Pharmacology & Pharmacy
Vol.06 No.03(2015), Article ID:54963,10 pages
10.4236/pp.2015.63020
Antidiabetic Effects of Omega-3 Polyunsaturated Fatty Acids: From Mechanism to Therapeutic Possibilities
Yuko Iwase, Noriyasu Kamei, Mariko Takeda-Morishita*
Laboratory of Drug Delivery Systems, Faculty of Pharmaceutical Sciences, Kobe Gakuin University, Kobe, Japan
Email: *mmtakeda@pharm.kobegakuin.ac.jp
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 February 2015; accepted 19 March 2015; published 24 March 2015
ABSTRACT
Diabetes mellitus (DM) is chronic disease characterized by hyperglycemia and insulin resistance caused by dysfunction of pancreatic b cells. Over the past few decades, epidemiological studies have suggested that dietary long-chain polyunsaturated fatty acids such as docosahexaenoic acid and eicosapentaenoic acid decrease the risk of metabolic diseases including DM. The mechanisms underlying the therapeutic efficacy of dietary long-chain polyunsaturated fatty acids in treating DM have been partly revealed. In this review, the authors describe the antidiabetic effects of long- chain polyunsaturated fatty acids and also discuss their possibilities as therapeutics for DM in the light of recent findings.
Keywords:
Omega-3 Polyunsaturated Fatty Acids, Diabetes, Insulin Secretion, Docosahexaenoic Acid

1. Introduction
Diabetes mellitus (DM) is a chronic disease in which the blood glucose level is too high because the body experiences insulin deficiency, decreased ability to use insulin, or both. The World Health Organization (WHO) has estimated that 347 million people worldwide have DM and projects that DM will be the seventh leading cause of death in 2030 [1] . According to the American Diabetes Association, most DM cases can be classified into two types: type 1 diabetes (T1DM) and type 2 diabetes (T2DM) [2] . T1DM is an immune-mediated disease characterized by an absolute deficiency of insulin secretion. T1DM patients have autoimmune destruction of pancreatic b cells, which leads to the absolute insulin deficiency. T2DM accounts for 90% - 95% of all DM cases. T2DM patients have both hyperglycemia and hyperinsulinemia. Recent reports have shown that insulin resistance in the brain correlates strongly with Alzheimer’s disease (AD) and that AD and DM are risk factors for each other. Because AD causes brain insulin resistance, oxidative stress, and cognitive impairment, it is sometimes called “type 3 DM” [3] -[7] .
Most T2DM patients are obese as a result excessive food intake, a high-fat diet, or lack of physical activity. Chronic inflammation caused by obesity has emerged as an important physiological mechanism linked to insulin resistance and T2DM. Obesity is associated with increased production of proinflammatory cytokines and activation of the inflammatory pathways in key metabolic tissues. Obesity itself causes insulin resistance. To cope with insulin resistance, pancreatic b cell mass increases to provide the required amount of insulin to maintain a normal blood glucose level in the early stages of DM [8] . However, hyperglycemia over a long period causes abnormal insulin secretion, which exhausts pancreatic b cells. Pancreatic b cell dysfunction leads to the accumulation of M1 macrophages in the pancreas and the secretion of inflammatory cytokines. Inflammatory cytokines cause inflammation and worsen insulin resistance.
Obesity also induces insulin resistance in adipose tissue [9] -[14] . Accumulated triacylglycerol in adipose tissue resulting from obesity increases both the size and number of adipocytes. Enlarged adipocytes secrete inflammatory cytokines such as tumor necrosis factor-alpha (TNF-a) and interleukin-6 (IL-6), and induce insulin resistance. Enlarged adipocytes also secrete the chemokine monocyte chemoattractant protein-1 (MCP-1) [13] [15] -[17] . M1 macrophages express MCP-1 receptors, and MCP-1 secretion causes M1 macrophage migration and accumulation in adipose tissue, leading to worsening of inflammation and insulin resistance. In addition, brain inflammation has been linked to obesity, and brain inflammation resulting from obesity inhibits leptin delivery into the brain (hypothalamus) [18] -[24] . Inflammation induces insulin resistance and aggravates DM. Therefore, suppressing inflammation is a promising approach to antidiabetic treatment. Administration of omega-3 polyunsaturated fatty acids (n-3 PUFAs) is one approach for suppressing inflammation. Some experiments have shown that n-3 PUFAs have anti-inflammatory effects in the hypothalamus [18] -[24] .
Fatty acids are organic acids with an aliphatic chain and a carboxyl group. Aliphatic acids with one double bond are monounsaturated fatty acids, and those with more than one double bond are PUFAs. PUFAs can be divided into two categories: the n-6 family (n-6 PUFA), which is derived from linolenic acid, and the n-3 family (n-3 PUFA), which is derived from a-linolenic acid [25] . Over the past few decades, epidemiological studies have suggested that n-3 PUFAs such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) decrease the risk of coronary heart disease, hypertension, and stroke, and improve mood disorders and cognitive function. Greenland Inuit who eat a diet rich in seafood containing a high level of n-3 PUFAs have low rates of coronary heart disease and DM compared with Danes who eat a typical Western diet [26] [27] .
As shown in Figure 1, in this review, the authors focus on the mechanisms underlying the hypoglycemic action and antidiabetic effects of n-3 PUFAs in terms of the roles of the G protein-coupled receptor 120 (GPR120) as well as their possibilities as therapeutics for DM in Section 2 (Insulin-sensitizing effects of n-3 PUFAs via GPR120), peroxisome proliferator-activated receptors (PPARs), and sterol regulatory element-binding proteins (SREBPs) in Section 3 (Insulin-sensitizing effects of n-3 PUFAs via SREBP and PPARs). Improvements in endothelial dysfunction induced by n-3 PUFAs and the use of n-3 PUFAs as a DM biomarker that reflects blood n-3 PUFA concentration are also described in Section 4 (Improvement of vascular endothelial dysfunction by n-3 PUFAs).
2. Insulin-Sensitizing Effects of n-3 PUFAs via GPR120
The antidiabetic effect of n-3 PUFAs is based on the secretion of glucagon-like peptide-1 (GLP-1), which is mediated partly by GPR120 [28] . Incretins, peptide hormones secreted in response to food intake, increase endogenous insulin secretion. GLP-1, a 30-amino acid peptide hormone derived from proglucagon, is the most potent incretin hormone and is secreted from lower intestinal L cells. Secreted GLP-1 is rapidly degraded by dipeptidyl peptidase-4 (DPP-4), which causes the short half-life for GLP-1 of <2 min [29] [30] . In cultured cells, GLP-1 secretion by GLUTag, STC-1, and NCI-H716 cells has been reported [31] -[33] . Food intake stimulates GLP-1 secretion by L cells [34] , and the direct administration of nutrients to the apical lumen of L cells also increases their secretion of GLP-1 [35] [36] . Elrick and co-workers reported in 1964 that insulin secretion induced by oral glucose administration was higher than that induced by intravenous administration [37] . It was proposed
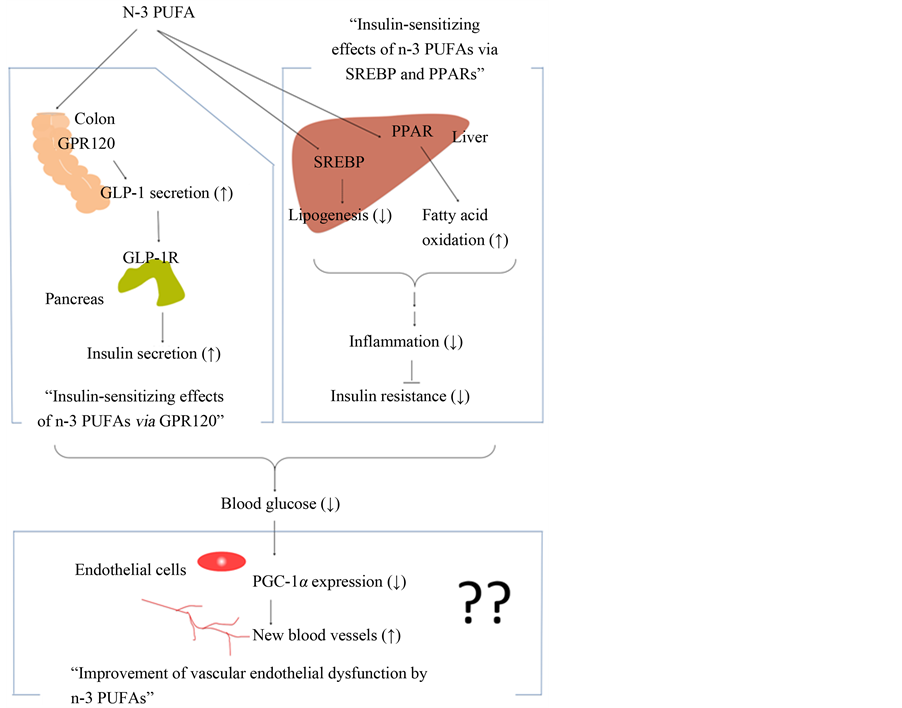
Figure 1. A schematic representation of the effects of n-3 PUFAs on the hypoglycemic action and antidiabetic effects in this review. The role of GPR120, and the role of SREBP and PPAR are described in Section 2 and Section 3, respectively. Improvements in endothelial dysfunction in- duced by n-3 PUFAs are described in Section 4.
that this phenomenon reflects the stimulation of insulin secretion by GLP-1 secreted by L cells in response to oral glucose administration.
Of note, GLP-1-induced insulin release requires hyperglycemia because GLP-1 causes only minimal stimulation of insulin secretion under normal circumstances. The insulinotropic effect of GLP-1 is linked to hyperglycemia, and GLP-1 does not cause hypoglycemia. Several studies have reported a significant reduction in GLP-1 secretion in response to food intake in T2DM patients [38] -[40] . However, the elimination rate of GLP-1 is similar in T2DM patients and in healthy individuals [41] .
Green and co-workers reported that exposure to 5 mM glucose increases GLP-1 secretion, glucose transporter 4 (GLUT4) protein content, and subsequent glycogen synthesis in myocytes. GLP-1 secretion increases the GLUT4 protein level in skeletal muscle or fat, which increases glucose uptake and glycogen synthesis [42] . Thus, stimulation of GLP-1 secretion is one promising approach to inducing antidiabetic effects. Many antidiabetic agents that work by stimulating GLP-1 secretion, such as the GLP-1 agonists exenatide, and liraglutide, have been reported. Studies have shown that the increase in GLP-1 secretion stimulated by mastication leads to suppression of insulin resistance [43] -[45] . GLP-1 secretion by long-chain monounsaturated fatty acids also has been reported in murine, rat, and human L cells [33] [46] [47] . Hirasawa and co-workers showed that stimulation of GPR120 by a-linolenic acid, which was one of the n-3 PUFAs, promotes the secretion of GLP-1 in vitro and in vivo, and increases circulating insulin. GPR120, one of the G-protein coupled receptors, is highly expressed in mature adipose tissue, inflammatory macrophages, and lower intestinal L cells [48] -[51] . Hirasawa and co-workers suggested that n-3 PUFA intake might be useful in the treatment of DM [49] . In fact, Morishita and co-workers reported that direct administration of n-3 PUFAs such as DHA, EPA, and EPA-ethyl esters into the intestine stimulates GLP-1 release in rats [52] -[55] . In their proof of concept study for DHA as DM therapeutics, the strong effects of intracolonic administration of DHA on blood glucose, plasma GLP-1 concentration, and pancreatic islets were clearly demonstrated (Figures 2(a)-(c)) [53] . The blood glucose concentrations were decreased by DHA intracolonic administration, and plasma GLP-1 concentrations tended to be higher in DHA intracolonic administrated mice. In addition, DHA treatment stimulated pancreatic b cells apoptosis and suppressed cell growth in DM mice. Furthermore, insulin sensitivity was improved by a diet containing DHA and EPA for 8 weeks compared with a diet containing linolenic acid [56] . The study suggested the high possibility of DHA or EPA as DM therapeutics as well as importance of their targeting to the lower intestine.
In GPR120-knockdown mice, DHA treatment did not suppress the secretion of inflammatory cytokines such as TNF-a and IL-6, and did not attenuate the release of the chemokine, MCP-1. These changes led to anti-in- flammatory effects and improved insulin sensitivity. These results show clearly that the anti-inflammatory effects and anti-insulin-resistance effects of DHA are GPR120 dependent [49] .
Dysfunctional GPR120 has been reported in both obese rats and humans [57] . A GPR120-specific agonist improved insulin sensitivity in obese mice [58] , indicating that the induction of GLP-1 secretion through GPR120 has antidiabetic effects. n-3 PUFA bound to GPR120 causes suppression of the toll-like receptor pathway and the TNF-a pathway, which leads to anti-inflammatory effects. b-Arrestin 2 plays an important role in these pathways. Luan and co-workers showed that b-arrestin 2 was strongly downregulated in diabetic mouse models and that knockdown of b-arrestin 2 exacerbated insulin resistance, whereas the administration of b-arrestin 2 restored insulin sensitivity in mice [59] . They also showed that insulin stimulated the formation of new b-arres- tin 2 signal complexes, in which b-arrestin 2 scaffolds Akt and Src to the insulin receptor. Loss or dysfunction
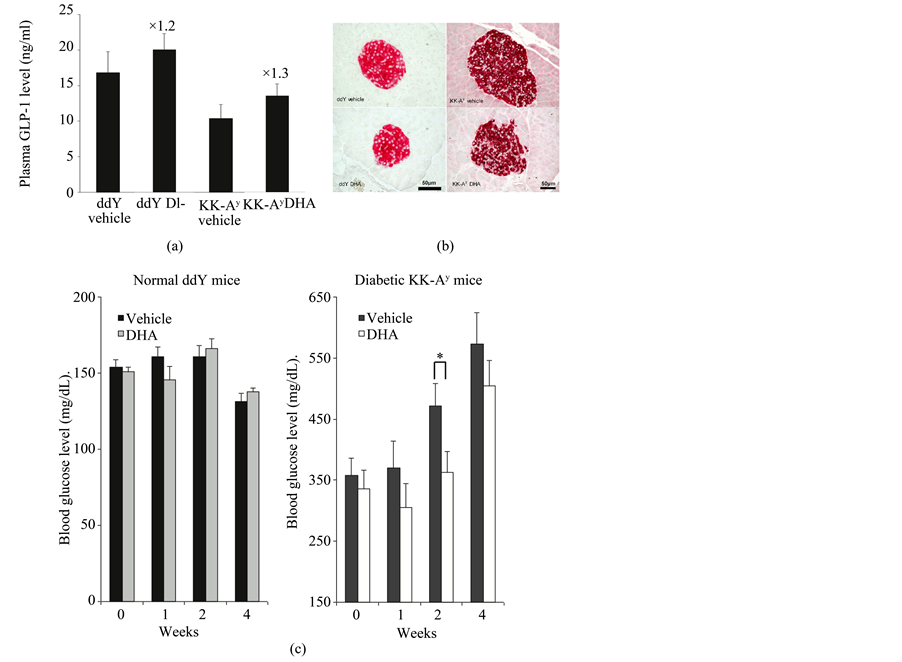
Figure 2. The effects of intracolonic administration of DHA on (a) blood glucose, (b) plasma GLP-1 secretion, and (c) pancreatic islets. Each data po- ints represents the mean ± S.E.M. (n = 9 - 10). *p < 0.05, significant difference between vehicle- and DHA-treatment group. Reproduced with per- mission from [53] .
of b-arrestin 2 causes a deficiency in this signal complex and disturbance of insulin signaling in vivo, thereby contributing to the development of insulin resistance and progression of T2DM.
Whether insulin resistance is improved by n-3 PUFAs is controversial [60] -[62] . Some reports have shown that fish oil intake alleviates insulin resistance by suppressing inflammation caused by macrophages in vitro but not in vivo. Shida and co-workers reported that the improvement in insulin resistance by n-3 PUFAs correlated strongly with the intestinal GPR120 location [53] . To be most effective in controlling the blood glucose level with n-3 PUFAs (or fish intake), n-3 PUFAs must reach the colon or lower intestine. They suggested that n-3 PUFAs must be administrated directly to the colon because orally ingested fatty acids do not easily reach the colon [53] . In addition, Ichimura and co-workers reported a human GPR120 variant [57] and that the risk of obesity or insulin resistance relates to the GPR120 variant. In T2DM, patients with a mutation in the gene encoding GPR120, an n-3 PUFA-rich diet intake did not always improve insulin sensitivity or DM.
Other genetic risk factors may also be involved in the absence of improvement in insulin resistance in response to n-3 PUFA supplementation. For instance, because the brain uptake of DHA is strongly influenced by the apolipoprotein E e4 allele (APOE4), DHA intake has no beneficial effects on cognition in people with APOE4, and this allele is a strong risk factor for AD [19] . Therefore, restoring insulin resistance by providing an n-3 PUFA-rich diet should be considered in future trials of genetic risk factors ideally before the first stage of DM.
3. Insulin-Sensitizing Effects of n-3 PUFAs via SREBP and PPARs
After a meal, the increased circulating levels of glucose and insulin promote de novo fatty acid synthesis and impair b-oxidation, leading to the development of hepatic steatosis. Lipid accumulation in the liver in the form of excess ectopic lipids is caused by elevated levels of circulating serum triacylglycerol and free fatty acids, and is preceded by inflammatory and endoplasmic reticulum stress, which leads to insulin resistance and impaired insulin secretion [63] . N-3 PUFAs suppress hepatic lipid synthesis through the suppression of hepatic SREBP-1 expression by accelerating its transcript decay [64] . For instance, dietary n-3 PUFAs decrease the transcription of the genes encoding hepatic lipogenic or glycolytic enzymes, such as fatty acid synthase, acetyl-CoA carboxylase, stearoyl-CoA desaturase, malic enzyme, l-pyruvate kinase, and glucokinase [65] . Mice fed a high-fat and high-glucose diet for 4 weeks gained weight and exhibited abnormal glucose tolerance, increased serum TNF-a and IL-6 concentrations, and increased expression of hepatic fatty acid synthetase [66] . However, mice fed the high-fat and high-glucose diet supplemented with 1% EPA exhibited normal body weight and levels of serum TNF-a and IL-6. Furthermore, hepatic mRNA of fatty acid synthetase, acetyl-CoA carboxylase, SREBP1c, and PPAR-g were decreased to the control levels.
The expression of lipogenic enzymes or fatty acid synthesis-regulating proteins is suppressed by EPA, which prevents the development of hepatic steatosis and ameliorates insulin resistance. Additionally, n-3 PUFAs stimulate hepatic lipid metabolism through binding to PPARs, which regulates the expression of genes associated with lipid metabolism and adipocyte differentiation. Experiments with PPAR-a-null mice showed mitigation of high-fat diet-induced insulin resistance and improvement in the efficacy of n-3 PUFAs [67] . Wild-type mice fed a high-fat diet containing 27% safflower oil for 2 weeks exhibited a decreased glucose infusion rate (GIR) to half of that of mice fed a normal diet and the emergence of insulin resistance. However, only a limited decrease in GIR was seen in wild-type mice fed the high-fat diet containing 27% safflower oil supplemented with 1% fish oil. By contrast, the high-fat diet containing 27% safflower oil and 1% fish oil fed to PPAR-a-null mice caused hepatic accumulation of triacylglycerol and insulin resistance.
Increased visceral fat deposition alters serum adiponectin level. Serum adiponectin secretion is lower in T2DM patients than in healthy individuals, and this difference is implicated in insulin resistance. Therefore, an increase in serum adiponectin level may indicate an antidiabetic effect. In mice, a diet containing n-3 PUFAs decreased adipose mass, suppressed systemic inflammation, and increased adiponectin transcription in adipose tissue and serum adiponectin concentration [68] -[71] . However, in PPAR-a-null mice, increasing the serum adiponectin concentration did not ameliorate insulin resistance [67] . Taken together, these data suggest that n-3 PUFA intake may attenuate inflammation and endoplasmic reticulum stress, thereby reducing insulin resistance and impaired insulin secretion.
4. Improvement of Vascular Endothelial Dysfunction by n-3 PUFAs
In this section, the authors focus on DM complications. WHO classifies DM complications into two categories: microvascular or damage to small blood vessels, and macrovascular or damage to larger blood vessels [72] . Microvascular complications include damage to the eyes, which can lead to blindness, damage to the kidneys, which can lead to renal failure, and neural damage, which can lead to diabetic foot disorders. Macrovascular complica- tions include cardiovascular diseases such as heart attack, stroke, and insufficient blood flow to the legs. There is evidence from large randomized controlled trials that good metabolic control in people with T1DM or T2DM can delay the onset and progression of these complications. These diabetic complications are based on vascular endothelial dysfunction, which is caused by PPAR-g coactivator 1a (PGC-1a), a member of the transcription coactivator family [73] .
Vascular endothelial dysfunction occurs frequently in DM. PGC-1a protein expression is elevated in vascular endothelial cells from diabetic model mice and DM patients. Sawada and co-workers reported that vascular endothelial cells from diabetic model mice or DM patients, which expressed high levels of PGC-1a protein, exhibited significantly less migration compared with control cells, suggesting that PGC-1a contributes to the decrease in cell migration [73] . Vascular endothelial cells that overexpress PGC-1a display significant repression of migration, as measured in migration assays. PGC-1a activates Notch signaling, which is a powerful inhibitor of endothelial migration and sprouting angiogenesis, and inhibits endothelial cell migration, leading to vascular endothelial growth factor (VEGF) resistance. Sawada and co-workers also showed that endothelial cells that overexpress PGC-1a exhibited significantly blunted formation of new blood vessels and strong inhibition of the rate of recovery of blood flow and reendothelialization in in vivo experiments compared with control cells. Another report has indicated that Notch signaling inhibition can rescue VEGF resistance in diabetic endothelial cells and improve blood flow recovery in the murine hind limb ischemia model [74] . By contrast, lack of PGC- 1a decreased blood flow recovery in the murine hind limb ischemia model.
In skeletal muscle, PGC-1a expression regulates mitochondrial biosynthesis, increases GLUT4 expression, and increases insulin secretion, all of which increase glucose uptake and insulin sensitivity. In other words, the increase in PGC-1a expression in skeletal muscle ameliorates insulin resistance. Conjugated linolenic acid and n-3 PUFAs increase mitochondrial biosynthesis and metabolic rate in skeletal muscle cells [12] . N-3 PUFAs increased PGC-1a expression in skeletal muscle cells by up to 165% of the level in control cells not exposed to n- 3 PUFAs [75] .
To date, there have been no reports on whether PGC-1a expression in endothelial cells is related to antidiabetic effects. In endothelial cells cultured in 25 mM glucose, the PGC-1a mRNA and protein expression levels were doubled compared with control levels, suggesting that a continuous high blood glucose level can increase PGC-1a protein expression in endothelial cells in DM. Because n-3 PUFAs have a hypoglycemic effect, the improvement in endothelial cell dysfunction based on the suppression of PGC-1a expression is expected upon normalization of blood glucose by n-3 PUFAs. The control of PGC-1a expression in endothelial cells by n-3 PUFAs might be a novel therapeutic target for preventing DM complications. In addition, the n-3 PUFA concentration might serve as a DM biomarker. For instance, studies have shown that higher serum n-3 PUFA concentration is associated with a long-term lower risk of T2DM [76] , increased serum and cerebrospinal fluid n-3 PUFA concentrations correlate strongly with a decrease in the phosphorylation of tau protein in cerebrospinal fluid [7] , and serum n-3 PUFA concentration is closely related to the antidiabetic effects of the DPP-4 inhibitors [77] .
5. Conclusions
In this review, the authors have focused on the hypoglycemic and antidiabetic effects of n-3 PUFAs from two points of view. The first is the insulin-sensitizing effect caused by n-3 PUFAs, which is based on GLP-1 secretion mediated by GPR120. Because this effect correlates strongly with the intestinal GPR120 location, targeted delivery of n-3 PUFAs to the colon is essential for the most effective control of blood glucose level by n-3 PUFAs. The second is the insulin-sensitizing effect of n-3 PUFAs mediated by SREBP and PPAR, which alter lipid metabolism and suppress inflammation, and can thereby ameliorate insulin resistance.
Further, the DM complication of blood vessel damage caused by endothelial dysfunction is improved by repressing PGC-1a expression in endothelial cells. We propose that controlling of PGC-1a expression in endothelial cells with n-3 PUFAs might provide a novel therapeutic approach to preventing blood vessel damage as a DM complication. In addition, n-3 PUFA concentration may be useful as a DM biomarker because the n-3 PUFA concentration correlates strongly with DM risk.
Recent human clinical trials with n-3 PUFAs in DM patients are listed in Table 1 as recent human clinical

Table 1. Recent human clinical trials with n-3 PUFAs in diabetes patients.
*ClinicalTrials.gov: available from http://clinicaltrials.gov/ct2/home.
trials with n-3 PUFAs in diabetes patients. Some of them are ongoing. Outcome of these researches is highly expected, and it will clarify the antidiabetic effects of n-3 PUFAs and the role of n-3 PUFAs in the treatment of DM.
References
- World Health Organization (2015) Diabetes. Fact Sheet No. 312. http://www.who.int/mediacentre/factsheets/fs312/en/
- American Diabetes Association (2012) Diagnosis and Classification of Diabetes Mellitus. Diabetes Care, 35, S64-S71.
- de la Monte, S.M. and Wands, J.R. (2008) Alzheimer’s Disease is Type 3 Diabetes-Evidenced Review. Journal of Di- abetes Science and Technology, 2, 1101-11138. http://dx.doi.org/10.1177/193229680800200619
- Duarte, A.I., Candeias, E., Correia, S.C., et al. (2013) Crosstalk between Diabetes and Brain: Glucagon-Like Peptide-1 Mimetics as a Promising Therapy against Neurodegeneration. Biochimica et Biophysica Acta, 1832, 527-541.
- Janson, J., Laedtk, T., Parisi J.E., et al. (2004) Increases Risk of Type 2 Diabetes in Alzheimer’s Disease. Diabetes, 53, 474-481. http://dx.doi.org/10.2337/diabetes.53.2.474
- Takalo, M., Haapasalo, A., Martiskainen, H., et al. (2014) High-Fat Diet Increases Tau Expression in the Brain of T2DM and AD Mice Independently of Peripheral Metabolic Status. Journal of nutritional Biochemistry, 25, 634-641. http://dx.doi.org/10.1016/j.jnutbio.2014.02.003
- Freund, L.Y., Vedin, I., Cederholm, T., et al. (2014) Transfer of Omega-3 Fatty Acids across the Blood-Brain Barrier after Dietary Supplementation with a Docosahexaenoic Acid-Rich Omega-3 Fatty Acid Preparation in Patients with Alzheimer’s Disease: The OmegAD Study. Journal of International Medicine, 275, 428-436. http://dx.doi.org/10.1111/joim.12166
- Weir, G.C., Laybutt, D.R., Kaneto, H., et al. (2001) Beta-Cell Adaptation and Decompensation during the Progression of Diabetes. Diabetes, 50, S154-S159. http://dx.doi.org/10.2337/diabetes.50.2007.S154
- Kanda, H., Tateya, S., Tamori, Y., et al. (2006) MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. Journal of Clinical Investigation, 116, 1494-1505. http://dx.doi.org/10.1172/JCI26498
- Kamei, N., Tobe, K., Suzuki, R., et al. (2006) Overexpression of Macrophage Chemoattractant Protein-1 in Adipose Tissue Cause Macrophage Recruitment and Insulin Resistance. The Journal of Biological Chemistry, 281, 26602-26614. http://dx.doi.org/10.1074/jbc.M601284200
- Flachs, P., Horakova, O., Brauner P., et al. (2005) Polyunsaturated Fatty Acids of Marine Origin up Regulate Mitochondrial Biogenesis and Induce β-Oxidation in White Fat. Diabetologia, 48, 2365-2375. http://dx.doi.org/10.1007/s00125-005-1944-7
- Vaughan, R.A., Garcia-Smith, R., Bisoffiet M., et al. (2012) Conjugated Linoleic Acid or Omega 3 Fatty Acids Increase Mitochondrial Biosynthesis and Metabolism in Skeletal Muscle Cells. Lipid in Health and Disease, 11, 142-152. http://dx.doi.org/10.1186/1476-511X-11-142
- Weisberg, S.P., McCann, D., Desai, M., et al. (2003) Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. Journal of Clinical Investigation, 112, 1796-1808. http://dx.doi.org/10.1172/JCI200319246
- Xu, H., Barnes, G.T., Yang, Q., et al. (2003) Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. Journal of Clinical Investigation, 112, 1821-1830. http://dx.doi.org/10.1172/JCI200319451
- Lazar, M.A. (2006) The Humoral Side of Insulin Resistance. Nature Medicine, 12, 43-44. http://dx.doi.org/10.1038/nm0106-43
- Sell, H. and Eckel, J. (2007) Monocyte Chemotactic Protein-1 and Its Role in Insulin Resistance. Current Opinion in Lipidology, 18, 258-262. http://dx.doi.org/10.1097/MOL.0b013e3281338546
- Sell, H. and Eckel, J. (2009) Chemotactic Cytokines, Obesity and Type 2 Diabetes: In Vivo and in Vitro Evidence for a Possible Causal Correlation? Proceedings of the Nutrition Society, 68, 378-384. http://dx.doi.org/10.1017/S0029665109990218
- Pimentel, G.D., Lira, F.S., Rosa, J.C., et al. (2013) High-Fat Fish Oil Diet Prevents Hypothalamic Inflammatory Profile in Rats. ISRN Inflammation, 2013, Article ID: 419823.
- Vandal, M., Alata, W., Tremblay, C., et al. (2014) Reduction in DHA Transport to the Brain of Mice Expressing Human APOE4 Compared to APOE2. Journal of Neurochemistry, 129, 516-526. http://dx.doi.org/10.1111/jnc.12640
- Afshordel, S., Hagl, S., Werner, D., et al. (2015) Omega-3 Polyunsaturated Fatty Acids Improve Mitochondrial Dysfunction in Brain Aging―Impact of Bcl-2 and NPD-1 Like Metabolites. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 92, 23-31.
- Eckert, G.P., Chang, S., Eckmann, J., et al. (2011) Liposome-Incorporated DHA Increases Neuronal Survival by Enhancing Non-Amyloidogenic APP Processing. Biochimica et Biophysica Acta, 1808, 234-243.
- Wellhauser, L. and Belsham, D.D. (2014) Activation of the Omega-3 Fatty Acid Receptor GPR120 Mediates Anti-In- flammatory Actions in Immortalized Hypothalamic Neurons. Journal of Neuroinflammation, 27, 60. http://dx.doi.org/10.1186/1742-2094-11-60
- Cintra, D.E., Ropelle, E.R., Moraes, J.C., et al. (2012) Unsaturated Fatty Acids Revert Diet-Induced Hypothalamic Inflammation in Obesity. PLoS ONE, 7, e30571. http://dx.doi.org/10.1371/journal.pone.0030571
- Zhao, Y., Calon, F., Julien, C., et al. (2011) Docosahexaenoic Acid-Derived Neuroprotectin D1 Induces Neuronal Survival via Secretase- and PPARγ-Mediated Mechanisms in Alzheimer’s Disease Models. PLoS ONE, 6, e15816. http://dx.doi.org/10.1371/journal.pone.0015816
- Wall, R., Ross, R.P., Fitzgerald, G.F. and Stanton, C. (2010) Fatty Acids from Fish: The Anti-Inflammatory Potential of Long-Chain Omega-3 Fatty Acids. Nutrition Reviews, 68, 280-289. http://dx.doi.org/10.1111/j.1753-4887.2010.00287.x
- Bang, H.O. and Dyerberg, J. (1972) Plasma Lipids and Lipoproteins in Greenlandic West Coast Eskimos. Acta Medica Scandinavica, 192, 85-94. http://dx.doi.org/10.1111/j.0954-6820.1972.tb04782.x
- Kromann, N. and Green, A. (1980) Epidemiological Studies in the Upernavik District, Greenland. Incidence of Some Chronic Diseases 1950-1974. Acta Medica Scandinavica, 208, 401-406. http://dx.doi.org/10.1111/j.0954-6820.1980.tb01221.x
- Lim, G.E. and Brubaker, P.L. (2006) Glucagon-Like Peptide 1 Secretion by the L-Cell. The View from Within. Diabetes, 55, S70-S77. http://dx.doi.org/10.2337/db06-S020
- Kieffer, T.J., McIntosh, C.H. and Pederson, R.A. (1995) Degradation of Glucose-Dependent Insulinotropic Polypeptide and Truncated Glucagon-Like Peptide 1 in Vitro and in Vivo by Dipeptidyl Peptidase IV. Endocrinology, 136, 3585- 3596.
- Holst, J.J. (2006) Glucagon-Like Peptide-1: From Extract to Agent: The Claude Bernard Lecture, 2005. Diabetologia, 49, 253-260. http://dx.doi.org/10.1007/s00125-005-0107-1
- Drucker, D.J., Jin, T., Asa, S.L., et al. (2006) Activation of Proglucagon Gene Transcription by Protein Kinase-A in a Novel Mouse Enteroendocrine Cell Line. Molecular Endocrinology, 8, 1646-1655.
- Abello, J., Ye, F., Bosshard, A., et al. (1994) Stimulation of Glucagon-Like Peptide-1 Secretion by Muscarinic Agonist in a Murine Intestinal Endocrine Cell Line. Endocrinology, 134, 2011-2017.
- Reimer, R.A., Darimont, C., Gremlich, S., et al. (2001) A Human Cellular Model for Studying the Regulation of Glucagon-Like Peptide-1 Secretion. Endocrinology, 142, 4522-4528. http://dx.doi.org/10.1210/endo.142.10.8415
- Drucker, D.J. (2006) The Biology of Incretin Hormones. Cell Metabolism, 3, 153-165. http://dx.doi.org/10.1016/j.cmet.2006.01.004
- Rocca, A.S. and Brubaker, P.L. (1999) Role of the Vagus Nerve in Mediating Proximal Nutrient-Induced Glucagon- Like Peptide-1 Secretion. Endocrinology, 140, 1687-1694.
- Roberge, J.N. and Brubaker, P.L. (1993) Regulation of Intestinal Proglucagon-Derived Peptide Secretion by Glucose- Dependent Insulinotropic Peptide in a Novel Enteroendocrine Loop. Endcrinology, 133, 233-240.
- Elrick, H., Stimmler, L., Hlad Jr., C.J. and Rai, Y. (1964) Plasma Insulin Responses to Oral and Intravenous Glucose Administration. Journal of Clinical Endocrinology Metabolism, 24, 1076-1082. http://dx.doi.org/10.1210/jcem-24-10-1076
- Toft-Nielsen, M.B., Damholt, M.B., Madsbad, S., et al. (2001) Determinants of the Impaired Secretion of Glucagon- Like Peptide-1 in Type 2 Diabetic Patients. Journal of Clinical Endocrinology Metabolism, 86, 3717-3723. http://dx.doi.org/10.1210/jcem.86.8.7750
- Vilsbøll, T., Krarup, T., Deacon, C.F., et al. (2001) Reduced Postprandial Concentrations of Intact Biologically Active Glucagon-Like Peptide 1 in Type 2 Diabetic Patients. Diabetes, 50, 609-613. http://dx.doi.org/10.2337/diabetes.50.3.609
- Muscelli, E., Mari, A., Casolaro, A., et al. (2008) Separate Impact of Obesity and Glucose Tolerance on the Incretin Effect in Normal Subjects and Type 2 Diabetic Patients. Diabetes, 57, 1340-1348. http://dx.doi.org/10.2337/db07-1315
- Vilsbøll, T., Agersø, H., Krarup, T. and Holst, J.J. (2003) Similar Elimination Rates of Glucagon-Like Peptide-1 in Obese Type 2 Diabetic Patients and Healthy Subjects. Journal of Clinical Endocrinology and Metabolism, 88, 220-224. http://dx.doi.org/10.1210/jc.2002-021053
- Green, C.J., Henriksen, T.I., Pedersen, B.K. and Solomon, T.P. (2012) Glucagon Like Peptide-1-Induced Glucose Metabolism in Differentiated Human Muscle Satellite Cells Is Attenuated by Hyperglycemia. PLoS ONE, 7, e44284. http://dx.doi.org/10.1371/journal.pone.0044284
- Sonoki, K., Iwase, M. Takata, Y., et al. (2013) Effect of Thirty-Times Chewing per Bite on Secretion of Glucagon- Like Peptide-11 in Health Volunteers and Type 2 Diabetic Patients. Endocrine Journal, 60, 311-319. http://dx.doi.org/10.1507/endocrj.EJ12-0310
- Tsuchiya, M., Niijima-Yaoita, F., Yoneda, H., et al. (2014) Long-Term Feeding on Powdered Food Causes Hyperglycemia and Signs of Systemic Illness in Mice. Life Science, 103, 8-14. http://dx.doi.org/10.1016/j.lfs.2014.03.022
- Yamazaki, T., Yamori, M., Asai, K., et al. (2013) Mastication and Risk for Diabetes in Japanese Population: A Cross-Sectional Study. PLoS ONE, 8, e4113. http://dx.doi.org/10.1371/journal.pone.0064113
- Rocca, A.S. and Brubaker, P.L. (1995) Stereospecific Effects of Fatty Acids on Proglucagon-Derived Peptide Secretion in Fetal Rat Intestinal Cultures. Endocrinology, 136, 5593-5599.
- Brubaker, P.L., Schloos, J. and Drucker, D.J. (1998) Regulation of Glucagon-Like Peptide-1 Synthesis and Secretion in the GLUTag Enteroendocrine Cell Line. Endocrinology, 139, 4108-4114.
- Oh, D.Y., Talukdar, S., Bae, E.J., et al. (2010) GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-In- flammatory and Insulin-Sensitizing Effects. Cell, 142, 687-698. http://dx.doi.org/10.1016/j.cell.2010.07.041
- Hirasawa, A., Tsumaya, K., Awaji, T., et al. (2005) Free Fatty Acids Regulate Gut Incretin Glucagon-Like Peptide-1 Secretion through GPR120. Nature Medicine, 11, 90-94. http://dx.doi.org/10.1038/nm1168
- Katsuma, S., Hatae, N., Yano, T., et al. (2005) Free Fatty Acids Inhibit Serum Deprivation-Induced Apoptosis through GPR120 in a Murine Enteroendocrine Cell Line STC-1. Journal of Biological Chemistry, 280, 19507-19515. http://dx.doi.org/10.1074/jbc.M412385200
- Adachi, T., Tanaka, T., Takemoto, K., et al. (2006) Free Fatty Acids Administrated into the Colon Promote the Secretion of Glucagon-Like Peptide-1 and Insulin. Biochemical and Biophysical Research Communications, 340, 332-357. http://dx.doi.org/10.1016/j.bbrc.2005.11.162
- Morishita, M., Tanaka, T., Shida, T. and Takayama, K. (2008) Usefulness of Colon Targeted DHA and EPA as Novel Diabetes Medications That Promote Intrinsic GLP-1 Secretion. Journal of Controlled Release, 132, 99-104. http://dx.doi.org/10.1016/j.jconrel.2008.09.001
- Shida, T., Kamei, N. and Takeda-Morishita, M. (2013) Colonic Delivery of Docosahexaenoic Acid Improves Impaired Glucose Tolerance via GLP-1 Secretion and Suppresses Pancreatic Islet Hyperplasia in Diabetic KK-Ay Mice. International Journal of Pharmacology, 450, 63-69. http://dx.doi.org/10.1016/j.ijpharm.2013.04.029
- Morishita, M., Kajita, M., Suzuki, A., et al. (2000) The Dose-Related Hypoglycemic Effects of Insulin Emulsions Incorporating Highly Purified EPA and DHA. International Journal of Pharmacology, 201, 175-185. http://dx.doi.org/10.1016/S0378-5173(00)00411-7
- Suzuki, A., Morishita, M., Kajita, M., et al. (1998) Enhanced Colonic and Rectal Absorption of Insulin Using a Multiple Emulsion Containing Eicosapentaenoic Acid and Docosahexaenoic Acid. Journal of Pharmaceutical Sciences, 87, 1196-1202. http://dx.doi.org/10.1021/js980125q
- Andersen, G., Harnack, K., Erbersdobler, H.F. and Somoza, V. (2008) Dietary Eicosapentaenoic Acid and Docosahexaenoic Acid Are More Effective than Alpha-Linolenic Acid in Improving Insulin Sensitivity in Rats. Annals of Nutrition and Metabolism, 52, 250-256. http://dx.doi.org/10.1159/000140518
- Ichimura, A., Hirasawa, A., Poulain-Godefroy, O., et al. (2012) Dysfunction of Lipid Sensor GPR120 Leads to Obesity in Both Mouse and Human. Nature, 483, 350-354. http://dx.doi.org/10.1038/nature10798
- Oh, D.Y., Walenta, E., Akiyama, T.E., et al. (2014) A GPR120-Selective Agonist Improves Insulin Resistance and Chronic Inflammation in Obese Mice. Nature Medicine, 20, 942-947. http://dx.doi.org/10.1038/nm.3614
- Luan, B., Zhao, J., Wu, H., et al. (2009) Deficiency of A Beta-Arrestin-2 Signal Complex Contributes to Insulin Re- sistance. Nature, 457, 1146-1149. http://dx.doi.org/10.1038/nature07617
- Spencer, M., Finlin, B.S., Unal, R., et al. (2013) Omega-3 Fatty Acids Reduce Adipose Tissue Macrophages in Human Subjects with Insulin Resistance. Diabetes, 62, 1709-17171. http://dx.doi.org/10.2337/db12-1042
- de Caterina, R., Madonna, R., Bertolotto, A. and Schmidt, E.B. (2007) N-3 Fatty Acids in the Treatment of Diabetic Patients. Diabetes Care, 30, 1012-1026. http://dx.doi.org/10.2337/dc06-1332
- Labonté, M.È., Couture, P., Tremblay, A.J., Hogue, J.C., Lemelin, V. and Lamarche, B. (2013) Eicosapentaenoic and Docosahexaenoic Acid Supplementation and Inflammatory Gene Expression in the Duodenum of Obese Patients with Type 2 Diabetes. Nutrition Journal, 12, 98. http://dx.doi.org/10.1186/1475-2891-12-98
- Brookheart, R.T., Michel, C.T. and Schaffer, J.F. (2009) As a Matter of Fat. Cell Metabolism, 10, 9-12. http://dx.doi.org/10.1016/j.cmet.2009.03.011
- Xu, J., Teran-Garcia, M., Park, J.H., et al. (2001) Polyunsaturated Fatty Acids Suppress Hepatic Sterol Regulatory Element-Binding Protein-1 Expression by Accelerating Transcript Decay. Journal of Biological Chemistry, 276, 9800- 9807. http://dx.doi.org/10.1074/jbc.M008973200
- Xu, J., Nakamura, M.T., Cho, H.P. and Clarke, S.D. (2007) Sterol Regulatory Element Binding Protein-1 Expression Is Suppressed by Dietary Polyunsaturated Fatty Acids. Journal of Biological Chemistry, 274, 23577-23583. http://dx.doi.org/10.1074/jbc.274.33.23577
- Liu, X., Xue, Y., Liu, C., et al. (2013) Eiocasapentaenoic Acid-Enriched Phospholipid Ameliorates Insulin Resistance and Lipid Metabolism in Diet-Induced-Obese Mice. Lipid in Health and Disease, 12, 109. http://dx.doi.org/10.1186/1476-511X-12-109
- Neschen, S., Morino, K., Dong, J., et al. (2007) N-3 Fatty Acids Preserve Insulin Sensitivity in Vivo in a Peroxisome Proliferator-Activated Receptor-α-Dependent Manner. Diabetes, 56, 1034-1041. http://dx.doi.org/10.2337/db06-1206
- Wu, J.H., Cahill, L.E. and Mozaffarian, D. (2013) Effects of Fish Oil on Circulating Adiponectin: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Journal of Endocrinology and Metabolism, 98, 2451-2459. http://dx.doi.org/10.1210/jc.2012-3899
- Flachs, P., Mohamed-Ali, V. and Horakova, O. (2006) Polyunsaturated Fatty Acids of Marine Origin Induce Adiponectin in Mice Fed a High-Fat Diet. Diabetologia, 49, 394-397. http://dx.doi.org/10.1007/s00125-005-0053-y
- Banga, A., Unal, R., Tripathi, P., et al. (2009) Adiponectin Translation Is Increased by the PPARgamma Agonist Pioglitazone and Omega-3 Fatty Acids. American Journal of Physiology, Endocrinology and Metabolism, 296, E480-E489. http://dx.doi.org/10.1152/ajpendo.90892.2008
- Tishinsky, J.M., Ma, D.W. and Robinson, L.F. (2011) Eicosapentaenoic Acid and Rosiglitazone Increase Adiponectin in an Additive and PPARγ-Dependent Manner in Human Adipocytes. Obesity, 19, 262-268. http://dx.doi.org/10.1038/oby.2010.186
- World Health Organization. Diabetes Programme [Article Online]. http://www.who.int/diabetes/action_online/basics/en/index3.html
- Sawada, N., Jiang, A., Takizawa, F., et al. (2014) Endothelial PGC-1α Mediates Vascular Dysfunction in Diabetes. Cell Metabolism, 19, 246-258. http://dx.doi.org/10.1016/j.cmet.2013.12.014
- Cao, L., Aran, P.R., Kim, J., et al. (2010) Modulating Notch Signaling to Enhance Neovascularization and Reperfusion in Diabetic Mice. Biomaterials, 31, 9048-9056. http://dx.doi.org/10.1016/j.biomaterials.2010.08.002
- Bryner, R.W., Woodworth-Hobbs, M.E., Williamson, D.L. and Always, S.E. (2012) Docosahexaenoic Acid Protects Muscle Cells from Palmitate-Induced Atrophy. ISRN Obesity, 2012, Article ID: 647348.
- Virtanen, J.K., Mursu, J., Voutilainen, S., Uusitupa, M. and Tuomainen, T.P. (2014) Serum Omega-3 Polyunsaturated Fatty Acids and Risk of Incident Type 2 Diabetes in Men: The Kuopio Ischemic Heart Disease Risk Factor Study. Diabetes Care, 37, 1189-1196. http://dx.doi.org/10.2337/dc13-1504
- Iwasaki, M., Hoshian, F., Tsuji, T., et al. (2012) Predicting Efficacy of Dipeptidyl Peptidase-4 Inhibitors in Patients with Type 2 Diabetes: Association of Glycated Hemoglobin Reduction with Serum Eicosapentaenoic Acid and Docosahexaenoic Acid Levels. Journal of Diabetes Investigation, 3, 464-467. http://dx.doi.org/10.1111/j.2040-1124.2012.00214.x
- Samimi, M., Jamilian, M., Asemi, Z. and Esmaillzadeh, A. (2014) Effects of Omega-3 Fatty Acid Supplementation on Insulin Metabolism and Lipid Profiles in Gestational Diabetes: Randomized, Double-Blind, Placebo-Controlled Trial. Clinical Nutrition, in press.
- Serhiyenko, V., Serhiyenko, A. and Segin, V. (2014) The Effect of Omega-3 Polyunsaturated Fatty Acids on N-Ter- minal Pro-Brain Natriuretic Peptide and Lipid Concentration in Patients with Type 2 Diabetes Mellitus and Cardiovascular Autonomic Neuropathy. Romanian Journal of Diabetes Nutrition and Metabolic Diseases, 21, 97-101. http://dx.doi.org/10.2478/rjdnmd-2014-0014
NOTES

*Corresponding author.