American Journal of Analytical Chemistry
Vol. 3 No. 3 (2012) , Article ID: 18273 , 9 pages DOI:10.4236/ajac.2012.33034
Multiresidue Method for Determination of 67 Pesticides in Water Samples Using Solid-Phase Extraction with Centrifugation and Gas Chromatography-Mass Spectrometry
1Analysis of Pesticides and Organic Pollutants Laboratory LAPPO, Lebanese Atomic Energy Commission LAEC, National Council for Scientific Research CNRS, Beirut, Lebanon
2Laboratory of Analysis of Organic Compounds (509), Faculty of Sciences I, Lebanese University, Beirut, Lebanon
3ISM-LPTC-UMR 5255 (Laboratory of Physicoand Toxico-Chemistry), CNRS, Université Bordeaux, Talence, France
Email: fjaber@cnrs.edu.lb
Received December 1, 2011; revised January 8, 2012; accepted February 17, 2012
Keywords: Multi-Residue; Pesticides; Solid-Phase Extraction; Centrifugation; Gas Chromatography-Mass Spectrometry
ABSTRACT
A new multi-residue method based on solid-phase extraction (SPE) with centrifugation was developed for determination and quantitation of 67 pesticides in water samples. Two SPE cartridges were tested: Chromabond C18 and Oasis HLB. Parameters that influence the extraction efficiency such as the eluent volume, the sample loading volume, the addition of organic solvent to water sample, sorbent drying and elute concentration were optimized. The innovation of this work was the examination of the use of a centrifugation technique in both the drying and elution steps. When combined with centrifugation, the volume of the elution solvent was reduced to 2 mL and the time for sorbent drying decreased also to 10 min under vacuum. Under the optimized conditions, this method showed good recoveries higher than 65% - 68% for the 67 analyzed pesticides using the C18 and HLB cartridges with relative standard deviations lower than 9.7% - 12.3%. Limits of quantification were between 2 and 20 ng∙L–1. The simplicity of the described method, use of less of organic solvent, short procedure time, and good recoveries demonstrate the advantages of this environmentally friendly approach for routine analysis of numerous samples.
1. Introduction
Organic contaminants present in the environment are a result of different sources of pollution from anthropogenic activities [1]. The pesticides, generated by the intensification of agriculture, are regarded as some of the most dangerous contaminants of the environment, despite their numerous merits. Not only are they toxic; they are also mobile and capable of bioaccumulation. On top of this, they can take part in various physical, chemical and biological processes. Many of these pesticides are characterized by a strong persistence which explains their wide presence in the different compartments of the environment [1,2]. Due to these physicochemical characteristics and their extensive use, many of these pesticides end-up in surface and ground water. They are found nowadays in all surface waters and in a growing number of aquifers. Their presence in water is considered as a potential risk not only for drinking water quality and human health, but also for ecosystems [3].
In this context, strict regulations for the control of pesticide residues concentration levels in environment have been established. According to the European Union Directive on drinking water quality (98/83/EC), the admissible concentration for pesticides is 0.1 µg∙L–1 for each individual substance and 0.5 µg∙L–1 for the sum of pesticides (including their metabolites) [4]. In the field of environmental water policy, the European Union adopted the Water Framework Directive (WFD) (2000/60/EC), whose objectives are to improve, protect and prevent further deterioration of water quality. The maximal concentrations authorized can vary from the ng∙L–1 order to few µg∙L–1 [5]. Such imposed regulations have driven the development of novel analytical techniques and improvement of existing ones, so that the largest possible number of compounds at low levels can be determined in one sensitive method.
For instrumental analysis, gas chromatography (GC) or liquid chromatography (LC) followed by mass spectrometry detection (MS) or tandem mass spectrometry (MS-MS) are the most commonly used techniques for the quantification of pesticides in water. However, in spite of the recent technical progress, the pesticide quantification in water requires a first step of extraction and preconcentration [6]. This sample preparation step is, in fact, the critical step of the whole analysis. The most common method is solidphase extraction (SPE) [7-9], which can be used to determine a broad range of pesticides in one analysis.
Modern trends in sample preparation are towards the simplification of the process and minimizing the organic solvent used. In our previous paper [10], we have reported a rapid and reliable method based on SPE with centrifugation for the extraction and followed by GC/MS detection for the determination of the 16 PAH identified by the US environmental protection agency (US-EPA) as priority pollutants in water samples. The focus of the present research is the development of a similar rapid multi-residue method for the analysis and ultra-trace quantification of 67 pesticides from different classes in waters. The potential factors affecting SPE extraction of the analytes were optimized and discussed in details. Then, the whole procedure was validated.
2. Experimental
2.1. Reagents, Standards and Small Apparatus
Acetonitrile (ACN), acetone, dichloromethane (DCM), hexane (Hex) and methanol (MeOH) (HPLC grade ≥ 99.9%) were purchased from Sigma-Aldrich (SigmaAldrich, St Louis, MO, USA). Pesticide standards of high purity level (98% - 99.8%) were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany) and ChemService (West Chester, PA, USA). Individual standard pesticide solutions were prepared in acetonitrile with a concentration of about 1000 mg∙L–1. Three standard mixture pesticide solutions were prepared by diluting each individual standard solution with acetonitrile, in order to get a concentration of about 35 mg∙L–1 for each compound. Working standards solutions were prepared by diluting the three standard mixture pesticide solutions with acetonitrile at a concentration of 1.0 mg∙L–1. All standard solutions were stored in dark below –20˚C.
An analytical balance (0.01 mg, Sartorius, Germany), an ultrasonic bath (Type T 760 DH, Prolabo, VWR International, France), a vortex shaker (Model VM-300P, Gemmy industrial corporation, Taiwan), a centrifuge (model U-320R, Boeco, Germany) and a nitrogen sample concentrator (Techne, UK) were used during experiments.
2.2. Solid-Phase Extraction
Two different SPE cartridges were evaluated: Chromabond C18ec, 200 mg, 3 mL, 45 µm from Machery-Nagel (Düren, Germany) and Oasis® HLB (hydrophilic-lipophilic balance) 60 mg, 3 mL, 50 µm from Waters (Milford, MA, USA). The SPE procedure was performed using a VacElut vacuum manifold from Varian (Santa Clara, CA, USA) with a Visiprep large-volume sampler from Supelco (Sigma-Aldrich, St. Louis, MO, USA).
For SPE optimization, blank water samples were spiked with the working standard mixed solution and with the surrogate standard (PCB 52) prepared in MeOH, and then they were shaken manually and with ultrasonic bath to enhance homogenization. The outlines of the SPE procedure used for the extraction of pesticides in water samples were described in our previous paper [10]. The Chromabond C18 and Oasis HLB cartridges were preconditioned and activated at the beginning with 3 mL of methanol and then 3 mL of water. Each water sample was percolated using a regulated vacuum through the SPE cartridge with a flow rate of 5 - 10 mL∙min–1, and then the cartridge was dried immediately either under vacuum or using centrifugation technique (2500 rpm for 2 min) followed by 10 min under vacuum. The analytes were eluted using the centrifugation technique, where 1 mL of the elution solvent was added to the cartridge with the valve closed, then the cartridge was centrifuged (2300 rpm for 1 min), and finally the elute was recovered (Figure 1). This step was repeated if necessary. The extract was collected and evaporated to about 100 μL with a weak nitrogen stream. The obtained extract was reconstituted in 200 μL of acetonitrile, and the solution of recovery internal standards (PCB 153) was added. Samples were stored in the dark at about –20˚C until GC-MS analysis.
For method application, groundwater samples were collected in 2.5 L dark glass bottles from three different geographic points located in North Lebanon (Denniyeh), East Bekaa (Terbol) and in South Lebanon (Abbassieh).
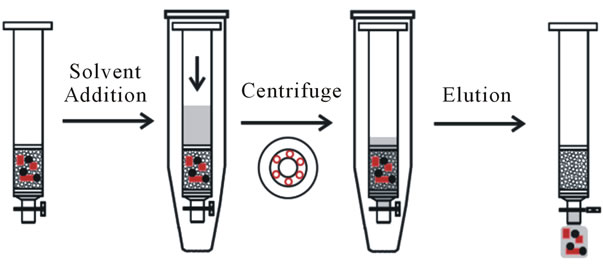
Figure 1. Schematic representation of SPE elution procedure using centrifugation.
Sampling was performed in November 2010 and samples were filtered immediately through a Whatman GF/F filter (0.7 μm porosity), extracted and analyzed.
2.3. GC-MS Analysis
An Agilent 6890N GC connected to an Agilent 5975 MS was used (Agilent technologies, USA). The GC-MS was equipped with Agilent 7683 B autosampler and split/ splitless injector with electronic pressure control. The column used was a capillary column (HP-5MS, 30 m, 0.25 mm i.d., 0.25 µm, Agilent J & W GC columns). The temperature program was the following: initial temperature 70˚C, held for 1 min, 10˚C∙min–1 ramp to 160˚C then held for 5 min, finally by 3˚C∙min–1 to 240˚C and held for 18.5 min. The total analysis time was 60.17 min and the equilibration time 0.5 min. The temperature of the injection port was 250˚C and a 1 µL volume was injected in splitless mode. Helium was used as carrier gas at a constant flow of 1 mL∙min–1. The mass spectrometer was operated in electron ionization mode with an ionizing energy of 70 eV, ion source temperature 230˚C, MS quadruple temperature 150˚C, and solvent delay 5.5 min.
Analysis was performed in the selected ion monitoring mode (SIM) based on the use of one target and two or three qualifier ions. Target and qualifier abundances were determined by injection of individual pesticide standards under the same chromatographic conditions in full-scan mode with the mass/charge ratio ranging from m/z 50 to 550. Pesticides were identified according to the retention times, the target and qualifier ions, and the qualifier to target abundance ratios.
2.4. Quantification
When using GC-MS analysis, samples were confirmed to contain a pesticide if the observed retention times of peaks of sample solutions were the same as the peaks for the standards in solvent and the observed abundance ratio of ions was identical to that of the standard ions. Instrumental and procedural blanks were analyzed to avoid laboratory contamination and analytical interferences, and the blank value must be deducted from the results. For the SPE optimization, the quantification was based on peak area ratio of the target ion divided by the internal standard (PCB 153) in order to evaluate the recoveries of the analyzed pesticides. For the method validation, the quantification was done according to the surrogate standard (PCB 52) added from the beginning of the analytical procedure.
3. Results and Discussion
3.1. Selection of Pesticides
After the study of the pesticides most commonly used in Lebanon, a total of 104 pesticides were initially considered for this study. Under the instrumental conditions given above, efforts were undertaken to optimize the tuning parameters of all the pesticides in the GC-MS in flow injection mode and then their analytical performances were tested in different scan modes. Out of the total pesticides selected, only 67 pesticides were finally found to be suitable for GC-MS analysis. The others were eliminated because either no peaks were observed or their sensitivity was extremely low and did not meet the requirements. Some pesticides were removed due to poor stability; they either decomposed rapidly after preparation of the mixed standard solutions or failed a series of experiments, for example recovery after fortification. Eventually, 67 pesticides from different chemical families including organochlorines, organophosphates, carbamates, pyrethroids, pyrimidines, azoles, triazoles and others, were chosen to be applicable to GC-MS determination. The analyzed pesticides cover a wide range of compounds employed in agriculture, like insecticides, herbicides, fungicides and acaricides. The studied pesticides are listed in Table 1.
3.2. Development of the Solid Phase Extraction Procedure
For the optimization of the SPE, several parameters were studied including sorbent selection 1), elution solvent 2), sample volume 3), addition of organic solvent to water sample 4), sorbent drying 5), and elute concentration 6). The complete extraction protocol is outlined in Figure 2.
3.2.1. Sorbent Selection (SPE Cartridge)
The pesticide compounds exhibit a wide range of polarities; they fall into the highly polar, medium polar and non-polar category with octanol-water partition coefficient (log Kow) between 0.7 and 7.3 for oxadixyl and bifenthrin respectively. With this in mind, two solid phase sorbents were chosen, Chromabond C18 and Oasis HLB. Both sorbents have been applied widely in the research of environmental pollutant chemistry [11,12], and they proved to be suitable for pesticides extraction from water samples [7,13-16].
Preliminary extraction experiments were first conducted with both sorbents to evaluate the efficiency of each cartridge in the analysis of the target pesticides. Three independent recovery assays were performed for each studied cartridge. The elution was done with solvents described in literature: DCM for C18 cartridges [17] and MeOH for HLB cartridges [18]. The volume used for elution was (×3) 3 mL. Recoveries showed that both cartridges allow good recovery percentage (higher than 50%) for most of the target compounds, except for tebuconazole, metalaxyl and oxadixyl which showed lower recoveries
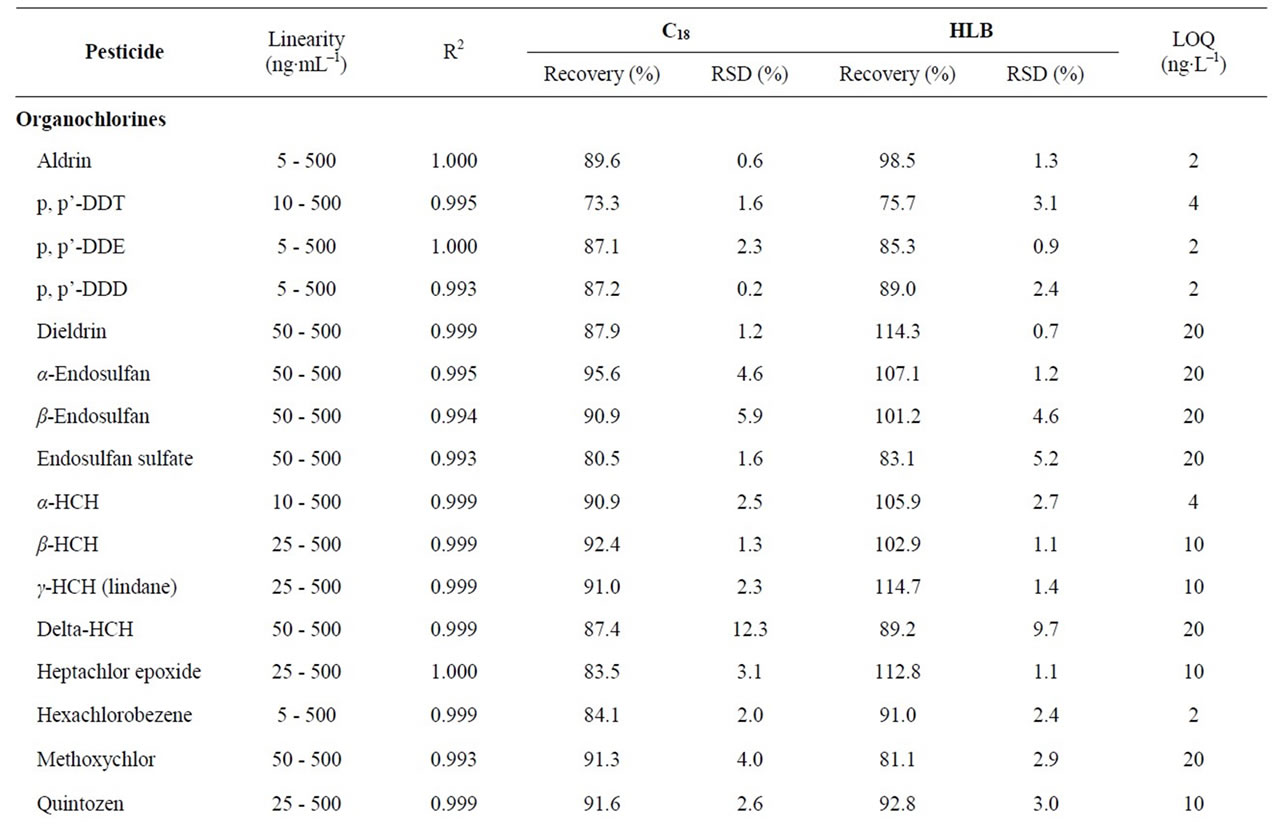
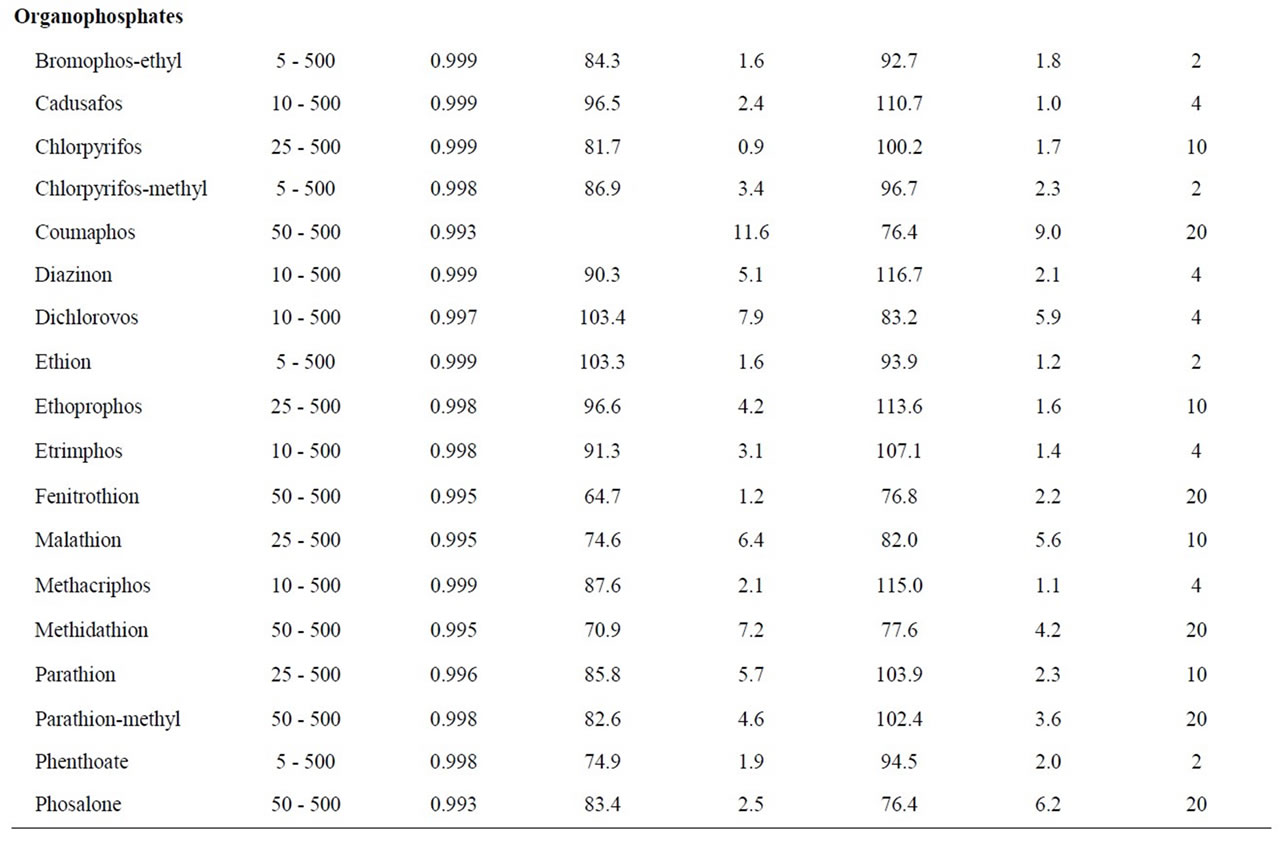
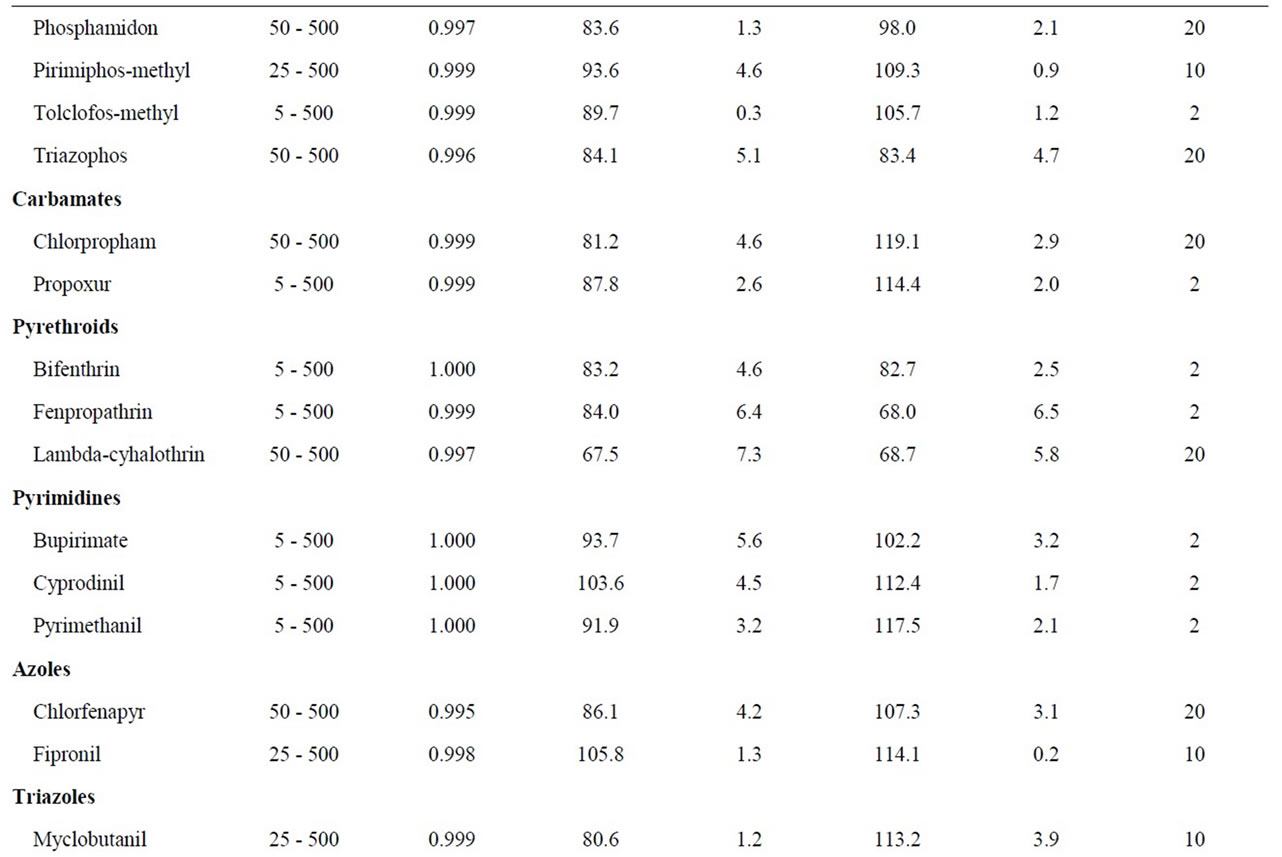
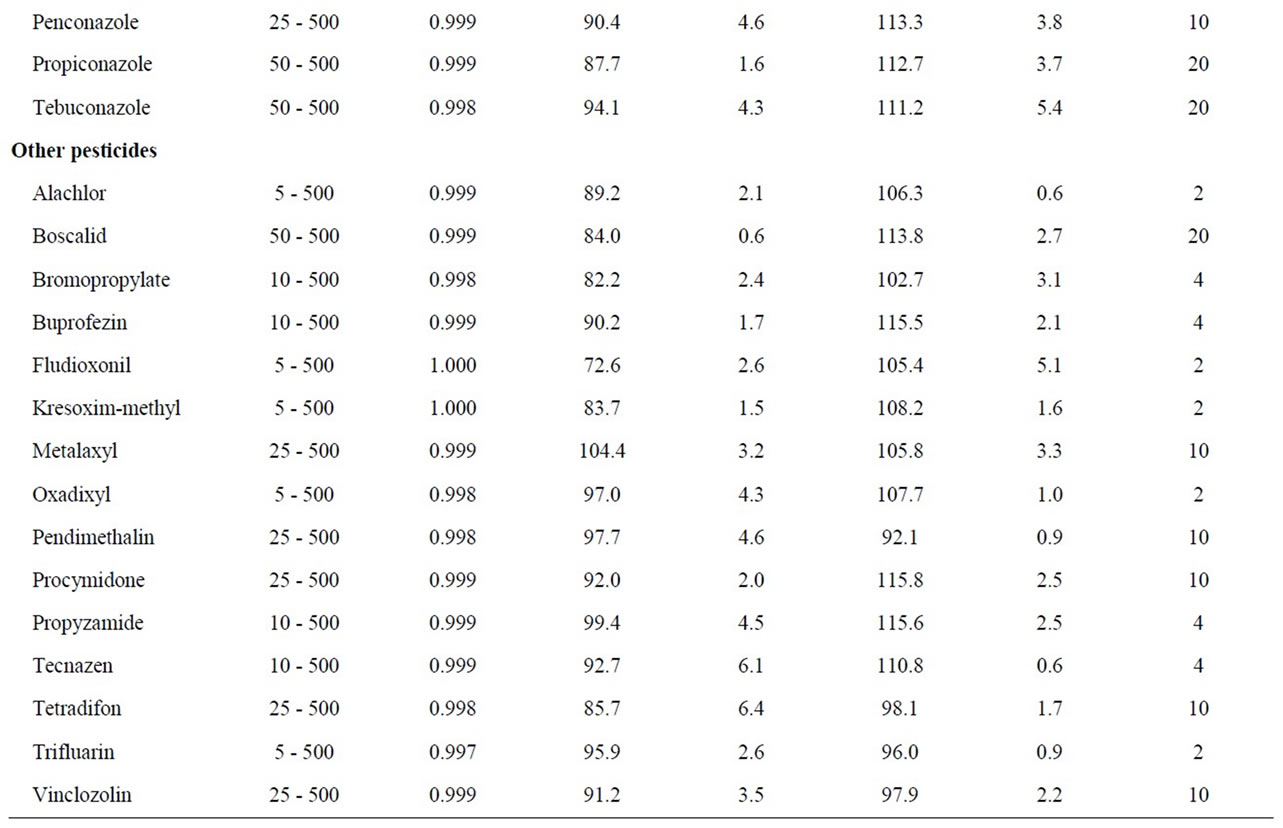
Table 1. Validation results: linear range, recoveries, repeatability and limits of quantitation (LOQs).
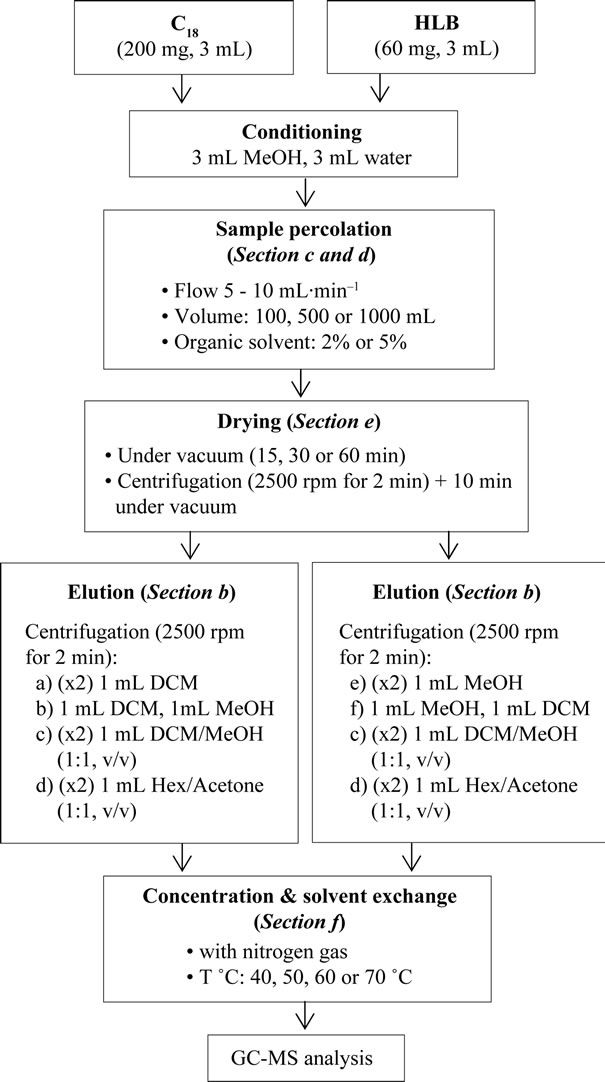
Figure 2. Schematic of the developed analytical method for the water sample extraction and analysis.
on C18 cartridges (<10%). The poor recovery for the three polar pesticides (log Kow between 0.7 and 3.7) is rather due to the elution step than an adsorption process. Therefore both cartridges, C18 and HLB, were found suitable for the extraction of the selected pesticides from water and so considered for further optimization in this study.
3.2.2. Solvent Selection for Analyte Elution
The elution strength of the organic solvent depends on the type of sorbent used, so different elution solvents and procedures were evaluated for Chromabond C18 and Oasis HLB cartridges. The organic solvents considered in elution procedures were from different polarities including acetone, DCM, Hex, and MeOH.
For traditional elution, a large volume of the elution phase is percolated slowly on SPE cartridge to ensure desorption of analytes from the stationary phase. In our work, the centrifugation technique was applied as an alternate to traditional elution, in order to reduce the volume of the elution phase required. The efficiency of this technique was proved in our previous paper [10], where recoveries obtained using a small volume of elution phase with centrifugation were comparable to those obtained with large volume of elution phase in the traditional elution. The rapid rotation of the centrifuge ensures that the adsorbent is well impregnated with the elution solvent, enabling a complete desorption of the analytes, even the strongly adsorbed, into the solvent. In this new procedure, a small volume (1 mL) of the elution phase was added to the cartridge with the valve closed, then the cartridge was centrifuged (2300 rpm for 1 min), and the elute was recovered. This step was repeated and the total volume of elute was concentrated and analyzed by GC-MS.
For C18, the elution procedures were: (2×) 1 mL of DCM (procedure a), 1 mL of DCM followed by 1 mL of MeOH (procedure b), (2×) 1 mL of DCM/MeOH (1:1, v/v) (procedure c), (2×) 1 mL of Hex/Acetone (1:1, v/v) (procedure d). For HLB, the elution procedures were: (2×) 1 mL of MeOH (procedure e), 1 mL of MeOH followed by 1 mL of DCM (procedure f), (2×) 1 mL of DCM/ MeOH (1:1, v/v) (procedure c), (2×) 1 mL of Hex/Acetone (1:1, v/v) (procedure d). The extracts from each procedure were concentrated, exchanged into 200 µL of acetonitrile and analyzed by GC-MS to determine the recoveries for 50 ng of each pesticide.Results showed that the procedure b (DCM followed by MeOH) provided higher recoveries on average for elution of analytes adsorbed on C18 cartridges (92% vs 83% - 84% for procedures a, c, and d; DCM, DCM/MeOH and Hex/Acetone respectively). For the elution of analytes adsorbed on HLB cartridges, similar recoveries of 80% - 83% were obtained for elution using MeOH only and using MeOH followed by DCM (procedures e and f). The procedure f was more preferable as it provides higher recoveries for non-polar analytes, Lambda-cyhalothrin for example. The recoveries for other procedures were lower with averages of 78% for procedure c (DCM/MeOH) and 62% for procedure d (Hex/Acetone).
3.2.3. Sample Loading Volume
In addition to the selectivity, a higher preconcentration factor is usually expected in the trace level sample analysis. One of the main approaches is changing the volume of water sample to reach the analytical goal. However, large volume of sample will not lead to the maximum extraction factor sometimes because of breakthrough problems, when the analyte is no longer retained by the sorbent due to elution by sample loading volume. Predicting the breakthrough volume based on the analogy between LC and SPE has largely been described [19-21]. The breakthrough volume can vary with the concentration of the target compounds, which is more likely to breakthrough at higher concentration. So the breakthrough volume should be evaluated at the conservatively highest predictive concentration of the target compound in the research area.
In our work, the influence of the sample loading volume was investigated for both cartridges, Chromabond C18 and Oasis HLB, where loading volume was varied from 10 to 1000 mL of water fortified with 200 ng∙L–1. The results for C18 cartridges and HLB cartridges indicated that no apparent differences were found among the enrichment efficiencies for most of the pesticides analyzed over the range of sample volume loaded on C18 and HLB cartridges, except for the two highly polar pesticides with high solubility in water, dichlorovos and propoxur (log Kow between 1.54 and 1.9, solubility between 18,000 and 1900 mg∙L–1 respectively). The recovery of dichlorovos decreased significantly by 34% - 45%, and the recovery of propoxur fell by 4% - 31% for sample volumes of 500 and 1000 mL loaded on C18 cartridges. This meant the breakthrough volumes of these two compounds were very low on this type of cartridge. By contrast, the breakthrough volumes of these two compounds and other pesticides in this study exceeded 1000 mL for HLB cartridges based on the increase of recovery with volume observed. Due to comprehensive consideration of enhancing the enrichment factor and avoid excessive sample-handling time and applicability for routine analysis, 500 mL was adopted as the water sample volume for C18 and HLB cartridges.
However, low recoveries (lower than 60%) were observed for some target compounds, organochlorines and pyrethroids (p, p’-DDT, p, p’-DDE and p, p’-DDD; bifenthrin, fenpropathrin and lambda-cyhalothrin) for water volumes exceeding 10 mL, which probably resulted from the losses of these hydrophobic compounds induced by the adsorption on the inside wall of the glass sample bottle. This problem is discussed in the next section.
3.2.4. Addition of Organic Solvent
As explained in the last section (5.3.2.c), the addition of organic solvent to water sample was considered in our study to enhance the extraction efficiencies of the high hydrophobic pesticides. Generally, organic solvent addition can increase the solubility of analytes adsorbed on glass bottles in the aqueous phase. MeOH was the organic solvent added to 500 mL of spiked water sample with different percentages (5%, 10% and 20%) and tests were performed on C18 and HLB cartridges.
When adding MeOH to water sample, recoveries for most of the analyzed pesticides decreased significantly, especially with a percentage of 10% and 20% of MeOH. This decrease in recoveries was less pronounced for HLB cartridges than for C18 cartridges. On the other hand, whatever the percentage of MeOH added, recoveries of the hydrophobic organochlorines and pyrethroids were not strongly improved. Therefore, we decided not to add MeOH to the water samples to prevent losses of analytes due to breakthrough. Low recoveries of the most hydrophobic analytes could be improved by adding the surrogate standard to water sample simultaneously with pesticide standards before SPE extraction. The quantification of pesticides recoveries using the surrogate standard can improve the recoveries of these analytes, and therefore used for the method validation.
3.2.5. Sorbent Drying
Initial sorbent drying after the sample loading was performed on extraction sorbents that had been dried under high vacuum before solvent elution. The influence of the drying time (from 10 to 60 min) was investigated Chromabond C18 and Oasis HLB cartridges, after the percolation of 500 mL of blank water fortified with the suite of pesticide compounds (50 ng each). In our previous paper [10], we have shown that the use of the centrifugation technique before vacuum drying ensures the complete remove of residual water from SPE cartridge and reduce the necessary drying time. Therefore, removing water from SPE cartridge using centrifugation technique (2500 rpm for 2 min) followed by a short time vacuum (10 min) was evaluated as well. Results showed that a drying time of 15 or 30 min was not sufficient for C18 and HLB cartridges and recoveries of most of analytes improved with drying time to 60 min. Even small amounts of residual water can apparently affect the elution of most of analytes adsorbed on SPE sorbent. When comparing recoveries for cartridges dried for 60 min under vacuum and those dried using centrifugation followed by 10 min under vacuum, we noticed that there was little difference between recoveries. However, it was advantageous to use the centrifugation before vacuum drying in order to reduce the total sample preparation time.
3.2.6. Elute Evaporation Temperature
The effect of elute evaporation temperature was studied to determine the optimum temperature for the organic solvents to vaporize quickly but with smaller amounts of target compounds being lost during the process. Duplicate samples of the elution phase (1 mL DCM + 1 mL MeOH) containing 50 ng of each analyte were concentrated under gentle flow of nitrogen at four temperature levels, including 40˚C, 50˚C, 60˚C and 70˚C. For temperatures above 40˚C, significant losses occurred for two compounds (fenpropathrin, lambda-cyhalothrin), and there was tend for rest of the analytes among the increasing temperature. Therefore, a temperature of 40˚C was used in the final method. For the most volatile analyte, dichlorovos, recoveries werearound 36% for all the evaporation temperature tested.
3.3. Method Validation
The performance of the optimized SPE-GC-MS analysis method was validated evaluating the linearity, recoveries, precision and limits of quantification (LOQs). The results are listed in Table 1.
Linearity was determined for the instrumental response. The range of concentration studied was 5 - 500 µg∙L–1 analyzing reference standard solution at seven concentration levels (5, 25, 50, 100, 250 and 500 µg∙L–1). The calibration curves were plotted using a least-square regression analysis. Each compound showed good linearity for the GC-MS analysis in the studied working range, with correlation coefficient (R2) greater than 0.993.
The recovery and precision of the developed analytical method were evaluated for C18 and HLB cartridges. Recovery studies of all pesticides were performed at 100 ng∙L–1, which is the maximal admissible concentration set in the EU directive for individual pesticides in drinking water [4]. Precision was described as the value of relative standard deviation (RSDs) of the areas obtained for each analyte after the replicate (n = 6) analyses of spiked water samples. Recoveries were calculated with the surrogate standard (PCB 52) and values were above 65% - 68% for all the analytes using the C18 and HLB cartridges.The overall method repeatability for all pesticides expressed as RSDs was satisfactory and in the range of 0.2% - 12.3% for C18 and 0.2% - 9.7% for HLB.
The LOQs were experimentally determined at a signal-to-noise ratio (S/N) of 6, by the spiking of blank water sample at four levels (2, 4, 10 and 20 ng∙L–1, n = 6). LOQs obtained were in the low ng∙L–1 level for all analyzed pesticides (2 - 10 ng∙L–1), except for few compounds where a value of 20 ng∙L–1 was necessary to achieve the LOQ. As a result, this analytical method can be applied in routine for the trace analysis of pesticides in water.
3.4. Application to Real Water Sample
The developed SPE-GC-MS method was applied to the analysis of 67 pesticides in real groundwater samples collected from three different places in Lebanon in November 2010. Water samples were extracted by the developed SPE method according to the optimized conditions described above. For the ground water samples analyzed, no high pesticide concentration was found. Only few compounds were detected at very low concentration in one or more groundwater samples, such as aldrin, DDD, DDE, hexachlorobenzene and diazinon. This means that the presence of these persistent compounds, mainly used in the past, is not critical nowadays and only few traces are still detected in the groundwater system. One pesticide, metalaxyl was detected in the Terbol groundwater sample at a level of 14.5 ng∙L–1. This compound, mainly used as systemic fungicide to control plant diseases or water-mold fungi, is used on many food and feed crops, and on non-food, residential and greenhouse crops such as tobacco, ornamental plants, trees, shrubs and vines, and lawns and turf. Metalaxyl, moderately stable under normal environmental conditions (with a half-life of 400 days), is persistent and mobile at the same time. His tendency to leach in many soils was demonstrated with a potential to reach groundwater. Metalaxyl has also been detected in ground water in five states in the US at levels typically reaching up to 3 µg∙L–1 [22].
4. Conclusions
This work presents a new SPE method using the centrifugation technique for the extraction of 67 pesticides from water samples. The proposed method shows practical environmental and economical advantages in terms of sample preparation time, simplicity, reduction in solvent use, and cost and is particularly suitable for routine applications requiring a high sample throughput. Optimized conditions include the percolation of a sample volume of 500 on either C18 or HLB cartridges and a drying step using centrifugation followed by 10 min under vacuum. The evaluation of the elution parameters demonstrated that 1 mL of DCM with centrifugation followed by 1 mL of MeOH was successfully used for the elution of analytes on both cartridges. The concentration was performed at a temperature of 40˚C. Under the optimized conditions, this method showed good recoveries higher than 65% - 68% for the 67 analyzed pesticides using the C18 and HLB cartridges with relative standard deviations lower than 9.7% - 12.3%. Limits of quantification were between 2 and 20 ng∙L–1. The developed method was successfully applied for the analysis of traces of pesticides in groundwater samples collected from three different regions in Lebanon.
REFERENCES
- T. Reemtsma and M. Jekel, “Organic Pollutants in the Water Cycle, Wiley-VCH VerlagGmBH& Co. KGaA, Weinheim,” Journal of Chromatography A, Vol. 1150, 2006, pp. 267-278.
- J. A. C. Barth, P. Grathwohl and K. C. Jones, “Introduction to Special Issue Aqua Terra: Pollutant Behavior in the Soil, Sediment, Ground, and Surface Water System,” Environmental Pollution, Vol. 148, No. 3, 2007, p. 693. doi:10.1016/j.envpol.2007.01.043
- H. Bailey, L. Deanovic, E. Reyes, T. Kimball, K. Larson, K. Cortright, V. Conner and D. E. Hinton, “Diazinon and Chlorpyrifos in Urban Waterways in Northern California, USA,” Environmental Toxicology and Chemistry, Vol. 19, No. 1, 2000, p. 82. doi:10.1002/etc.5620190109
- European Union (EU), “Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption,” 1998.
- European Union (EU), “Directive 2000/60/EC of the European Parliament and of the Council Establishing a Framework for the Community Action in the Field of Water Policy,” 2000.
- J. J. Vreuls, A. J. H. Louter and U. A. Th. Brinkman, “On-Line Combination of Aqueous-Sample Preparation and Capillary Gas Chromatography,” Journal of Chromatography A, Vol. 856, 1999, pp. 279-314. doi:10.1016/S0021-9673(99)00432-X
- M. E. Baez, M. Rodriguez, O. Lastra and P. Contreras, “Solid-Phase Extraction of Organophosphorus, Triazine, and Triazole-Derived Pesticides from Water Samples. A Critical Study,” Journal of High Resolution Chromatography, Vol. 20, No. 11, 1997, pp. 591-596. doi:10.1002/jhrc.1240201105
- H. Sabik, R. Jeannot and B. Rondeau, “Multiresidue Methods Using Solid-Phase Extraction Techniques for Monitoring Priority Pesticides, Including Triazines and Degradation Products, in Ground and Surface Waters,” Journal of Chromatography A, Vol. 885, No. 1-2, 2000, pp. 217-236. doi:10.1016/S0021-9673(99)01084-5
- M. J. M. Wells and L. Z. Yu, “Solid-Phase Extraction of Acidic Herbicides,” Journal of Chromatography A, Vol. 885, No. 1-2, 2000, pp. 237-250. doi:10.1016/S0021-9673(00)00206-5
- A. Kouzayha, M. Al Iskandarani, S. Mokh, A. R. Rabaa, H. Budzinski and F. Jaber, “Optimization of a Solid- Phase Extraction Method Using Centrifugation for the Determination of 16 Polycyclic Aromatic Hydrocarbons in Water,” Journal of Agricultural and Food Chemistry, Vol. 59, No. 14, 2011, pp. 7592-7600. doi:10.1021/jf200123v
- M. Asi, A. Hussain and S. Muhmood, “Solid Phase Extraction of Pesticide Residues in Water Samples: Ddt and Its Metabolites,” International Journal of Environmental Research, Vol. 2, No. 1, 2008, pp. 43-48.
- E. Concha-Grana, M. Turnes-Carou, S. MuniateguiLorenzo, P. Lopez-Mahia, D. Prada-Rodriguez and D. Fernandez-Fernandez, “Evaluation of HCH Isomers And Metabolites in Soils, Leachates, River Water and Sediments of a Highly Contaminated Area,” Chemosphere, Vol. 64, 2006, p. 588. doi:10.1016/j.chemosphere.2005.11.011
- C. De la Colina, A. P. Heras, G. D. Cancela and F. Sanchez Rasero, “Determination of Organophosphorus and Nitrogen-Containing Pesticides in Water by Solid-Phase Extraction with Gas Chromatography with Nitrogen— Phosphorus Detection,” Journal of Chromatography A, Vol. 655, No. 1, 1993, pp. 127-132. doi:10.1016/0021-9673(93)87020-M
- United States Environmental Protection Agency (US-EPA), “Method 525: Determination of Organic Compounds in Drinking-Water by Liquid—Solid Extraction and GC-MS”.
- M. Forcada, J. Beltran, F. J. Lopez and F. Hernandez, “Multiresidue Procedures for Determination of Triazine and Organophosphorus Pesticides in Water by Use of Large-Volume PTV Injection in Gas Chromatography,” Chromatographia, Vol. 51, No. 5-6, 2000, pp. 362- 368. doi:10.1007/BF02490618
- Waters Corporation, “Organophosphorus Pesticides in Drinking Water and Fruit. Environmental and Agrochemical Applications Notebook,” 2002, p. 9.
- M. Tahara, R. Kubota, H. Nakazawa, H. Tokunaga and T. Nishimura, “Analysis of Active Oxon Forms of Nine Organophosphorus Pesticides in Water Samples Using Gas Chromatography with Mass Spectrometric Detection,” Journal of Health Science, Vol. 52, 2006, pp. 313-319. doi:10.1248/jhs.52.313
- A. M. Rodrigues, V. Ferreira, V. V. Cardoso, E. Ferreira and M. J. Benoliel, “Determination of Several Pesticides in Water by Solid-Phase Extraction, Liquid Chromatography and Electrospray Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1150, No. 1-2, 2007, pp. 267-278. doi:10.1016/j.chroma.2006.09.083
- M. C. Hennion, C. Cau-Dit-Coumes and V. Pichon, “Trace Analysis of Polar Organic Pollutants in Aqueous samples. Tools for the Rapid Prediction and Optimization of the Solid-Phase Extraction Parameters,” Journal of Chromatography. A, Vol. 823, No. 1-2, 1998, pp. 147- 161. doi:10.1016/S0021-9673(98)00479-8
- M. C. Hennion, “Solid-Phase Extraction: Method Development, Sorbents, and Coupling with Liquid Chromatography,” Journal of Chromatography A, Vol. 856, No. 1-2, 1999, pp. 3-54. doi:10.1016/S0021-9673(99)00832-8
- V. Pichon, “Solid-Phase Extraction for Multiresidue Analysis of Organic Contaminants in Water,” Journal of Chromatography A, Vol. 885, No. 1-2, 2000, pp. 195-215. doi:10.1016/S0021-9673(00)00456-8
- United States Environmental Protection Agency (USEPA), “Metalaxyl. Prevention, Pesticides and Toxic Substances (7508W) EPA-738-F-94-013,” 1994.