Journal of Biophysical Chemistry
Vol.2 No.3(2011), Article ID:6743,11 pages DOI:10.4236/jbpc.2011.23029
More stable, more estrogenic: the SERM-ERα LBD complex
1Department of Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology, Stockholm, Sweden; 2School of Chemistry, National University of Ireland, Galway, Ireland; *Corresponding author: leif.eriksson@nuigalway.ie
Received 21 June 2011; revised 12 July 2011; accepted 25 July 2011.
Keywords: Breast Cancer; Tamoxifen Resistance; Molecular Dynamics Simulations; Dihydrobenzoxathiin; SERM
ABSTRACT
Many synthetic selective estrogen receptor modulators (SERMs) have been cocrystallized with the human estrogen receptor α ligand binding domain (ERα LBD). Despite stabilizing the same canonical inactive conformation of the LBD, most SERMs display different ligand-dependent pharmacological profiles. We show here that increased partial agonism of SERMs is associated with increased conformational stability of the SERM-LBD complexes, by investigation of dihydrobenzoxathiin-based SERMs using molecular modelling techniques. Analyses of tamoxifen (TAM) and 4-hydroxytamoxifen (OHT) in complex with the LBD furthermore indicates that the conversion of TAM to OHT increases both the affinity to ERα and the partial agonism of the anti-cancer drug, which provides a plausible explanation of the counterintuitive results of TAM therapy.
1. INTRODUCTION
It has been widely asserted that the incidence of breast cancer is 1 in 8 women over the course of lifetime in the United States. Approximately 70% of all breast cancers are hormone receptor positive and the patients will likely be treated by tamoxifen (TAM), the most commonly used selective estrogen receptor modulator (SERM), which exerts tissue-selective effects [1]. TAM works as an estrogen antagonist in breast cells, significantly reducing the risk of breast cancer recurrence [2,3]. At the same time, it works as an estrogen agonist in bone, in the cardiovascular system and in uterine tissue [4], with the undesirable utertropic side-effect of increased incidence of endometrial cancer [5]. A generally accepted mechanism is that TAM, and most SERMs, stabilize the estrogen receptor α ligand binding domain (ERα LBD) in the canonical inactive conformation, which differs from the estradiol induced active conformation. TAM and its metabolites competitively bind to the tumor ER, thereby blocking the binding of estrogen and inhibiting estrogen-induced tumor cell growth.
TAM is extensively metabolized by cytochrome P450 (CYP) enzymes (Scheme 1) and the biotransformation differs dramatically between individuals depending on the persons specific gene variants. The polymorphic variants of CYP enzymes can either increase or decrease the rate and amount of TAM that is metabolized. The 4-hydroxyl metabolites, OHT and endoxifen, are potent antiestrogens with much higher affinity to ER than TAM itself, as shown by the cocrystallized OHT-LBD complex (PDB code 3ERT [10]) visualized in Figure 1. OHT and endoxifen bind to ERα forming hydrogen bonds between the additional 4-hydroxy group and the two
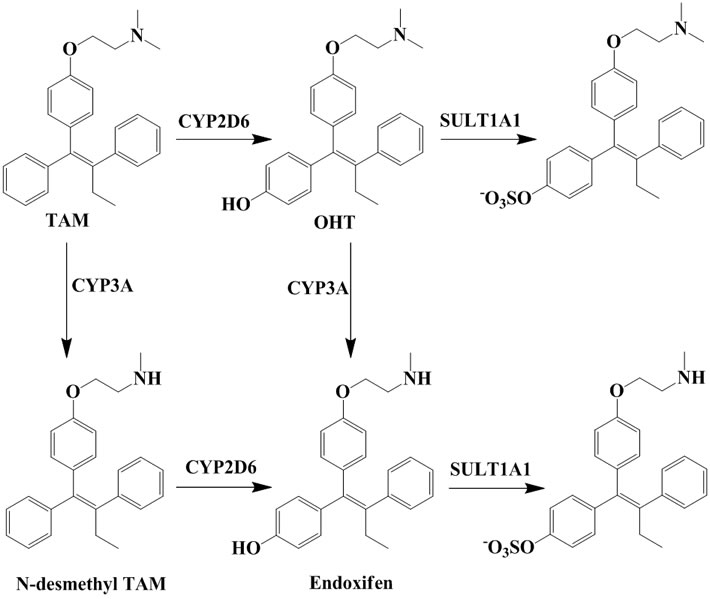
Scheme 1. Important metabolites of TAM and the major CYP isoforms responsible for the biotransformation [6,7] and clearance of active 4-hydroxyl metabolites [8,9].
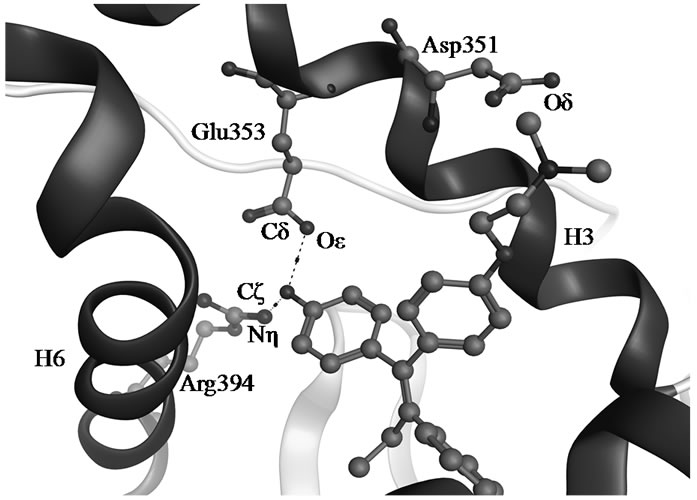
Figure 1. Hydrogen bonds between OHT and ERα LBD (3ERT).
residues Glu353 and Arg394 in ERα, resulting in 30- to 100-fold higher potency than the TAM parent drug [11-13]. The conversion of TAM to 4-hydroxy metabolites is rationalized as an advantage for antiestrogenic activity and predominantly catalyzed by CYP2D6 [6,7]. The active 4-hydroxy metabolites are subsequently sulphated by sulphotransferase (SULT1A1) and rapidly excreted [8,9]. An intuitive clinical implication is thus that patients carrying functional CYP2D6 alleles and less active SULT1A1 genotype may stand better chances for therapeutic success with TAM.
The association between genetic variants and clinical response to TAM therapy has been extensively investigated by retrospective studies, but results are nonetheless controversial. It was reported that TAM-treated patients lacking CYP2D6 enzymatic activity necessary for the final conversion to 4-hydroxy metabolites had significantly more recurrences of breast cancer compared to carriers of functional alleles [14-19]. However this suggestion was questioned in other studies, which reported that CYP2D6 activity had no significant association with recurrence [20,21]. Even more counterintuitive research results were obtained in that patients expressing inactive CYP2D6 and active SULT1A1, which results in less OHT and endoxifen, had better clinical outcomes [22- 24]. The connection between CYP2D6 and SULT1A1 enzymatic activities and the efficacy of TAM-treatment in individuals remain unresolved until more conclusive studies have been reported. A considerable number of patients suffer from recurrence despite the endocrine therapy, due to a developed resistance towards TAM. The acquired TAM-resistance in tumors is associated with isomerization of the potent trans-OHT to the less potent cis form, and a less understood mechanism with regards to markedly reduced cellular concentrations of TAM [25,26]. The findings therefore need to be investigated and explained in terms other than that TAM itself is a parent drug with fairly week affinity to ER.
There has been considerable interest in the development of new SERMs, which could provide more potent anti breast cancer drugs with less complex and unfavourable profiles. Many SERMs have failed during clinical trials because of low efficacy or side effects, due to their metabolism in vivo, or mechanisms that are as yet poorly understood [27]. Raloxifene is a recently approved SERM for the treatment of osteoporosis, although originally being developed for breast cancer therapy. It is well documented to be less estrogenic than TAM [28,29], resulting in lower risk of developing endometrial cancer in raloxifene treated patients compared to those treated with TAM [30]. The ligand-dependent bioactivity has been rationalized by the capacity of charge-neutralization. For most SERMs, a tertiary amine is presented in the side chain. This basic amine is assumed to interact with the negatively charged residue Asp351 in helix H3 of ERα LBD, thereby neutralizing the charge of Asp351, and this has been proposed to be important for SERM anti-estrogenicity [29]. The cocrystallized structures of raloxifene-LBD (1ERR [31]) and OHT-LBD (3ERT [10]) show that the nitrogen of raloxifene shields the charge of Asp351 through a hydrogen bond that is around 1 Å shorter compared with OHT (Figure S1). This charge-neutralizing capacity of the salt bridge was considered as consistent with the relative anti-estrogenicity of raloxifene vs TAM. However, this hypothesis has been greatly challenged through other cocrystallized structures of OHT-LBD (2JF9 [32] and 2BJ4 [33]), in which the interaction of OHT with Asp351 closely mirrors that seen in the structure of raloxifen-LBD (Table S1 and Figure S2).
The typical SERM TAM works as an antagonist in breast and conversely an agonist in uterus. What makes the agonism/antagonism tissue-specific? The mechanisms are hitherto unknown, which may depend on the various pathways for SERM signalling in different cells. But, for the same cell type, such as in uterine tissue, raloxifene is confirmed to be less estrogenic than TAM. The mechanism of mixed agonism/antagonism may differ depending on the chemical structure of the SERM ligand itself. An understanding of the intrinsic features that govern the estrogenic or anti-estrogenic properties of SERM-LBD complexes is important for developing improved therapeutic agents. Although much effort has been made, there is no unified theory that has explained the ligand-dependent biological profiles when the cocrystallized structures are almost identical. In the current work, force field based computations were used to characterize the structural properties of the ligand-LBD complexes. The conformational dynamic stability was found to correlate well with SERM signalling as seen in the cellular response to treatment with the various compounds.
2. COMUTATIONAL METHODS
In order to gain further insight regarding ligand induced bioactivity of ERα, molecular modelling and molecular dynamics (MD) simulations were performed. Cocrystallized structures of SERMs with the ERα LBD were obtained from the RCSB Protein Data Bank (PDB). The ligand-LBD complexes with PDB code 3ERT [10], 1XP1, 1XP6, 1XP9, 1XPC [34], and 1ERR [31] were used in the current study. The Molecular Operating Environment (MOE) program, version 2010.10 [35] was used for visualizations, structural alignments, and the calculations of root-mean-square deviation (RMSD) of Cα atoms for the crystal structures.
Independent MD simulations were conducted for each system as outlined below. The ligand-LBD complex was solvated in a periodic box with TIP3P [36] water molecules extending 10 Å outside the protein. MD simulations were carried out for each ligand-LBD complex for 20 ns at 298 K using the program YASARA structure [37], after initial energy minimization procedures. The AMBER03 [38] force field was used for the protein and the general amber force field [39] (GAFF) for the ligands throughout this work. Partial atomic charges of the ligands were calculated using the AM1-BCC model [40]. The net charge was +1 for SERMs, with the nitrogen atom protonated. The cocrystallized structures of ligand-LBD complexes were used as starting points. TAM, being the only ligand lacking a cocrystallized structure, was introduced by removing the 4-hydroxyl group of OHT in 3ERT. All systems were neutralized by adding counter ions [41], and additional water molecules were randomly replaced by Na+ or Cl– to give a total NaCl concentration of 0.9% (corresponding to physiological solution). Long-range Coulomb interactions were included using particle-mesh Ewald (PME) summation for the electrostatics with a cut-off of 7.86 Å. The simulations were carried out in their entirety with multiple time steps, 1.25 fs for intramolecular and 2.5 fs for intermolecular forces, using a predefined macro (run_md) within the YASARA package. The MD trajectories were sampled every 50 ps, resulting in 400 simulation snapshots per run, which were evaluated to derive statistical averages and properties of the corresponding geometries.
The RMSD of Cα atoms for the protein was monitored for each simulation trajectory. The RMSD values were calculated from the cartesian atomic coordinates with regard to the initial conformation according to the formula (R is the vector linking the corresponding n atom pairs in space):
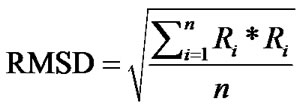
3. RESULTS AND DISCUSSION
3.1. Dihydrobenzoxathiin SERMs
The biological activity of a series of dihydrobenzoxathiin-based SERMs (Scheme 2) has been rigorously tested by scientists at Merck, with particular focus on the estrogenic and antiestrogenic properties in uterine tissue [34,42-48]. Of special interest is the stereochemistry of the isomers and how this correlates with uterine agonism/antagonism. Compounds 15, 16, 18 and 19 in Table 1 (numbering from reference [34]), cocrystallized with ERα LBD, were analysed further in the current work in order to better understand the relation between structures, properties and SERM bioactivities.
The X-ray structures of complexes of the enantiomeric pairs 15/16 and 18/19 bound to ERα LBD are compared by structural alignment. The geometrical comparisons are shown in Figure 2 for 15 vs 16, and 18 vs 19, respectively, and are summarized in Table 1 along with the experimental biodata [34].
The RMSDs, calculated for Cα atoms of the LBD, of 0.16 Å (15 vs 16) and 0.25 Å (18 vs 19) for the superposed crystal structures suggest that there is no difference in 3D geometries for either enantiomeric pair in complex with the LBD. Visualization of the ligand binding pocket (Figures 2(b), 2(d)) show identical binding mode and interactions for the enantiomeric pairs contacting the receptor in the solid state. Experimentally, the enantiomeric pairs exhibited equivalent affinity to the receptor and potency to inhibit MCF-7 cell line proliferation in vitro. The geometrical similarities of the cocrystallized structures presented are in full agreement with the IC50 trends generated in assays of ERα binding and MCF-7 breast cancer cell proliferation (Table 1).
However, the stereochemistry has a strong impact on uterine activity in the in vivo assays. The positioning of the methyl substituents provide extraordinary uterine antagonism of 15 and 18, while partial agonism of 16 and 19. The distances from Asp351 to the basic amine of the ligands, 2.6 - 2.8 Å measured between Oδ351 and Nlig,
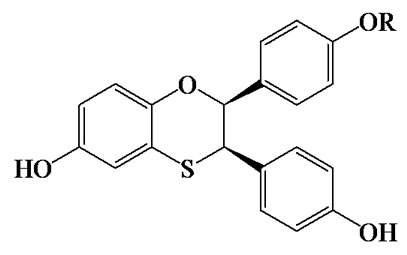
Scheme 2. Dihydrobenzoxathiin.
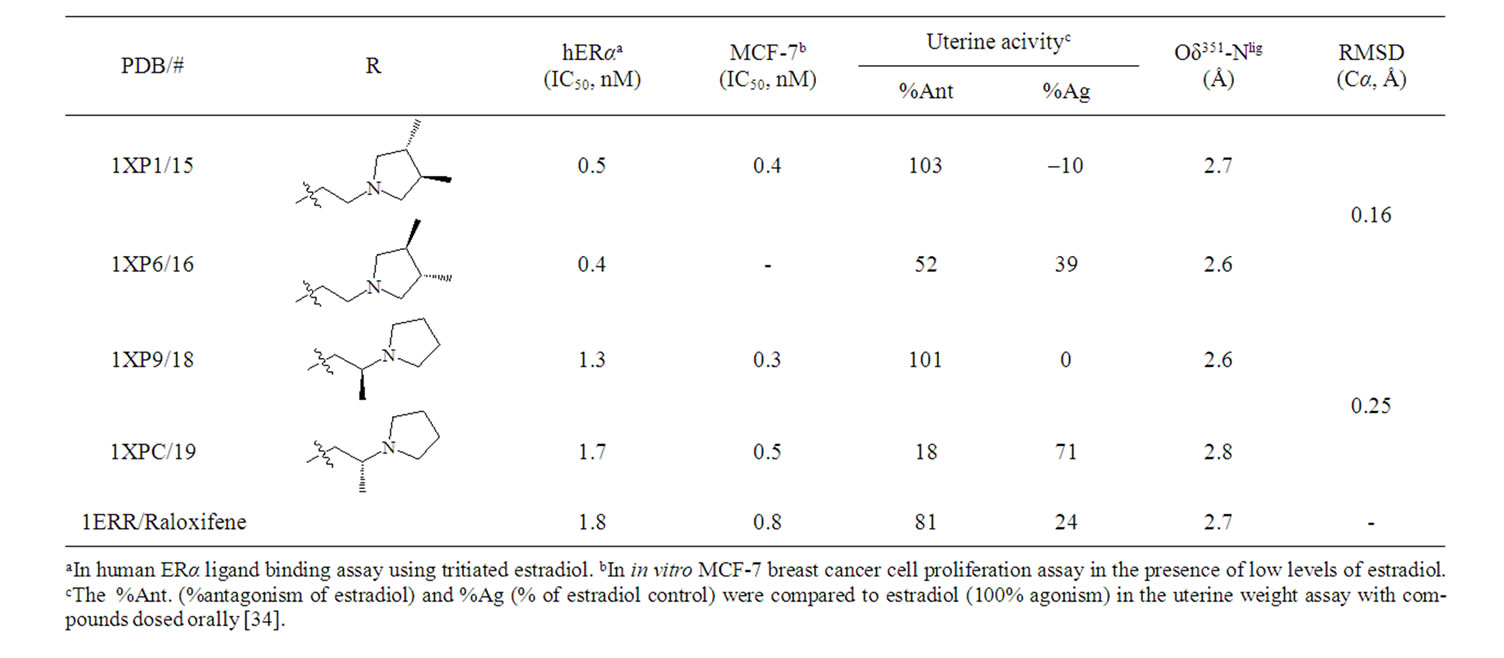
Table 1. Biodata [34] of SERMs and geometry comparisons of the cocrystallized structures.
 (a)
(a)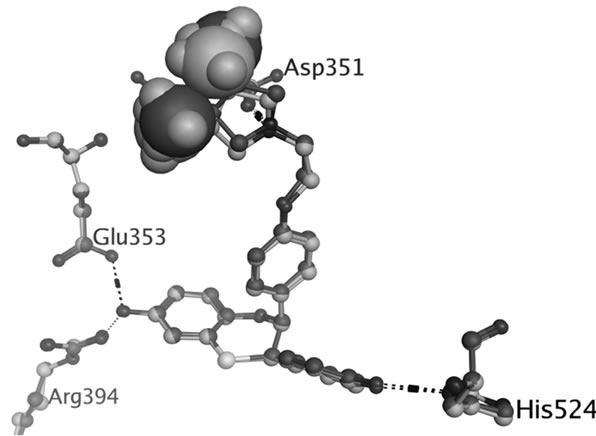 (b)
(b) (c)
(c)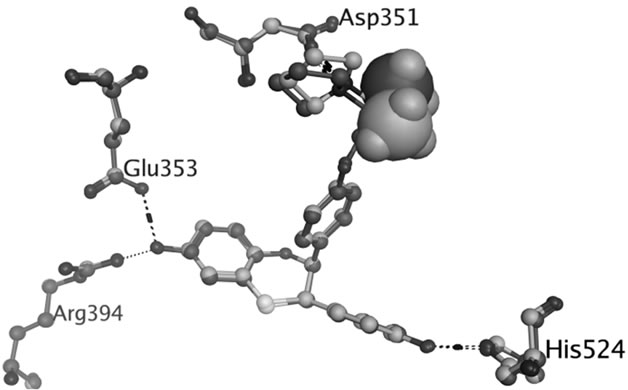 (d)
(d)
Figure 2. Superposed structures of the enantiomeric pairs 15 vs 16 (A,B) and 18 vs 19 (C,D) in complex with ERα LBD. The methyl substituents are shown as space filling models. The backbone and carbon atoms of 15-LBD (1XP1) and 18-LBD (1XP9) are coloured green, 16-LBD (1XP6) and 19-LBD (1XPC) purple. Nitrogen atoms are coloured blue, oxygen red, and sulphur yellow.
show that there is no difference in the salt bridge formed by each enantiomer. The charge-neutralizing capacity of the basic side chain is thus a questionable explanation for the bioactivity profiles of SERMs; at least it is not applicable for the dihydrobenzoxathiin derivatives. The only difference between the enantiomers is the orientation of the methyl groups, as seen in Figure 2. The methyl groups of 16 and 19 more closely approach the L11-12 loop region than what is the case in 15 and 18, respectively. This slight geometric shift caused by the stereochemistry is consistent with the activity profiles that 16 and 19 were assigned as more estrogenic than 15 and 18. However, the methyl groups do not form any strong interaction with the LBD, and as seen from Figure 2 show no obvious effect on the receptor conformations in the solid state.
Molecular mechanics based computations in solution were used to gain insight into the effects on the receptor caused by the stereochemistry of the side chain. Unconstrained simulations were conducted with explicit solvents to mimic the 18-LBD and 19-LBD complexes under physiological conditions. The trajectories were monitored during the entire simulations, as illustrated in Figure 3. RMSDs of the Cα atoms were calculated with respect to the starting conformations, which indicates the stability of the ligand-LBD complexes. Figure 3(a) shows the RMSDs of the full LBD. For neither system do the RMSDs exceed 4 Å throughout the 20 ns simulation time, which indicates that both ligands stabilize the inactive conformation of LBD. Apparent ligand-dependent dynamic behaviour is noted by comparing the trajectories of the two enantiomers in complex with the LBD, in which 18 stabilizes the receptor better than 19, based on the lower RMSD during most part of the simulation time. Since the only difference between 18 and 19 is the placement of the methyl group, the conformational changes in the region from L11-12 to the C-termini were monitored in order to identify the ligand induced effects (Figure 3(b)). Similar trends were found as for the full LBD. Thus, the conformational drift is largely a result of changes in the region from L11-12 to the C-termini. The lower dynamic flexibility of the 19-LBD complex is in agreement with the characteristics seen in the crystal structures. It can be argued from the comparisons in Figures 2(a) and 2(c), that the methyl groups of 16 and 19 project closer to the loop region and thus sterically support the loop in a better way. Compared with the biodata, this conformational stability coincides with the partial agonist properties of the SERMs, following the principle of more stable, more estrogenic.
The principle of the charge-neutralizing capacity as a measure of anti-estrogenic capability was also analyzed during the simulations. As seen in Figure 3(c), the at
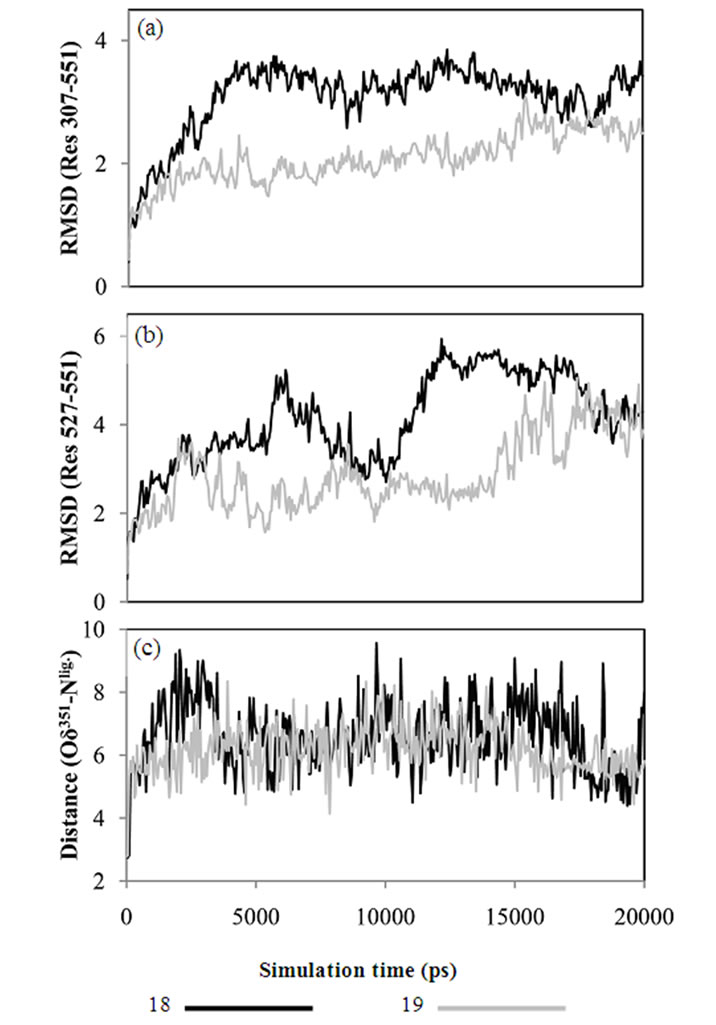
Figure 3. RMSD calculated from MD trajectories for (a) Cα atoms of LBD, Res 307-551, (b) Cα atoms from L11-12 to C-terminals, Res 527-551. (c) Distances between Asp351 and the tertiary amine of ligand, monitored through the distance between Oδ351 and Nlig.
tractive interaction between Asp351 and Nlig is very week as manifested in that the salt bridge is broken at the very beginning of the simulations, and has an average interatomic distance of around 6 Å. This is much larger than the distance required for a salt bridge, and indicates that the negatively charged side chain of Asp351 is solvent exposed rather than interacting with the basic tail of the ligands. Similar dynamic behaviour of Asp351 was found for TAM-LBD and OHT-LBD complexes (Figure 4(c)). The findings from the current MD simulations are supported by the experimental results [49] that TAM does not interact with Asp351 in an optimal manner. Furthermore, our simulation results provide a reasonable explanation of the experimental facts [49-51] that mutating Asp351 into hydrophobic residues (Ala, Val and Gly) reduces the estrogenic activity of the SERMs. The charged side chain of Asp351 is solvent exposed in the SERM-LBD complex, and facili
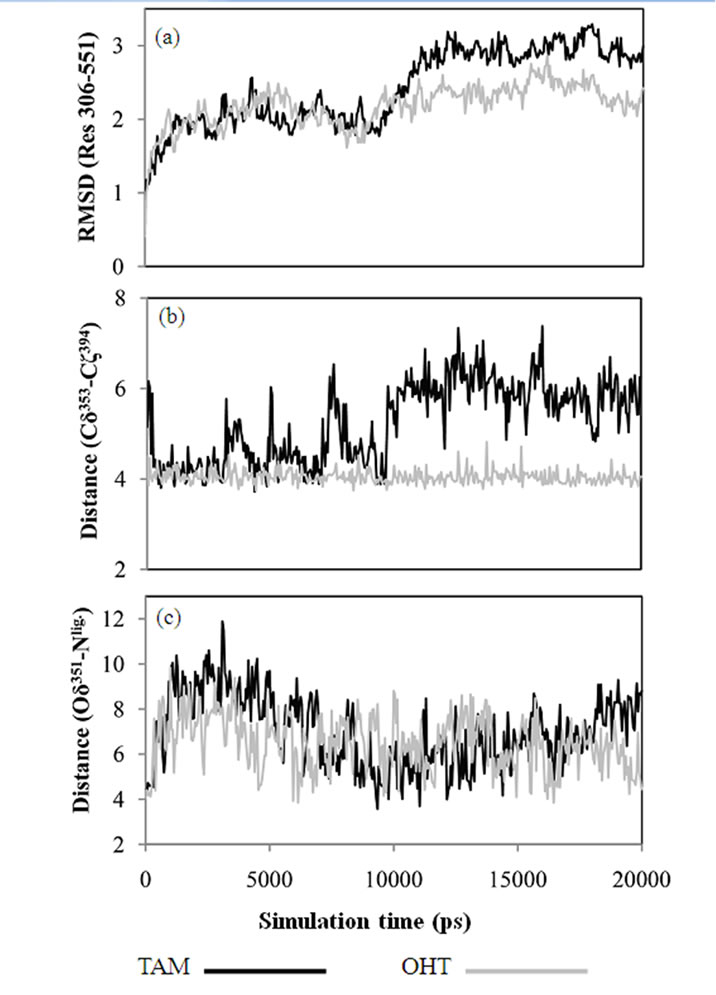
Figure 4. (a) RMSD of Cα atoms of the ERa LBD, Res 306-551, calculated from MD trajectories. (b) Distances between Glu353 and Arg394 calculated between Cδ353 and Cζ394. (c) Distances between Asp351 and the tertiary amine of ligand, calculated as the minimum distance between Oδ351 and Nlig.
tates interactions between LBD and other domains of ERα or coactivators which may activate ERα and result in partial agonism of the SERMs. Oppositely, if Asp351 is mutated into non-charged residues like Ala, Val and Gly, the interaction with other elements is decreased, and therefore reduces the estrogenic activity of the SERMs.
3.2. TAM/OHT-LBD
OHT stabilizes the canonical inactive conformation of the receptor, and is in Figure 5 compared to the dihydrobenzoxathiin derivatives bound ensembles. The RMSD of 0.75 Å for the five superposed crystal structures is largely a result of deviations in the L11-12 loop region. The location of TAM within the LBD is apparently associated with the same mechanisms as those of OHT that gives rise to its anti-breast cancer profile. OHT binds to ERα with much higher affinity than TAM, while
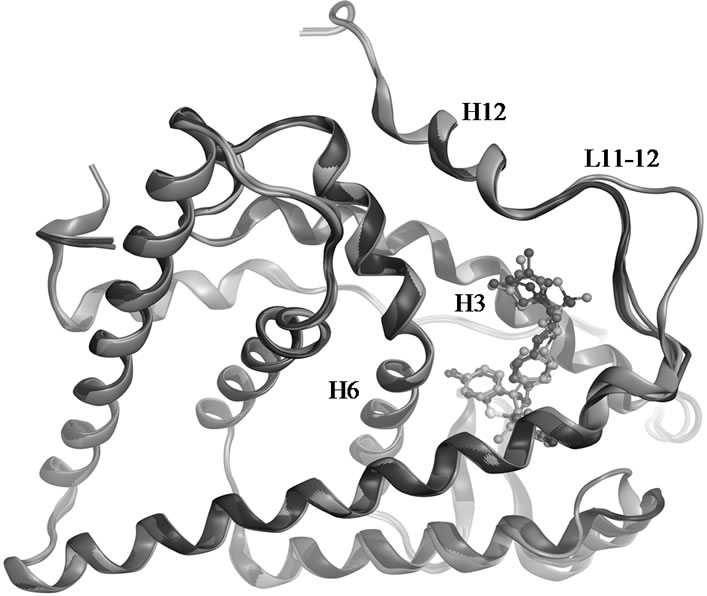
Figure 5. Superposed crystal structures of 15 (1XP1 in green), 16 (1XP6 in purple), 18 (1XP9 in blue), 19 (1XPC in red) and OHT (3ERT in gold) in complex with ERα LBD.
at the same time getting excreted rapidly through the sulfoxylation by SULT1A1. It is therefore difficult to compare the SERM profiles of TAM and OHT in in vivo assays as TAM also converts to OHT by the actions of CYP2D6. However, TAM has been assumed to have the same SERM profile as OHT but with less potency, due to a lower affinity to the receptor. Therefore, some clinical results reported are considered as counterintuitive, for instance that patients expressing inactive CYP2D6 and active SULT1A1, which results in reduced affinity towards OHT and endoxifen, nonetheless displayed better clinical response to TAM therapy [22-24]. Thus, assuming that TAM and OHT only differ in affinity, does not provide a reasonable explanation to resolve the CYP2D6 debate or the clinical implications of SULT1A1 activity.
MD simulations of OHT and TAM in complex with the LBD were performed in order to determine the influence of the 4-hydroxy group on the receptor. The conformational stability of the ligand-LBD complexes were again estimated by calculating RMSDs. Special attention was paid to the dynamics of the charged residues Glu353 and Arg394 located on H3 and H6, respectively, and their interactions with OHT. The results are shown in Figure 4.
The conformational drift in the TAM-LBD complex is statistically higher than that of the OHT-LBD one. The 4-hydroxy group of OHT forms hydrogen bonds with the side chains of both Glu353 and Arg394, as seen in Figure 1, whereas these interactions are lacking for the TAM-LBD system. To identify the effects of the interactions mediated by the 4-hydroxy group, the distances between Cδ353 and Cζ394 are displayed as a function of time in Figure 4(b), which reveal significant differences. The Cδ353-Cζ394 distances stabilize at around 4 Å through the entire simulation time of the OHT-LBD complex, but fluctuate dramatically in the TAM-LBD system. This indicates that the charged residues Glu353 and Arg394 can not form a stable salt bridge, as supported by the minimum distance of 4 Å seen in the crystal structures. Instead, the interaction between Glu353 and Arg394 is mediated by the hydroxyl group of the ligand, resulting in increased inter-helix interaction between H3 and H6, which stabilizes the tertiary structure of the OHT-LBD complex. Hence, lack of this inter-helix interaction leads to larger conformational flexibility and thus increased RMSDs in the TAM-LBD complex.
To enable more statistically meaningful comparisons, repeating simulations were conducted for each system. Statistical averages are summarized from three parallel simulations in Table 2. Similar interactions between Asp 353 and Arg394, mediated by the hydroxyl groups, were found for 18, 19 and OHT bound to LBD. Larger conformational changes were found for 18 and TAM in complex with the LBD due to different effects of the ligands, i.e., steric effects from the basic side chain and reduced inter-helix interaction, respectively. Lower conformational drift was found for the 19-LBD and OHTLBD complexes. The extreme antagonism of 18 and partial agonism of 19 has been proven directly by in vivo experiments. The simulation behaviour indicates that OHT gives a more stable complex and is thus more estrogenic than TAM, i.e., that 4-hydroxylation increases the ligand affinity to ERα, but at the same time increases the estrogenic activity. Therefore, reduced concentration of TAM is a disadvantage in TAM therapy. The actions of CYP2D6 and SULT1A1 have paradoxical effects due to the partial agonism of OHT. To this end, the MD simulations provide a reasonable explanation for the observed clinical results.
4. CONCLUSIONS
The crystal structures demonstrate that SERMs stabilize a canonical inactive conformation of the ERα LBD.
At the same time, the “inactive” SERM-LBD complexes exert biological activity depending on cell type, which results in tissue-specific effects. Despite the essentially identical conformation of the LBDs, the SERM profiles differ considerably from each other. All-atom, explicit solvent, MD simulations of the SERM-LBD complexes were performed which disclose the contradictive ER ligand-dependent estrogenic or antiestrogenic properties, which have puzzled scientists for decades. This may provide useful information for the development of improved therapeutic agents.
In earlier studies, the charge-neutralizing capacity of the basic side chain of the SERMs has been considered essential for their antiestrogenic activity. The positively charged nitrogen atom on the SERM tail forms a salt bridge with the negatively charged residue Asp351 of ERα in most cocrystallized SERM-LBD structures. However, as this interaction is almost identical for most SERM bound LBD crystal structures, it cannot be associated with the differential bioactivity of SERMs. In addition, contrary to the solid state crystal structures, in all the simulation trajectories in condensed phase, the side chain of Asp351 was shown to be exposed to the solvent other than interacting with the tertiary amine of the SERMs, with an average distance of around 6 Å between Oδ351 and Nlig.
Comparisons were made between dihydrobenzoxathiin-based SERMs with regard to their bioactivities, cocrystallized structures and dynamical behaviour in solvent. We conclude that the SERM profiles of the synthetic ligands is correlated to the conformational stability of the ligand-LBD complexes rather than the chargeneutralizing capacity, following the principle of more stable, more estrogenic. As seen in the crystal structures, the position of the methyl substituents for compounds 16 and 19 provide better stabilization of the L11-12 region than is the case for 15 and 18, respectively. This is consistent with the uterine agonist/antagonist effects. As a result of the steric effects exerted, the 19-LBD complex displays less conformational drift during the 20 ns simulations than the 18-LBD complex, and is confirmed as a
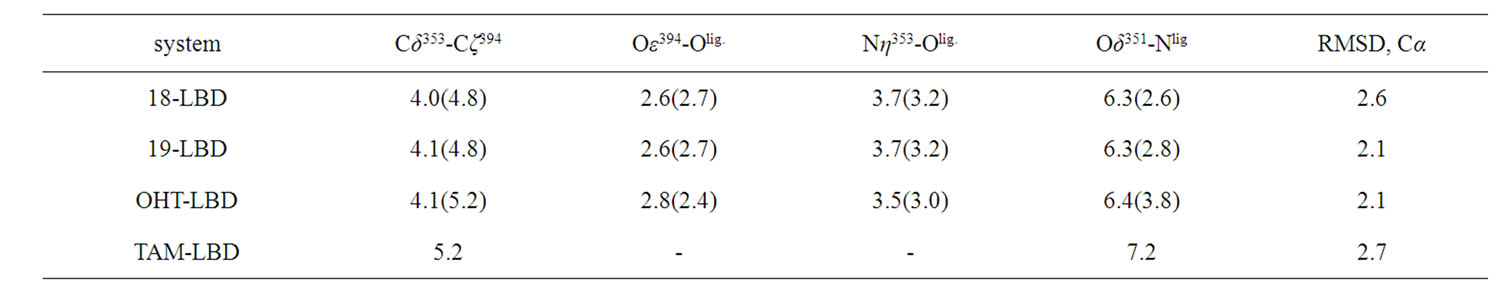
Table 2. Statistical averages of selected distances and RMSDs obtained from three parallel simulations (in Å). The distances in the crystal structures are displayed in parentheses, and averages are calculated from the distances between selected atoms. Olig is the oxygen atom of the ligand interacting with Asp353 and Arg394.
more stable structure, resulting in higher estrogenic activity in good agreement with experimental results.
The stability of the tertiary structures of TAM and OHT in complex with the ERα LBD were also investigated, and the results provide a reasonable explanation to the CYP2D6 debate and various related clinical observations. The 4-hydroxy group of OHT forms salt bridges with both Glu353 on H3 and Arg394 on H6, and hence mediate inter-helix interactions between these, which in turn stabilizes the OHT-LBD complex. Lacking these two hydrogen bonds, TAM binds to ERα with less affinity than OHT, and provides less potent antagonism; at the same time, the resulting less stable ligand-LBD complex also makes for less potent agonism. The simulation results thus indicate that TAM is less estrogenic than OHT, and that therefore the conversion of TAM to 4-hydroxy metabolites has contradictory effects on TAM therapy, as seen through the CYP2D6 debate. The results are also in good agreement with the clinical observations that decreased SULT1A1 activity and decreased cellular concentrations of TAM are associated with increased risk of recurrence.
The synthetic SERMs function as partial agonists to different extent, and their potency depends not only on the affinity to ERα, but also on how potent the ligand is in inducing the allostery and stabilizing the required conformation. Therefore, the stability and flexibility of SERM-LBD complex play essential roles for the SERM profiles.
5. ACKNOWLEDGEMENTS
We are grateful for financial support from the Swedish Science Research Council, the Faculty of Science and Technology at Örebro University, and the National University of Ireland, Galway.
6. SUPPORTING INFORMATION
Visualized structures and geometric comparison of OHT-LBD and raloxfiene-LBD are available via http://www.scirp.journal/JBPC.
REFERENCES
- Riggs, B.L. and Hartmann, L.C. (2003) Drug therapy: Selective estrogen-receptor modulators—Mechanisms of action and application to clinical practice. New England Journal of Medicine, 348, 618-629. doi:10.1056/NEJMra022219
- Clarke, M., Collins, R., Davies, C., Godwin, J., Gray, R., Peto, R. and Grp, E.B.C.T.C. (1998) Tamoxifen for early breast cancer: An overview of the randomised trials. The Lancet, 351, 1451-1467. doi:10.1016/S0140-6736(97)11423-4
- Jordan, V.C. (2003) A most unlikely pioneering medicine. Nature Reviews Drug Discovery, 2, 205-213. doi:10.1038/nrd1031
- Sato, M., Rippy, M.K. and Bryant, H.U. (1996) Raloxifene, tamoxifen, nafoxidine, or estrogen effects on reproductive and nonreproductive tissues in ovariectomized rats. FASEB Journal, 10, 905-912.
- Assikis, V.J., Neven, P., Jordan, V.C. and Vergote, I. (1996) A realistic clinical perspective of tamoxifen and endometrial carcinogenesis. European Journal of Cancer, 32A, 1464-1476. doi:10.1016/0959-8049(96)00184-0
- Jin, Y., Desta, Z., Stearns, V., Ward, B., Ho, H., Lee, K. H., Skaar, T., Storniolo, A.M., Li, L., Araba, A., Blanchard, R., Nguyen, A., Ullmer, L., Hayden, J., Lemler, S., Weinshilboum, R.M., Rae, J.M., Hayes, D.F. and Flockhart, D.A. (2005) CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. Journal of the National Cancer Institute, 97, 30-39. doi:10.1093/jnci/dji005
- Desta, Z., Ward, B.A., Soukhova, N.V. and Flockhart, D.A. (2004) Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. Journal of Pharmacology and Experimental Therapeutics, 310, 1062-1075. doi:10.1124/jpet.104.065607
- Seth, P., Lunetta, K.L., Bell, D.W., Gray, H., Nasser, S.M., Rhei, E., Kaelin, C.M., Iglehart, D.J., Marks, J.R., Garber, J.E., Haber, D.A. and Polyak, K. (2000) Phenol sulfotransferases: Hormonal regulation, polymorphism, and age of onset of breast cancer. Cancer Research, 60, 6859-6863.
- Falany, C.N., Wheeler, J., Oh, T.S. and Falany, J.L. (1994) Steroid sulfation by expressed human cytosolic sulfotransferases. Journal of Steroid Biochemistry and Molecular Biology, 48, 369-375. doi:10.1016/0960-0760(94)90077-9
- Shiau, A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A. and Greene, G.L. (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell, 95, 927-937. doi:10.1016/S0092-8674(00)81717-1
- Fabian, C., Tilzer, L. and Sternson, L. (1981) Comparative binding affinities of tamoxifen, 4-hydroxytamoxifen, and desmethyltamoxifen for estrogen-receptors isolated from human-breast carcinoma—Correlation with bloodlevels in patients with metastatic breast-cancer. Biopharmaceutics & Drug Disposition, 2, 381-390. doi:10.1002/bdd.2510020407
- Johnson, M.D., Zuo, H., Lee, K.H., Trebley, J.P., Rae, J.M., Weatherman, R.V., Desta, Z., Flockhart, D.A. and Skaar, T.C. (2004) Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Research and Treatment, 85, 151-159. doi:10.1023/B:BREA.0000025406.31193.e8
- Coezy, E., Borgna, J.L. and Rochefort, H. (1982) Tamoxifen and metabolites in Mcf7 cells—Correlation between binding to estrogen-receptor and inhibition of cell-growth. Cancer Research, 42, 317-323.
- Goetz, M.P., Rae, J.M., Suman, V.J., Safgren, S.L., Ames, M.M., Visscher, D.W., Reynolds, C., Couch, F.J., Lingle, W.L., Flockhart, D.A., Desta, Z., Perez, E.A. and Ingle, J.N. (2005) Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. Journal of Clinical Oncology, 23, 9312-9318. doi:10.1200/JCO.2005.03.3266
- Schroth, W., Antoniadou, L., Fritz, P., Schwab, M., Muerdter, T., Zanger, U.M., Simon, W., Eichelbaum, M. and Brauch, H. (2007) Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. Journal of Clinical Oncology, 25, 5187-5193. doi:10.1200/JCO.2007.12.2705
- Goetz, M.P., Knox, S.K., Suman, V.J., Rae, J.M., Safgren, S.L., Ames, M.M., Visscher, D.W., Reynolds, C., Couch, F.J., Lingle, W.L., Weinshilboum, R.M., Fritcher, E.G.B., Nibbe, A.M., Desta, Z., Nguyen, A., Flockhart, D.A., Perez, E.A. and Ingle, J.N. (2007) The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Research and Treatment, 101, 113-121. doi:10.1007/s10549-006-9428-0
- Kiyotani, K., Mushiroda, T., Sasa, M., Bando, Y., Sumitomo, I., Hosono, N., Kubo, M., Nakamura, Y. and Zembutsu, H. (2008) Impact of CYP2D6*10 on recurrence-free survival in breast cancer patients receiving adjuvant tamoxifen therapy. Cancer Science, 99, 995-999. doi:10.1111/j.1349-7006.2008.00780.x
- Xu, Y., Sun, Y., Yao, L., Shi, L., Wu, Y., Ouyang, T., Li, J., Wang, T., Fan, Z., Fan, T., Lin, B., He, L., Li, P. and Xie, Y. (2008) Association between CYP2D6*10 genotype and survival of breast cancer patients receiving tamoxifen treatment. Annals of Oncology, 19, 1423-1429. doi:10.1093/annonc/mdn155
- Newman, W.G., Hadfield, K.D., Latif, A., Roberts, S.A., Shenton, A., McHague, C., Lalloo, F., Howell, S. and Evans, D.G. (2008) Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clinical Cancer Research, 14, 5913-5918. doi:10.1158/1078-0432.CCR-07-5235
- Nowell, S.A., Ahn, J.Y., Rae, J.M., Scheys, J.O., Trovato, A., Sweeney, C., MacLeod, S.L., Kadlubar, F.F. and Ambrosone, C.B. (2005) Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Research and Treatment, 91, 249-258. doi:10.1007/s10549-004-7751-x
- Okishiro, M., Taguchi, T., Kim, S.J., Shimazu, K., Tamaki, Y. and Noguchi, S. (2009) Polymorphisms of CYP2D6*10 and CYP2C19*2,*3 are not associated with prognosis, endometrial thickness, or bone mineral density in Japanese breast cancer patients treated with adjuvant tamoxifen. Cancer, 115, 952-961. doi:10.1002/cncr.24111
- Nowell, S., Sweeney, C., Winters, M., Stone, A., Lang, N. P., Hutchins, L.F., Kadlubar, F.F. and Ambrosone, C.B. (2002) Association between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapy. Journal of the National Cancer Institute, 94, 1635-1640.
- Wegman, P., Vainikka, L., Stal, O., Nordenskjold, B., Skoog, L., Rutqvist, L.E. and Wingren, S. (2005) Genotype of metabolic enzymes and the benefit of tamoxifen in postmenopausal breast cancer patients. Breast Cancer Research, 7, R284-R290. doi:10.1186/bcr993
- Wegman, P., Elingarami, S., Carstensen, J., Stal, O., Nordenskjold, B. and Wingren, S. (2007) Genetic variants of CYP3A5, CYP2D6, SULT1A1, UGT2B15 and tamoxifen response in postmenopausal patients with breast cancer. Breast Cancer Research, 9, R7. doi:10.1186/bcr1640
- Osborne, C.K., Coronado, E., Allred, D.C., Wiebe, V. and Degregorio, M. (1991) Acquired tamoxifen resistance— Correlation with reduced breast-tumor levels of tamoxifen and isomerization of trans-4-hydroxytamoxifen. Journal of the National Cancer Institute, 83, 1477-1482. doi:10.1093/jnci/83.20.1477
- Osborne, C.K., Wiebe, V.J., Mcguire, WL., Ciocca, D.R. and Degregorio, M.W. (1992) Tamoxifen and the isomers of 4-hydroxytamoxifen in tamoxifen-resistant tumors from breast-cancer patients. Journal of Clinical Oncology, 10, 304-310.
- Hermkens, P.H., Kamp, S., Lusher, S. and Veeneman, G.H. (2006) Non-steroidal steroid receptor modulators. IDrugs, 9, 488-494.
- Grese, T.A., Sluka, J.P., Bryant, H.U., Cullinan, G.J., Glasebrook, A.L., Jones, C.D., Matsumoto, K., Palkowitz, A.D., Sato, M., Termine, J.D., Winter, M.A., Yang, N.N. and Dodge, J.A. (1997) Molecular determinants of tissue selectivity in estrogen receptor modulators. Proceedings of the National Academy of Sciences of the United States of America, 94, 14105-14110. doi:10.1073/pnas.94.25.14105
- Liu, H., Park, W.C., Bentrem, D.J., McKian, K.P., De los Reyes, A., Loweth, J.A., Schafer, J.M., Zapf, J.W. and Jordan, V.C. (2002) Structure-function relationships of the raloxifene-estrogen receptor-alpha complex for regulating transforming growth factor-alpha expression in breast cancer cells. Journal of Biological Chemistry, 277, 9189-9198. doi:10.1074/jbc.M108335200
- DeMichele, A., Troxel, A.B., Berlin, J. A., Weber, A.L., Bunin, G.R., Turzo, E., Schinnar, R., Burgh, D., Berlin, M., Rubin, S.C., Rebbeck, T.R. and Strom, B.L. (2008) Impact of raloxifene or tamoxifen use on endometrial cancer risk: A population-based case-control study. Journal of Clinical Oncology, 26, 4151-4159. doi:10.1200/JCO.2007.14.0921
- Brzozowski, A.M., Pike, A.C., Dauter, Z., Hubbard, R.E., Bonn, T., Engstrom, O., Ohman, L., Greene, G.L., Gustafsson, J.A. and Carlquist, M. (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389, 753-758. doi:10.1038/39645
- Heldring, N., Pawson, T., McDonnell, D., Treuter, E., Gustafsson, J.A. and Pike, A.C. (2007) Structural insights into corepressor recognition by antagonist-bound estrogen receptors. Jounal of Biological Chemistry, 282, 10449-10455. doi:10.1074/jbc.M611424200
- Kong, E.H., Heldring, N., Gustafsson, J.A., Treuter, E., Hubbard, R.E. and Pike, A.C. (2005) Delineation of a unique protein-protein interaction site on the surface of the estrogen receptor. Proceedings of the National Academy of Sciences of the United States of America, 102, 3593-3598. doi:10.1073/pnas.0407189102
- Blizzard, T.A., DiNinno, F., Morgan, J.D., Chen, H.Y., Wu, J.Y., Kim, S., Chan, W., Birzin, E.T., Yang, Y.T., Pai, L.Y., Fitzgerald, P.M.D., Sharma, N., Li, Y., Zhang, Z.P., Hayes, E.C., DaSilva, C.A., Tang, W., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2005) Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side chains and linkers. Bioorganic & Medicinal Chemistry Letters, 15, 107-113. doi:10.1016/j.bmcl.2004.10.036
- Chemical Computing Group (2010) Molecular Operating Environment, Montreal, Canada.
- Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W. and Klein, M.L. (1983) Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 79, 926-935. doi:10.1063/1.445869
- Krieger, E., Darden, T., Nabuurs, S.B., Finkelstein, A. and Vriend, G. (2004) Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins-Structure Function and Bioinformatics, 57, 678-683. doi:10.1002/prot.20251
- Duan, Y., Wu, C., Chowdhury, S., Lee, M.C., Xiong, G., Zhang, W., Yang, R., Cieplak, P., Luo, R., Lee, T., Caldwell, J., Wang, J. and Kollman, P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of Computational Chemistry, 24, 1999-2012. doi:10.1002/jcc.10349
- Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A. and Case, D.A. (2004) Development and testing of a general amber force field. Journal of Computational Chemistry, 25, 1157-1174. doi:10.1002/jcc.20035
- Jakalian, A., Jack, D.B. and Bayly, C.I. (2002) Fast, efficient generation of high-quality atomic charges. AM1- BCC model: II. Parameterization and validation. Journal of Computational Chemistry, 23, 1623-1641. doi:10.1002/jcc.10128
- Krieger, E., Nielsen, J.E., Spronk, C.A. and Vriend, G. (2006) Fast empirical pKa prediction by Ewald summation. Jounal of Molecular Graphics & Modelling, 25, 481-486. doi:10.1016/j.jmgm.2006.02.009
- Blizzard, T.A., Morgan, J.D., Chan, W.D., Birzin, E.T., Pai, L.Y., Hayes, E.C., DaSilva, C.A., Mosley, R.T., Yang, Y.T., Rohrer, S.P., DiNinno, F. and Hammond, M.L. (2005) Estrogen receptor ligands. Part 14: Application of novel antagonist side chains to existing platforms. Bioorganic & Medicinal Chemistry Letters, 15, 5124-5128. doi:10.1016/j.bmcl.2005.08.084
- Blizzard, T.A., DiNinno, F., Chen, H.Y., Kim, S., Wu, J.Y., Chan, W.D., Birzin, E.T., Yang, Y.T., Pal, L.Y., Hayes, E.C., DaSilva, C.A., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2005) Estrogen receptor ligands. Part 13: Dihydrobenzoxathiin SERAMs with an optimized antagonist side chain. Bioorganic & Medicinal Chemistry Letters, 15, 3912-3916. doi:10.1016/j.bmcl.2005.05.089
- Tan, Q., Blizzard, T.A., Morgan, J.D., Birzin, E.T., Chan, W.D., Yang, Y.T., Pai, L.Y., Hayes, E.C., DaSilva, C.A., Warrier, S., Yudkovitz, J., Wilkinson, H.A., Sharma, N., Fitzgerald, P.M.D., Li, S., Colwell, L., Fisher, J.E., Adamski, S., Reszka, A.A., Kimmel, D., DiNinno, F., Rohrer, S.P., Freedman, L.P., Schaeffer, J.M. and Hammond, M.L. (2005) Estrogen receptor ligands. Part 10: Chromanes: Old scaffolds for new SERAMs. Bioorganic & Medicinal Chemistry Letters, 15, 1675-1681. doi:10.1016/j.bmcl.2005.01.046
- Liu, J., Birzin, E.T., Chan, W.D., Yang, Y.T., Pai, L.Y., DaSilva, C., Hayes, E.C., Mosley, R.T., DiNinno, F., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2005) Estrogen receptor ligands. Part 11: Synthesis and activity of isochromans and isothiochromans. Bioorganic & Medicinal Chemistry Letters, 15, 715-718. doi:10.1016/j.bmcl.2004.11.018
- Blizzard, T.A., DiNinno, F., Morgan, J.D., Chen, H.Y., Wu, J.Y., Gude, C., Kim, S., Chan, W.D., Birzin, E.T., Yang, Y.T., Pai, L.Y., Zhang, Z.P., Hayes, E.C., DaSilva, C.A., Tang, W., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2004) Estrogen receptor ligands. Part 7: Dihydrobenzoxathiin SERAMs with bicyclic amine side chains. Bioorganic & Medicinal Chemistry Letters, 14, 3861-3864. doi:10.1016/j.bmcl.2004.05.074
- Blizzard, T.A., DiNinno, F., Morgan, J.D., Wu, J.Y., Chen, H.Y., Kim, S., Chan, W.D., Birzin, E.T., Yang, Y.T., Pai, L.Y., Zhang, Z.P., Hayes, E.C., DaSilva, C.A., Tang, W., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2004) Estrogen receptor ligands. Part 8: Dihydrobenzoxathiin SERAMs with heteroatom-substituted side chains. Bioorganic & Medicinal Chemistry Letters, 14, 3865-3868. doi:10.1016/j.bmcl.2004.05.073
- Tan, Q., Birzin, E.T., Chan, W., Yang, Y.T., Pai, L.Y., Hayes, E.C., DaSilva, C.A., DiNinno, F., Rohrer, S.P., Schaeffer, J.M. and Hammond, M.L. (2004) Estrogen receptor ligands. Part 5: The SAR of dihydrobenzoxathiins containing modified basic side chains. Bioorganic & Medicinal Chemistry Letters, 14, 3747-3751. doi:10.1016/j.bmcl.2004.04.100
- Dayan, G., Lupien, M., Auger, A., Anghel, S.I., Rocha, W., Croisetiere, S., Katzenellenbogen, J.A. and Mader, S. (2006) Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor alpha. Molecular Pharmacology, 70, 579-588. doi:10.1124/mol.105.021931
- MacGregor Schafer, J., Liu, H., Bentrem, D.J., Zapf, J.W. and Jordan, V.C. (2000) Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Research, 60, 5097-5105
- Anghel, S.I., Perly, V., Melancon, G., Barsalou, A., Chagnon, S., Rosenauer, A., Miller, W.H. Jr. and Mader, S. (2000) Aspartate 351 of estrogen receptor alpha is not crucial for the antagonist activity of antiestrogens. Jounal of Biological Chememistry, 275, 20867-20872. doi:10.1074/jbc.M002098200
SUPPORTING INFORMATION
 (a)(b)
(a)(b)
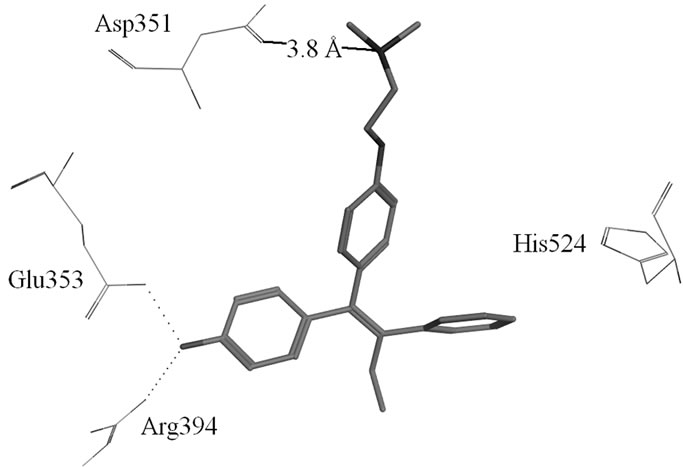
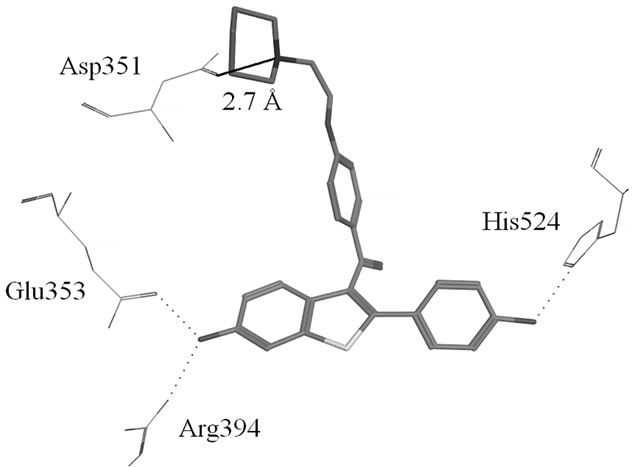
Figure S1. (a) Hydrogen-bonds between OHT and ERα LBD (3ERT); (b) Hydrogen-bonds between Raloxifene and ERα LBD (1ERR).
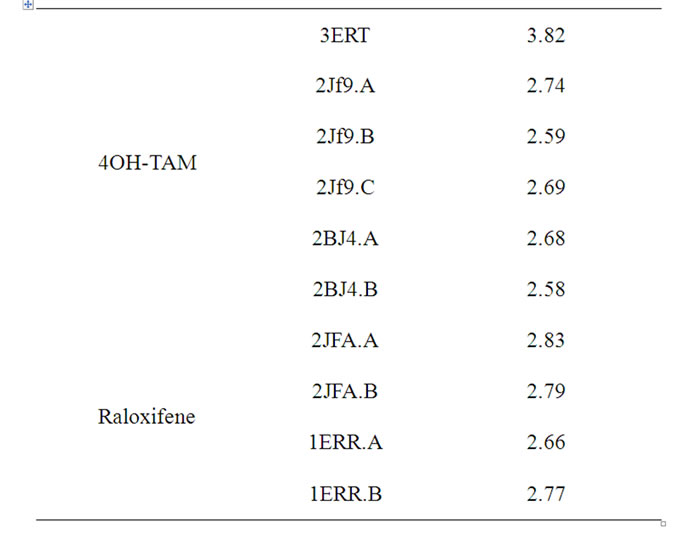
Table S1. Distances (Å) between Asp351 and the tertiary amine of OHT and Raloxifene in a variety of crystallographic structures.
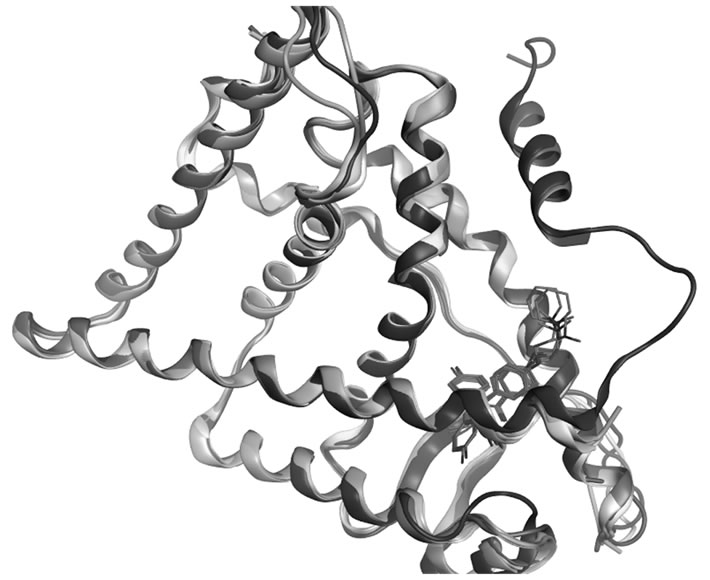
Figure S2. Superposed crystallographic structures of OHT and raloxifene in complex with ERα LBD, 3ERT (red), 2JF9.A (gray), 2BJ4.A (yellow), 1ERR.A (blue), and 2JFA.A (green).