficit associated with the prolonged use of BZDs [2,7,9,58]. As shown in Figure 1, mice treated with BZD showed no significant difference in motoric performance compared to vehicle-treated mice during the early stage of BZD injection (day 1 to 8). However, BZD-injected mice began to show poorer performance than vehicletreated mice upon the further treatment of BZD (from day 9). The motoric impairment seen in BZD-injected mice is unlikely due to the sedative effects of BZD for at least two reasons. Firstly, the motoric behavior of these mice did not differ from control mice during the initial several days of BZD injection. Secondly, mice were tested on Rotarod 20 hours after the injection of lorazepam of which half-life is 70 minutes [44]. The motoric impairment induced by repeated BZD injection may suggest that chronic BZD provokes permanent or long lasting changes in the brain. Our data indicate that one of the BZD-induced changes occurs at the level of Purkinje neuronal p38; BZD-treated mice show a higher number of Purkinje neurons containing p38 than vehicle-treated mice.
We chose Purkinje neurons among multiple types of cerebellar neurons. Purkinje neurons constitute the sole output of the cerebellar cortex, governing all movement [56] and coordination [61]. Damage to these neurons inevitably provokes the impairment of motor behavior and coordination [55,56,62]. Our recent study reveals that motoric deficit occurs in ethanol withdrawn rats accompanied by p38 accumulation in Purkinje neurons. This finding prompted us to test whether BZD-associated motoric deficit is mediated through p38 accumulation in this neuronal population. Our behavioral and immunohistochemical tests demonstrate that mice treated with chronic BZD show poor motoric performance and a significantly increased number of p38-positive Purkinje neurons. However, this observation is only correlational and does not necessarily indicate that the accumulation of Purkinje-specific p38 is responsible for the motoric deficit. We, therefore, generated mice that lack p38 specifically in Purkinje neurons. These mice indeed show substantially better motoric performance than wild-type mice under chronic BZD therapy, proving that Purkinje p38 contributes to BZD-induced motoric impairment. A previous study showed that a p38 inhibitor protects immature, vulnerable Purkinje neurons from neurotoxic stress [63]. Mice with Purkinje neuron degeneration have shown a spatial learning deficit in the water maze test [61,64,65]. These studies along with our current findings suggest that aberrant p38 activation/accumulation perturbs the movement-controlling function of Purkinje neurons.
Mitochondria are the main source of energy supply for cellular and neuronal survival. Mitochondria consume most of inhaled O2, and such O2-consuming mitochondrial respiration is obligatorily coupled with ATP production. In humans or experimental animals, mitochondrial defects are often observed in a variety of CNS disorders and brain aging [66]. BZD has been reported for both beneficial [67] and adverse [68,69] effects on mitochondria. BZD (diazepam) blocked the apoptotic release of cytochrome c from mitochondria [67], suggesting that BZD has a mito-protective effect. In contrast, several BZDs including diazepam, suppressed mitochondrial respiratory control ratio (State III/State IV) in rat kidney [70]. Diazepam also induced mitochondrial swelling [71], suggesting that BZDs have adverse effects on mitochondria. It is not clear what underlies the beneficial and adverse effects of BZD on mitochondria. One possibility is that BZD exerts differential effects on mitochondria depending upon the target area or function of mitochondria. It is also possible that the effects of BZD on mitochondria vary depending upon the duration or dose of BZD treatment. As far as the effect of chronic BZD on mitochondrial respiration is concerned, our current study demonstrates that chronic BZD hinders mitochondrial respiration accompanied by motoric impairment.
Although p38 is mainly located in cytosol, studies have demonstrated the effects of p38 on mitochondria. For instance, p38 inhibits the adverse effect of a nuclear receptor (PPARγ) coactivator on mitochondrial respiretion in muscle [72]. p38 activation is associated with free radical production [53]. p38 inhibitor (SB203580) mitigates the swelling of mitochondrial membrane induced by ethanol withdrawal stress (unpublished observation). These studies suggest that there is a crosstalk between cytosolic p38 and mitochondria. Our results strengthen this view in that the down-regulation of Purkinje p38 attenuates the suppressing effect of BZD on mitochondrial respiration (Figure 5). How the inhibition of cytosolic protein p38 mediates mitochondrial respiratory enhancement is an open question. p38 activation induces BAX (apoptotic protein) translocation to mitochondria, and BAX subsequently impedes mitochondrial respiration [73]. Therefore, p38 depletion would result in an opposite effect, the enhancement of mitochondrial respiration. Mitochondria are one of the major targets of estrogen, a neuroprotective steroid that directly acts on cytosol and mitochondria [74]. Thus, it is also possible that estrogen may interfere between p38 and BAX, protecting mitochondria. Alternatively, BZD inhibits ventilatory response [75,76], reducing the amount of O2 available for mitochondrial respiration. Although this possibility cannot be ruled out, this is a remote possibility because in general, BZD-induced ventilatory inhibition occurs at a much higher dose than an anxiolytic dose used in the current study. Unfortunately, we were not able to determine a direct relationship among BZD-induced mitochondrial respiratory deficits, motoric impairment, and excessive p38. The administration of mitochondrial respiratory inhibitors to brain could test whether the inhibitors blunts the motor-improving effects of p38-down-regulation. However, we encountered a technical limitation of delivering mitochondrial respiratory inhibitors to brain without creating the toxic or non-specific effects of the inhibitors.
In conclusion, our findings provide empirical evidence that chronic BZD impedes motoric function at least partly through the accumulation of p38 in Purkinje neurons. The current study may provide a new mechanistic insight involving p38 into BZD-induced motoric deficit.
5. ACKNOWLEDGEMENTS
This work has been supported by funding from NIH/NIAAA (AA- 018747). We wish to thank Xiaohua Ju for her technical assistance.
REFERENCES
- Bogunovic, O.J. and Greenfield, S.F. (2004) Practical geriatrics: Use of benzodiazepines among elderly patients. Psychiatric Services, 55, 233-235. doi:10.1176/appi.ps.55.3.233
- Buffett-Jerrott, S.E. and Stewart, S.H. (2002) Cognitive and sedative effects of benzodiazepine use. Current Pharmaceutical Design, 8, 45-58. doi:10.2174/1381612023396654
- Leipzig, R.M., Cumming, R.G. and Tinetti, M.E. (1999) Drugs and falls in older people: A systematic review and meta-analysis: I. Psychotropic drugs. Journal of the American Geriatrics Society, 47, 30-39.
- Leipzig, R.M., Cumming, R.G. and Tinetti, M.E. (1999) Drugs and falls in older people: A systematic review and meta-analysis: II. Cardiac and analgesic drugs. Journal of the American Geriatrics Society, 47, 40-50.
- Wagner, A.K., Zhang, F., Soumerai, S.B., Walker, A.M., Gurwitz, J.H., Glynn, R.J. and Ross-Degnan, D. (2004) Benzodiazepine use and hip fractures in the elderly: Who is at greatest risk? Archives of Internal Medicine, 164, 1567-1572. doi:10.1001/archinte.164.14.1567
- Hemmelgarn, B., Suissa, S., Huang, A., Boivin, J.F. and Pinard, G. (1997) Benzodiazepine use and the risk of motor vehicle crash in the elderly. JAMA, 278, 27-31. doi:10.1001/jama.1997.03550010041037
- Cumming, R.G. and Le Couteur, D.G. (2003) Benzodiazepines and risk of hip fractures in older people: A review of the evidence. CNS Drugs, 17, 825-837. doi:10.2165/00023210-200317110-00004
- Dawson, J., Boyle, J., Stanley, N., Johnsen, S., Hindmarch, I. and Skene, D.J. (2008) Benzodiazepine-induced reduction in activity mirrors decrements in cognitive and psychomotor performance. Human Psychopharmacology, 23, 605-613. doi:10.1002/hup.961
- Gray, S.L., Penninx, B.W., Blough, D.K., Artz, M.B., Guralnik, J.M., Wallace, R.B., Buchner, D.M. and LaCroix, A.Z. (2003) Benzodiazepine use and physical performance in community-dwelling older women. Journal of the American Geriatrics Society, 51, 1563-1570. doi:10.1046/j.1532-5415.2003.51502.x
- Korpi, E.R., Koikkalainen, P., Vekovischeva, O.Y., Makela, R., Kleinz, R., Uusi-Oukari, M. and Wisden, W. (1999) Cerebellar granule-cell-specific GABAA recaptors attenuate benzodiazepine-induced ataxia: Evidence from alpha 6-subunit-deficient mice. European Journal of Neuroscience, 11, 233-240. doi:10.1046/j.1460-9568.1999.00421.x
- Stanley, J.L., Lincoln, R.J., Brown, T.A., McDonald, L.M., Dawson, G.R. and Reynolds, D.S. (2005) The mouse beam walking assay offers improved sensitivity over the mouse rotarod in determining motor coordination deficits induced by benzodiazepines. Journal of Psychopharmacology, 19, 221-227. doi:10.1177/0269881105051524
- Licata, S.C., Jensen, J.E., Penetar, D.M., Prescot, A.P., Lukas, S.E. and Renshaw, P.F. (2009) A therapeutic dose of zolpidem reduces thalamic GABA in healthy volunteers: A proton MRS study at 4 T. Psychopharmacology, 203, 819-829. doi:10.1007/s00213-008-1431-1
- Malcolm, R., Myrick, H., Roberts, J., Wang, W., Anton, R.F. and Ballenger, J.C. (2002) The effects of carbamazepine and lorazepam on single versus multiple previous alcohol withdrawals in an outpatient randomized trial. Journal of General Internal Medicine, 17, 349-355.
- Korpi, E.R., Mattila, M.J., Wisden, W. and Luddens, H. (1997) GABA(A)-receptor subtypes: Clinical efficacy and selectivity of benzodiazepine site ligands. Annals of Internal Medicine, 29, 275-282. doi:10.3109/07853899708999348
- Greenblatt, D.J., Harmatz, J.S., von Moltke, L.L., Wright, C.E. and Shader, R.I. (2004) Age and gender effects on the pharmacokinetics and pharmacodynamics of triazolam, a cytochrome P450 3A substrate. Clinical Pharmacology & Therapeutics, 76, 467-479. doi:10.1016/j.clpt.2004.07.009
- Rijcken, C.A., Knegtering, H., Bruggeman, R., Tobi, H. and de Jong-van den Berg, L.T. (2005) Sex differences in concomitant medication with benzodiazepines or antidepressants in first-break schizophrenic patients treated with antipsychotic medication. Psychiatry Research, 134, 143-150. doi:10.1016/j.psychres.2003.06.005
- Blazer, D., Hybels, C., Simonsick, E. and Hanlon, J.T. (2000) Sedative, hypnotic, and antianxiety medication use in an aging cohort over ten years: A racial compareson. Journal of the American Geriatrics Society, 48, 1073-1079.
- Gleason, P.P., Schulz, R., Smith, N.L., Newsom, J.T., Kroboth, P.D., Kroboth, F.J. and Psaty, B.M. (1998) Correlates and prevalence of benzodiazepine use in community-dwelling elderly. Journal of General Internal Medicine, 13, 243-250. doi:10.1046/j.1525-1497.1998.00074.x
- Hohmann, A.A. (1989) Gender bias in psychotropic drug prescribing in primary care. Medical Care, 27, 478-490. doi:10.1097/00005650-198905000-00004
- Mayer-Oakes, S.A., Kelman, G., Beers, M.H., De Jong, F., Matthias, R., Atchison, K.A., Lubben, J.E. and Schweitzer, S.O. (1993) Benzodiazepine use in older, community-dwelling southern Californians: Prevalence and clinical correlates. The Annals of Pharmacotherapy, 27, 416-421.
- Van der Waals, F.W., Mohrs, J. and Foets, M. (1993) Sex differences among recipients of benzodiazepines in Dutch general practice. BMJ, 307, 363-366. doi:10.1136/bmj.307.6900.363
- Verhaeghe, W., Mets, T. and Corne, L. (1996) Benzodiazepine use among elderly patients presenting at the emergency room. Archives of Gerontology and Geriatrics, 22, 55-62. doi:10.1016/0167-4943(95)00677-X
- Boyd, A.R., Shivshankar, P., Jiang, S., Berton, M.T. and Orihuela, C.J. (2012) Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Experimental Gerontology, 47, 507-518. doi:10.1016/j.exger.2012.04.004
- Horstmann, S., Kahle, P.J. and Borasio, G.D. (1998) Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. Journal of Neuroscience Research, 52, 483-490. doi:10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4
- Skaper, S.D. and Walsh, F.S. (1998) Neurotrophic molecules: Strategies for designing effective therapeutic molecules in neurodegeneration. Molecular and Cellular Neuroscience, 12, 179-193. doi:10.1006/mcne.1998.0714
- Lee, J.C., Laydon, J.T., McDonnell, P.C., Gallagher, T.F., Kumar, S., Green, D., McNulty, D., Blumenthal, M.J., Heys, J.R., Landvatter, S.W., et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature, 372, 739-746. doi:10.1038/372739a0
- Jiang, Y., Chen, C., Li, Z., Guo, W., Gegner, J.A., Lin, S. and Han, J. (1996) Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). The Journal of Biological Chemistry, 271, 17920-17926. doi:10.1074/jbc.271.30.17920
- Stein, B., Yang, M.X., Young, D.B., Janknecht, R., Hunter, T., Murray, B.W. and Barbosa, M.S. (1997) p38-2, a novel mitogen-activated protein kinase with distinct properties. The Journal of Biological Chemistry, 272, 19509-19517. doi:10.1074/jbc.272.31.19509
- Lechner, C., Zahalka, M.A., Giot, J.F., Moller, N.P. and Ullrich, A. (1996) ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proceedings of the National Academy of Sciences of the United States of America, 93, 4355-4359. doi:10.1073/pnas.93.9.4355
- Li, Z., Jiang, Y., Ulevitch, R.J. and Han, J. (1996) The primary structure of p38 gamma: A new member of p38 group of MAP kinases. Biochemical and Biophysical Research Communications, 228, 334-340. doi:10.1006/bbrc.1996.1662
- Jiang, Y., Gram, H., Zhao, M., New, L., Gu, J., Feng, L., Di Padova, F., Ulevitch, R.J. and Han, J. (1997) Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. The Journal of Biological Chemistry, 272, 30122-30128. doi:10.1074/jbc.272.48.30122
- Lee, S.H., Park, J., Che, Y., Han, P.L. and Lee, J.K. (2000) Constitutive activity and differential localization of p38alpha and p38beta MAPKs in adult mouse brain. Journal of Neuroscience Research, 60, 623-631. doi:10.1002/(SICI)1097-4547(20000601)60:5<623::AID-JNR7>3.0.CO;2-4
- Nonaka, Y., Miyajima, M., Ogino, I., Nakajima, M. and Arai, H. (2008) Analysis of neuronal cell death in the cerebral cortex of H-Tx rats with compensated hydrocephalus. Journal of Neurosurgery: Pediatrics, 1, 68-74. doi:10.3171/PED-08/01/068
- Xiong, W., Kojic, L.Z., Zhang, L., Prasad, S.S., Douglas, R., Wang, Y. and Cynader, M.S. (2006) Anisomycin activates p38 MAP kinase to induce LTD in mouse primary visual cortex. Brain Research, 1085, 68-76. doi:10.1016/j.brainres.2006.02.015
- Moriguchi, T., Toyoshima, F., Gotoh, Y., Iwamatsu, A., Irie, K., Mori, E., Kuroyanagi, N., Hagiwara, M., Matsumoto, K. and Nishida, E. (1996) Purification and identification of a major activator for p38 from osmotically shocked cells. Activation of mitogen-activated protein kinase kinase 6 by osmotic shock, tumor necrosis factor-alpha, and H2O2. The Journal of Biological Chemistry, 271, 26981-26988. doi:10.1074/jbc.271.43.26981
- Aydin, M.V., Sen, O., Kayaselcuk, F., Bolat, F., Tufan, K., Caner, H. and Altinors, N. (2005) Analysis and prevalence of inflammatory cells in subtypes of lumbar disc herniations under cyclooxygenase-2 inhibitor therapy. Journal of Neurology Research, 27, 609-612. doi:10.1179/016164105X49210
- Hensley, K., Floyd, R.A., Zheng, N.Y., Nael, R., Robinson, K.A., Nguyen, X., Pye, Q.N., Stewart, C.A., Geddes, J., Markesbery, W.R., Patel, E., Johnson, G.V. and Bing, G. (1999) p38 kinase is activated in the Alzheimer’s disease brain. Journal of Neurochemistry, 72, 2053-2058. doi:10.1046/j.1471-4159.1999.0722053.x
- Vereker, E., O’Donnell, E. and Lynch, M.A. (2000) The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. The Journal of Neuroscience, 20, 6811-6819.
- Jung, M.E., Ju, X., Metzger, D.B. and Simpkins, J.W. (2011) Ethanol withdrawal hastens the aging of cytochrome c oxidase. Neurobiology Aging, 33, 618.e21-32.
- Kulisz, A., Chen, N., Chandel, N.S., Shao, Z. and Schumacker, P.T. (2002) Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. American Journal of Physiology. Lung Cellular and Molecular Physiology, 282, L1324-L1329.
- Kumar, C.N., Andrade, C. and Murthy, P. (2009) A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. Journal of Studies on Alcohol and Drugs, 70, 467-474.
- Greenblatt, D.J., Von Moltke, L.L., Ehrenberg, B.L., Harmatz, J.S., Corbett, K.E., Wallace, D.W. and Shader, R.I. (2000) Kinetics and dynamics of lorazepam during and after continuous intravenous infusion. Critical Care Medicine, 28, 2750-2757. doi:10.1097/00003246-200008000-00011
- Papini, O., da Cunha, S.P., da Silva Mathes Ado, C., Bertucci, C., Moises, E.C., de Barros Duarte, L., de Carvalho Cavalli, R. and Lanchote, V.L. (2006) Kinetic disposition of lorazepam with focus on the glucuronidation capacity, transplacental transfer in parturients and racemization in biological samples. Journal of Pharmaceutical and Biomedical Analysis, 40, 397-403. doi:10.1016/j.jpba.2005.07.021
- Bourin, M., Hascoet, M., Mansouri, B., Colombel, M.C. and Bradwejn, J. (1992) Comparison of behavioral effects after single and repeated administrations of four benzodiazepines in three mice behavioral models. Journal of Psychiatry and Neuroscience, 17, 72-77.
- Naassila, M., Legrand, E., d’Alche-Biree, F. and Daoust, M. (1998) Cyamemazine decreases ethanol intake in rats and convulsions during ethanol withdrawal syndrome in mice. Psychopharmacology, 140, 421-428. doi:10.1007/s002130050785
- Wieland, S., Belluzzi, J., Hawkinson, J.E., Hogenkamp, D., Upasani, R., Stein, L., Wood, P.L., Gee, K.W. and Lan, N.C. (1997) Anxiolytic and anticonvulsant activity of a synthetic neuroactive steroid Co 3-0593. Psychopharmacology, 134, 46-54. doi:10.1007/s002130050424
- Rewal, M., Jung, M.E. and Simpkins, J.W. (2004) Role of the GABA-A system in estrogen-induced protection against brain lipid peroxidation in ethanol-withdrawn rats. Alcoholism: Clinical and Experimental Research, 28, 1907-1915. doi:10.1097/01.ALC.0000148100.78628.E7
- Guo, G. and Bhat, N.R. (2007) p38alpha MAP kinase mediates hypoxia-induced motor neuron cell death: A potential target of minocycline’s neuroprotective action. Neurochemical Research, 32, 2160-2166. doi:10.1007/s11064-007-9408-8
- Sudo, T., Yagasaki, Y., Hama, H., Watanabe, N. and Osada, H. (2002) Exip, a new alternative splicing variant of p38 alpha, can induce an earlier onset of apoptosis in HeLa cells. Biochemical and Biophysical Research Communications, 291, 838-843. doi:10.1006/bbrc.2002.6529
- Guan, J., Luo, Y. and Denker, B.M. (2005) Purkinje cell protein-2 (Pcp2) stimulates differentiation in PC12 cells by Gbetagamma-mediated activation of Ras and p38 MAPK. Biochemical Journal, 392, 389-397. doi:10.1042/BJ20042102
- Ju, X., Wen, Y., Metzger, D. and Jung, M. (2013) The role of p38 in mitochondrial respiration in male and female mice. Neuroscience Letters, 544, 152-156. http://authors.elsevier.com/sd/article/S0304394013003534
- Bruchas, M.R., Schindler, A.G., Shankar, H., Messinger, D.I., Miyatake, M., Land, B.B., Lemos, J.C., Hagan, C.E., Neumaier, J.F., Quintana, A., Palmiter, R.D. and Chavkin, C. (2011) Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron, 71, 498-511. doi:10.1016/j.neuron.2011.06.011
- Park, G.B., Kim, Y.S., Lee, H.K., Song, H., Kim, S., Cho, D.H. and Hur, D.Y. (2011) Reactive oxygen species and p38 MAPK regulate Bax translocation and calcium redistribution in salubrinal-induced apoptosis of EBV-transformed B cells. Cancer Letters, 313, 235-248. doi:10.1016/j.canlet.2011.09.011
- Mausset, A.L., De Seze, R., Montpeyroux, F. and Privat, A. (2001) Effects of radiofrequency exposure on the GABAergic system in the rat cerebellum: Clues from semi-quantitative immunohistochemistry. Brain Research, 912, 33-46. doi:10.1016/S0006-8993(01)02599-9
- Jung, M.E., Yang, S.H., Brun-Zinkernagel, A.M. and Simpkins, J.W. (2002) Estradiol protects against cerebellar damage and motor deficit in ethanol-withdrawn rats. Alcohol, 26, 83-93. doi:10.1016/S0741-8329(01)00199-9
- Watanabe, M. (2008) Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. The Tohoku Journal of Experimental Medicine, 214, 175- 190. doi:10.1620/tjem.214.175
- Sankar, R. (2011) GABA(A) receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs, 26, 229-244. doi:10.2165/11599020-000000000-00000
- Verster, J.C., Volkerts, E.R. and Verbaten, M.N. (2002) Effects of alprazolam on driving ability, memory functioning and psychomotor performance: A randomized, placebo-controlled study. Neuropsychopharmacology, 27, 260-269. doi:10.1016/S0893-133X(02)00310-X
- Ashton, H. (1994) The treatment of benzodiazepine dependence. Addiction, 89, 1535-1541. doi:10.1111/j.1360-0443.1994.tb03755.x
- [61] Savic, M.M., Huang, S., Furtmuller, R., Clayton, T., Huck, S., Obradovic, D.I., Ugresic, N.D., Sieghart, W., Bokonjic, D.R. and Cook, J.M. (2008) Are GABAA receptors containing alpha5 subunits contributing to the sedative properties of benzodiazepine site agonists? Neuropsychopharmacology, 33, 332-339. doi:10.1038/sj.npp.1301403
- [62] Lalonde, R. and Strazielle, C. (2003) Motor coordination, exploration, and spatial learning in a natural mouse mutation (nervous) with Purkinje cell degeneration. Behavior Genetics, 33, 59-66. doi:10.1023/A:1021003600900
- [63] Rewal, M., Jung, M., Wen, Y., Brun-Zinkernagel, A. and Simpkins, J. (2003) Role of the GABAA system in behavioral, motoric, and cerebellar protection by estrogen during ethanol withdrawal. Alcohol, 31, 49-61. doi:10.1016/j.alcohol.2003.07.005
- [64] Rakotomamonjy, J., Levenes, C., Baulieu, E.E., Schumacher, M. and Ghoumari, A.M. (2011) Novel protective effect of mifepristone on detrimental GABAA receptor activity to immature Purkinje neurons. The FASEB Journal, 25, 3999-4010. doi:10.1096/fj.11-183384
- [65] Garrard, P., Martin, N.H., Giunti, P. and Cipolotti, L. (2008) Cognitive and social cognitive functioning in spinocerebellar ataxia: A preliminary characterization. Journal of Neurology, 255, 398-405. doi:10.1007/s00415-008-0680-6
- [66] Lalonde, R. and Strazielle, C. (2003) The effects of cerebellar damage on maze learning in animals. Cerebellum, 2, 300-309. doi:10.1080/14734220310017456
- [67] Madamanchi, N.R. and Runge, M.S. (2007) Mitochondrial dysfunction in atherosclerosis. Circulation Research, 100, 460-473. doi:10.1161/01.RES.0000258450.44413.96
- [68] Sarnowska, A., Beresewicz, M., Zablocka, B. and Domanska-Janik, K. (2009) Diazepam neuroprotection in excitotoxic and oxidative stress involves a mitochondrial mechanism additional to the GABAAR and hypothermic effects. Neurochemistry International, 55, 164-173. doi:10.1016/j.neuint.2009.01.024
- [69] Davis, L.F., Gatz, E.E. and Jones, J.R. (1971) Effects of chlordiazepoxide and diazepam on respiration and oxidative phosphorylation in rat brain mitochondria. Biochemical Pharmacology, 20, 1883-1887. doi:10.1016/0006-2952(71)90387-X
- [70] Roald, O.K., Steen, P.A., Milde, J.H. and Michenfelder, J.D. (1986) Reversal of the cerebral effects of diazepam in the dog by the benzodiazepine antagonist Ro15-1788. Acta Anaesthesiologica Scandinavica, 30, 341-345. doi:10.1111/j.1399-6576.1986.tb02427.x
- [71] Hirsch, J.D., Beyer, C.F., Malkowitz, L., Beer, B. and Blume, A.J. (1989) Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Molecular Pharmacology, 35, 157-163.
- [72] Chelli, B., Falleni, A., Salvetti, F., Gremigni, V., Lucacchini, A. and Martini, C. (2001) Peripheral-type benzodiazepine receptor ligands: Mitochondrial permeability transition induction in rat cardiac tissue. Biochemical Pharmacology, 61, 695-705. doi:10.1016/S0006-2952(00)00588-8
- [73] Fan, M., Rhee, J., St-Pierre, J., Handschin, C., Puigserver, P., Lin, J., Jaeger, S., Erdjument-Bromage, H., Tempst, P. and Spiegelman, B.M. (2004) Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: Modulation by p38 MAPK. Genes & Development, 18, 278-289. doi:10.1101/gad.1152204
- [74] Appaix, F., Guerrero, K., Rampal, D., Izikki, M., Kaambre, T., Sikk, P., Brdiczka, D., Riva-Lavieille, C., Olivares, J., Longuet, M., Antonsson, B. and Saks, V.A. (2002) Bax and heart mitochondria: Uncoupling and inhibition of respiration without permeability transition. Biochimica et Biophysica Acta, 1556, 155-167. doi:10.1016/S0005-2728(02)00358-4
- [75] Simpkins, J.W., Wang, J., Wang, X., Perez, E., Prokai, L. and Dykens, J.A. (2005) Mitochondria play a central role in estrogen-induced neuroprotection. Current Drug Targets-CNS & Neurological Disorders, 4, 69-83. doi:10.2174/1568007053005073
- [76] Blouin, R.T., Conard, P.F., Perreault, S. and Gross, J.B. (1993) The effect of flumazenil on midazolam-induced depression of the ventilatory response to hypoxia during isohypercarbia. Anesthesiology, 78, 635-641. doi:10.1097/00000542-199304000-00004
- [77] Megarbane, B., Lesguillons, N., Galliot-Guilley, M., Borron, S.W., Trout, H., Decleves, X., Risede, P., Monier, C., Boschi, G. and Baud, F.J. (2005) Cerebral and plasma kinetics of a high dose of midazolam and correlations with its respiratory effects in rats. Toxicology Letters, 159, 22-31. doi:10.1016/j.toxlet.2005.04.003