American Journal of Analytical Chemistry
Vol. 3 No. 2 (2012) , Article ID: 17290 , 6 pages DOI:10.4236/ajac.2012.32021
RP-HPLC Method for the Simultaneous Determination of Lisinopril and NSAIDs in API, Pharmaceutical Formulations and Human Serum
1United Biotechnologies, Karachi, Pakistan
2Fatima Jinnah Medical and Dental College, Karachi, Pakistan
3Jinnah University for Women, Karachi, Pakistan
Email: *drsafila@gmail.com, safila117@gmail.com
Received December 1, 2011; revised January 7, 2012; accepted January 15, 2012
Keywords: Lisinopril; NSAIDs; Method Validation; HPLC Determination
ABSTRACT
High performance liquid chromatographic method was developed valdated and applied for the simultaneous determination of lisinopril and NSAIDs in bulk, pharmaceuticals formulations and human serum. A Purospher star C18 (5 µm, 25 × 0.46 cm) column was used with mobile phase consisting of methanol: water: acetonitrile (80:17.5:2.5 v/v, pH 3.0) and quantitative evaluation was performed at 225 nm with a flow rate of 1.0 mL·min–1. The retention time of lisinopril was 2.2 min while naproxen, flurbiprofen, diclofenac sodium and mefenamic acid were found to be 4.0, 4.5, 5.0 and 6.7 min respectively. Suitability of this method for the quantitative determination of the drugs was proved by validation in accordance with the requirements laid down by International Conference on Harmonization (ICH) guidelines. The method is selective, precise, accurate and can be used for analysis of pharmaceutical preparations in quality control and clinical laboratories.
1. Introduction
Lisinopril (Figure 1) [1,2] a lysine-derivative, is a potent angiotensin converting enzyme inhibitor that mimics the structure of its substrate. It is primarily used in treatment of hypertension, congestive heart failure and also in preventing renal and retinal complications of diabetes [3].
Non-steroidal anti-inflammatory drugs (NSAIDs) usually indicated for the treatment of acute or chronic pain and inflammation are nonselective inhibitors of cyclooxygenase, inhibiting both cyclooxygenase-1 and cyclooxygenase-2 isoenzymes, which catalyzes the formation of prostaglandins and thromboxane from arachidonic acid [4].
Several studies have been reported on the interaction between ACE inhibitors and NSAIDs [5-11]. Likewise, there are number of possible drug interactions of lisinopril with NSAIDs [12-15].The study of potential drug interactions of lisinopril with various NSAIDs requires the quantitation of both of these drugs in various samples containing both of these types of drugs. A number of RP-HPLC methods are reported for the determination of lisinopril in dosage forms and spiked human plasma through derivatization [16] and in human plasma with fluorescence detection [17]. We earlier [18] reported a simultaneous method for the determination of lisinopril and statins in API, pharmaceutical formulations and human serum by RP-HPLC. However, in literature no HPLC method for separation and simultaneous determination of lisinopril and NSAIDs has been reported. The aim of the present study was to develop an efficient, reliable, accurate
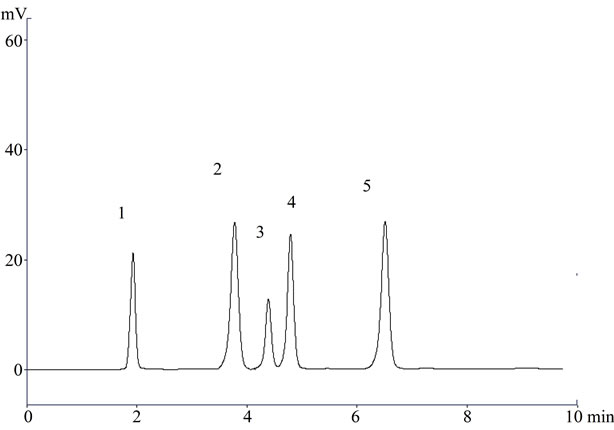
Figure 1. A representative chromatogram (1) Lisinopril; (2) Naproxen; (3) Flurbiprofen; (4) Diclofenac sodium; and (5) Mefenamic acid in raw material.
and sensitive method for the separation and quantitative determination of lisinopril and NSAIDs simultaneously as this would allow more efficient generation of clinical data at more modest cost than separate assays.
In present paper, we report a simple, easy, quick and inexpensive isocratic RP-HPLC method with ultraviolet detection at 225 nm for the simultaneous determination of LIS and four NSAIDs, i.e., diclofenac sodium, flurbiprofen, naproxen and mefenamic acid The method is equally valid for the determination in bulk materials, pharmaceutical dosage formulations and human serum. This method can be used for the quantitative analysis of NSAIDs and lisinopril alone or in combination. The low LOD and LOQ values merit the method for the determination of these drugs in clinical samples.
2. Experimental
2.1. Instrumentation
A Shimadzu HPLC system equipped with LC-10 AT VP pump and SPD-10 A VP UV–VIS detector was utilized. Chromatographic system was integrated via Shimadzu model CBM-102 to P-IV computer loaded with Shimadzu CLASS-VP software (Version 5.03) for data acquisition and mathematical calculations. Rheodyne manual injector was fitted with a 20 μL loop, Purospher® star C18 (5 µm, 25 × 0.46 cm) column a Hiber®, pre-packed Column RT 250-4.6 and DGU-14 AM on-line degasser. In addition, Mettler Toledo electronic balance, micro-liter syringe and micropore filtration assembly were used in this study.
2.2. Material and Reagents
Lisinopril, (Lisinopril® 5 mg) was kind gift from Atco Laboratories (Pvt) Ltd, NSAIDs used were diclofenac sodium (Voltral® 50 mg) from Novartis Pharmaceuticals (Pakistan) Ltd, flurbiprofen (Synalgo® 100 mg) from Platinum Pharmaceuticals (Pvt) Ltd., naproxen (Anex 275 mg) PharmEvo (Pvt) Ltd. and mefenamic acid (Ponstan® 250 mg) were obtained from Parke-Davis & Co Ltd. All these drugs had an expiry of not less than one year at the time of study. All reagents used were of HPLC grade. Acetonitrile, methanol and phosphoric acid 85% (Merk, Germany) and HPLC grade deionized filtered water were used to prepare the mobile phase. Stock solutions of lisinopril and NSAIDs were prepared in the mobile phase. Fresh working solutions were prepared daily. All solutions were filtered (0.45 μm) and degassed using sonicator.
2.3. Preparation of Solutions
Standard solutions of lisinopril and NSAIDs were prepared by dissolving appropriate amounts of each in mobile phase methanol: water: acetonitrile (80:17.5:2.5 v/v, pH 3.0) to obtain final drug concentrations of 100 μg·mL–1. For the calibration standards, seven calibrators of each drug were prepared by making serial dilutions from stock solutions. For the assay of pharmaceutical preparations, the content of 20 tablets were powdered, weighed and portion of the powder equivalent to the suitable amount of drug (according to the labeled claimed) was dissolved in mobile phase, sonicated and filtered in order to remove excepients and transferred into a 50 mL volumetric flask and diluted to the mark. Seven dilutions of each drug were prepared, portion of this solution was filtered through a disposable 0.45 μm filter and then injected to the rheodyne.
2.4. Serum Drug Analysis
Blood samples were collected from healthy volunteers and after cogulation centrifuged at 3000 rpm for 10 minutes. The supernatant (serum) obtained was stored at –20˚C. After thawing, serum was deprotinated by acetonitrile and spiked daily with working solutions to produce desired concentrations of lisinopril and NSAIDs. 20 μL volume of each sample was injected and chromatographed under above conditions.
2.5. Chromatographic Conditions
The chromatographic analysis was performed at ambient temperature with isocratic elution. The mobile phase consisted of methanol: water: acetonitrile (80:17.5:2.5 v/v) with pH adjusted to 3.0 with phosphoric acid (85%). The pump was set at a flow rate of 1.0 mL·min–1, sample volume of 20 μL was injected in triplicate onto the HPLC column and elute was monitored at 225 nm.
2.5.1. Method Development
In order to select a proper mobile phase for the separation of lisinopril and NSAIDs isocratic elution was applied. The optimization of the analytical procedure has been carried out by varying the mobile phase composition, flow rate and pH of the mobile phase. Preliminary the mobile phases investigated were methanol and water, broad peaks were recorded, therefore we could not use methanol and water as mobile phase. Optimal retention time for lisinopril was 2.2 min and for naproxen, flurbiprofen, diclofenac sodium and mefanimic acid was found to be 4.0, 4.5, 5 and 6.7 min and best resolution were achieved when mobile phase was methanol: water: acetonitrile (80:17.5:2.5 v/v) having pH adjusted to 3.0 with phosphoric acid. Mobile phase selection was based on peak parameters, ease of preparation and cost.
2.5.2. Validation Procedure
All validation steps were carried out according to the ICH guidelines. Method validation establishes that the method performance characteristics are suitable for the intended use. Various parameters of the method such as system suitability, selectivity, specificity, linearity (concentration-detector response relationship), accuracy, precision, sensitivity, detection and quantification limit recovery from the matrix were considered.
The system suitability was assessed by five replicate analyses of the drug at a concentration of 250 ng·mL–1. System suitability of the method was evaluated by analyzing the repeatability, peaks symmetry (symmetry factor), theoretical plates of the column, resolution between the peaks of NSAIDs and lisinopril, mass distribution ratio (capacity factor) and relative retention.
Specificity is the ability of a method to discriminate between the analyte of interest and other components that are present in the sample.
The specificity of the method was evaluated to ensure separation of lisinopril and NSAIDs. For demonstrating the specificity of the method for drug formulation, the drug and the excipients used in formulation products were spiked. The linearity of the method was evaluated at seven different concentrations that ranged from 2.5 - 100 μg·mL–1 for lisinopril and NSAIDs. Here the peak area using absorbance detection was studied for each drug. The accuracy of the method was evaluated from recovery assay which was made on the formulation samples. Thus, known amounts of each drug were prepared in triplicate at three levels (80%, 100% and 120%) and spiked into their corresponding formulation and the average recovery was calculated as the mean value obtained. To test the precision of the method, analysis were carried out on two different non-consecutive days. LOD and LOQ were calculated by the equation given in ICH guidelines. Ruggedness of this method was evaluated in two different labs with two different instruments. Lab 1 was in the Research Institute of Pharmaceutical Sciences, Faculty of Pharmacy University of Karachi, while Lab 2 was in the Department of Chemistry, Faculty of Science, University of Karachi.
3. Results and Discussion
The development of HPLC method for the determination of drugs has received considerable attention in recent years because of their importance in routine quality control analysis. HPLC methods generally requires complex and expensive equipment, provision for use and disposal of solvents, labor-intensive sample preparation procedure and personal skills in chromatographic techniques. The goal of this study was to develop a rapid, more accurate, precise reliable least time consuming HPLC method for the simultaneous determination of lisinopril and NSAIDs in the form of bulk drug samples, its formulations and human serum using the most commonly employed C-18 column with UV detector.
3.1. Method Development
In the present investigation the best separation of lisinopril and NSAIDs was achieved using a Purospher® star C18 (5 µm, 25 × 0.46 cm) column which provides efficient and reproducible separation of the components. Using other type of column under similar experimental condition, the separation lasted about 15 minutes. A mobile phase of methanol: water: acetonitrile (80:17.5:2.5 v/v) having pH adjusted with phosphoric acid to 3.0 provided a reproducible, baseline resolved peak. Small changes in pH of the mobile phase had a great influence to the chromatographic behavior of these substances, higher pH of the mobile phase also results in peak tailing and a lower pH retention time of NSAIDs and lisinopril was delayed. It is obvious from the chromatogram (Figures 1, 2 and 3) that NSAIDs and lisinopril eluted out having symmetrical peaks and were well separated from each other. The method was found to be rapid as lisinopril and NSAIDs eluted out at 2.2, 4.0, 4.5, 5 and 6.7 minutes respectively, which is important for routine analysis. In comparison with other published methods for determination of lisinopril and NSAIDs the advantages of this
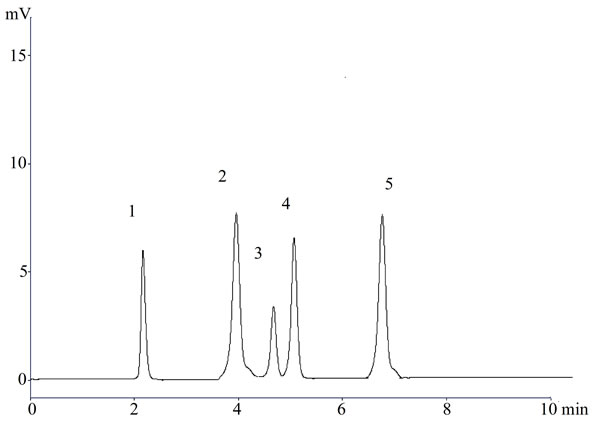
Figure 2. A representative chromatogram (1) Lisinopril; (2) Naproxen; (3) Flurbiprofen; (4) Diclofenac sodium; and (5) Mefenamic acid in dosage form.
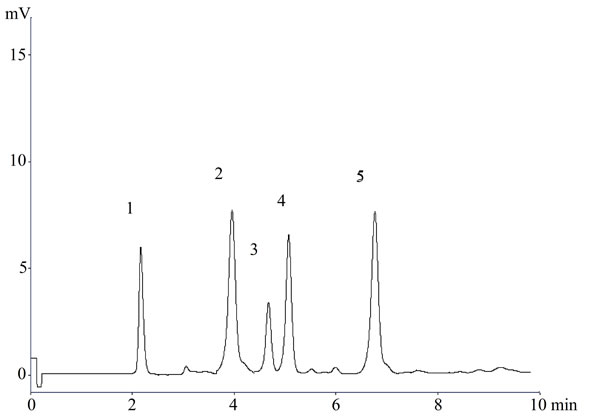
Figure 3. A representative chromatogram (1) Lisinopril; (2) Naproxen; (3) Flurbiprofen; (4) Diclofenac sodium; and (5) Mefenamic acid in serum.
method are ease of operation, short analysis time (total run time <10 minutes), utilization of readily available costeffective solvents, no matrix interferences, and satisfactory limit of quantification to enable pharmacokinetic studies of lisinopril and NSAIDs. Rapidness, sensitivity, simplicity, economical nature, acceptable resolution, good recovery and precision of this method gives it an advantage over the other reported HPLC methods for the determination of lisinopril and NSAIDs.
3.2. Method Validation
The newly developed method has been validated and holds well for the determination of drug in raw materials, dosage formulations and serum. For validation of analytical methods, the guidelines of the International Conference on the Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use have recommended the accomplishment of selectivity, specificity, linearity, accuracy test, precision, sensitivity, limit of detection and quantification of the method.
3.2.1. Selectivity and Specificity
The selectivity and specificity of the method was established through the study of resolution factor of the peak of lisinopril from that of NSAIDs. The method demonstrated good resolutions and was found to be free of interference from the excipients Figure 1 used in formulation products and thus, the method is specific for lisinopril and NSAIDs.
3.2.2. Range and Linearity
Table 1 shows the regression statistics of concentration analytical response, the standard deviation of the regression line and the optimum linear range (2.5 - 100 μg·mL–1) for each compound. Calibration curves were constructed in the range of expected concentrations (2.5 - 100 μg·mL–1) and were found to be linear within the quantification ranges for all the assayed drugs using a linear regression, excellent linearity was obtained in all cases with correlation coefficients <0.999.
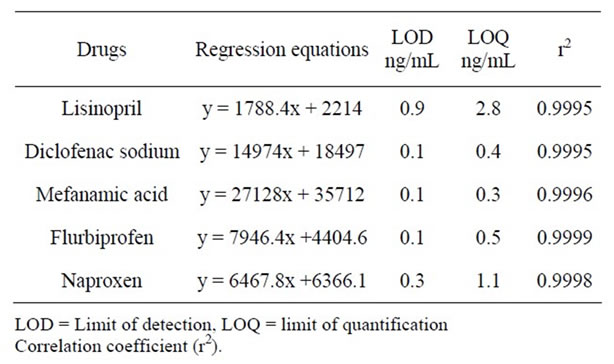
Table 1. Regression equations with LOD, LOQ.
3.2.3. Accuracy and Recovery
Data corresponding to these recovery assays for the studied analytes are presented in Table 2. The accuracy ranged from 99.9% - 100.03%, at low, medium and high levels for all investigated analytes. The data given in Table 2 shows that there is no significant difference between the amount of drug spiked in serum and the amount recovered. Thus, serum did not interfere with the estimation.
3.2.4. Precision
The intra-and inter-batch precision was evaluated by assaying the samples (Table 3). In this assay, the intrabatch precision and the inter-batch precision was ≤0.60 in bulk materials and 0.9% or less in human serum. The results demonstrated that the values were within the acceptable range and the method was sufficiently accurate and precise.
3.2.5. Ruggedness
The assay results indicated that the method was capable with high precision did not show any notable deviations from acceptable limits.
3.2.6. Limit of Detection and Quantification
The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be detected but not necessarily quantitated as an exact value. The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy. The LOD and LOQ are calculated as reported in literature given in Table 1.
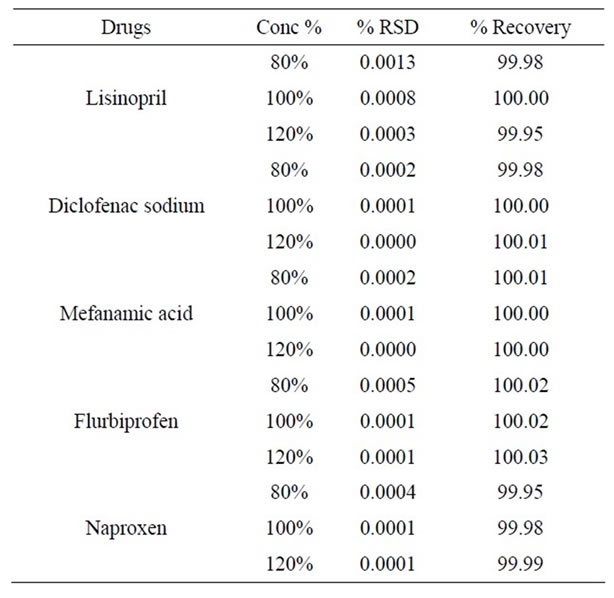
Table 2. Accuracy of lisinopril and NSAIDs.
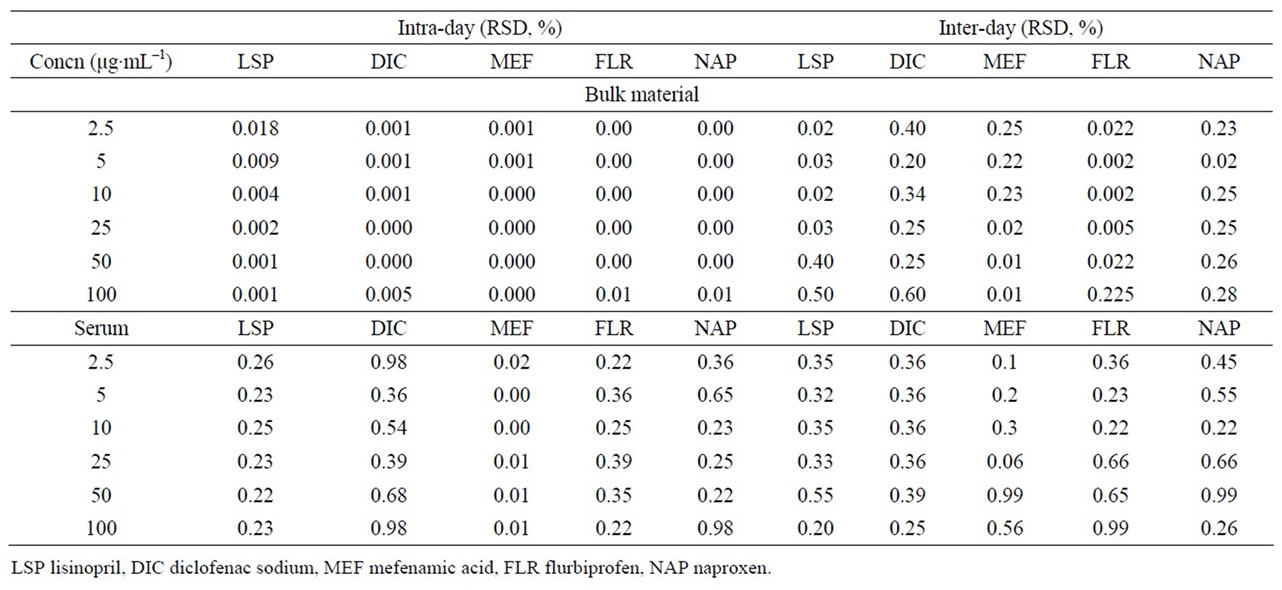
Table 3. Intra-day and inter-day precision of the method.
4. Conclusion
The new HPLC method described in this paper provides a simple, universal, convenient and reproducible approach for the simultaneous identification and quantification that can be used to determine lisinopril and any of the four NSAIDs. In summary, the proposed method can be used for the drug analysis in routine quality control. In addition, this method has the potential application to clinical research of drug combination, multi-drug pharmacokinetics and interactions.
REFERENCES
- A. A. Patchett, E. Harris, E. W. Tristram, M. J. Wyvratt, M. T. Wu, D. Taub, E. R, Peterson, T. J. Ikeler, J. Broeke, L. G. Payne, D. L. Ondeyka, E. D. Thorsett, W. J. Greenlee, N. S Lohr, R. D. Hoffsommer, H. Joshua, W. E. Ruyle, J. W. Rothrock, S. D. Aster, A. L. Maycock, F. M. Robinson, R. Hirschmann, C. S Sweet, E. H. Ulm, D. L. Gross, T. C. Vassil and C. A. Stone, “A New Class of Angiotensin-Converting Enzyme Inhibitors,” Nature, Vol. 288, No. 5788, 1980, pp. 280-283. doi:10.1038/288280a0
- The American Society of Health-System Pharmacists, “Lisinopril,” Retrieved 14 July 2011. http://www.drugs.com/monograph/lisinopril.html
- N. Chaturvedi, A. K. Sjolie, J. M. Stephenson, H. Abrahamian, M. Keipes, A. Castellarin, Z. Rogulja-Pepeonik and J. H. Fuller, “Effect of Lisinopril on Progression of Retinopathy in Normotensive People with Type 1 Diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus,” Lancet, Vol. 351, No. 9095, 1998, pp. 28-31.
- B. Hinz, O. Cheremina and K. Brune, “Acetaminophen (Paracetamol) Is a Selective Cyclooxygenase-2 Inhibitor in Man,” The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, Vol. 22, No. 2, 2008, pp. 383-390.
- N. Sultana, M. S. Arayne and R. U. Quraishi, “In vitro Interactions of Captopril with NSAID’s,” Pakistan Journal of Pharmaceutical Sciences, Vol. 19, No. 3, 2006, pp. 202-207.
- E. A. Chrischilles and R. B. Wallace, “Nonsteroidal AntiInflammatory Drugs and Blood Pressure in an Elderly Population,” Journals of Gerontology, Vol. 48, No. 3, 1993, pp. M91-M96.
- J. H. Gurwitz, D. E. Everitt, M. Monane, R. J. Glynn, I. Choodnovskiy, M. P. Beaudet and J. Avorn, “The Impact of Ibuprofen on the Efficacy of Antihypertensive Treatment with Hydrochlorothiazide in Elderly Persons,” The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, Vol. 51, No. 2, 1996, pp. M74-M79. doi:10.1093/gerona/51A.2.M74
- M. D .Guazzi, J. Campodonico, F. Celeste, M. Guazzi, G. Santambrogio, M. Rossi, D. Trabattoni and M. Alimento, “Antihypertensive Efficacy of Angiotensin Converting Enzyme Inhibition and Aspirin Counteraction,” Clinical Pharmacology & Therapeutics, Vol. 63, No. 1, 1998, pp. 79-86. doi:10.1016/S0009-9236(98)90123-0
- J. H. Gurwitz, J. Avorn, R. L. Bohn, R. J. Glynn, M. Monane and H. Mogun, “Initiation of Antihypertensive Treatment during Nonsteroidal Anti-Inflammatory Drug Therapy,” Journal of the American Medical Association (JAMA), Vol. 272, No. 10, 1994, pp. 781-786. doi:10.1001/jama.1994.03520100043031
- J. Polónia, I. Boaventura, G. Gama, I. Camões, F. Bernardo, P. Andrade, J. P. Nunes, F. Brandão and M. Cerqueira-Gomes, “Influence of Non-Steroidal Anti-Inflammatory Drugs on Renal Function and 24 h Ambulatory Blood Pressure-Reducing Effects of Enalapril and Nifedipine Gastrointestinal Therapeutic System in Hypertensive Patients,” Journal of Hypertension, Vol. 13, No. 8, 1995, pp. 925-931. doi:10.1097/00004872-199508000-00014
- W. Linz, G. Wiemer, P. Gohlke, T. Unger and B. A. Schölkens, “Contribution of Kinins to the Cardiovascular Actions of Angiotensin-Converting Enzyme Inhibitors,” Pharmacological Reviews, Vol. 47, No. 1, 1995, pp. 25- 49.
- V. Kumar, S. Malik and S. Singh, “Polypill for the Treatment of Cardiovascular Diseases Part 2. LC-MS/ TOF Characterization of Interaction/Degradation Products of Atenolol/Lisinopril and Aspirin, and Mechanisms of Formation Thereof,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 48, No. 3, 2008, pp. 619-628. doi:10.1016/j.jpba.2008.06.003
- K. Dubey, D. K. Balani, C. B. Tripathi, R. Singh, R. Bajaj and K. K. Pillai, “Adverse Interactions of Rofecoxib with Lisinopril in Spontaneously Hypertensive Rats,” Clinical toxicology, Vol. 43, No. 5, 2005, pp. 361-373. doi:10.1081/CLT-200066053
- R. Fogari, A. Zoppi, R. Carretta, F. Veglio, A. Salvetti and Italian Collaborative Study Group, “Effect of Indomethacin on the Antihypertensive Efficacy of Valsartan and Lisinopril: A Multicentre Study,” Journal of Hypertension, Vol. 20, No. 5, 2002, pp. 1007-1014. doi:10.1097/00004872-200205000-00037
- C. H. Brown, “Effect of Rofecoxib on the Antihypertensive Activity of Lisinopril,” The Annals of Pharmacotherapy, Vol. 34, No. 12, 2000, pp. 1486.
- A. A. El-Emam, S. H. Hansen, M. A. Moustafa, S. M. El-Ashry and D. T. El-Sherbiny, “Determination of Lisinopril in Dosage Forms and Spiked Human Plasma through Derivatization with 7-Chloro-4-nitrobenzo-2- oxa-1,3-diazole (NBD-Cl) Followed by Spectrophotometry or HPLC with Fluorimetric Detection,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 34, No. 1, 2004, pp. 35-44. doi:10.1016/j.japna.2003.08.021
- S. Olcay and E. Lale, “An HPLC Method for the Determination of Lisinopril in Human Plasma and Urine with Fluorescence Detection,” Journal of Chromatography B, Vol. 809, No. 1, 2004, pp. 159-165. doi:10.1016/j.jchromb.2004.06.014
- N. Sultana, M. S. Arayne and S. Naveed, “Validated Method for the Simultaneous Determination of Lisinopril, Pravastatin, Atorvastatin and Rosuvastatin in API, Formulations and Human Serum by RP-HPLC,” Chinese Journal of Chemistry, Vol. 29, No. 6, 2011, pp. 1216-1220. doi:10.1002/cjoc.201190226
NOTES
*Corresponding author.